
Abstract
Metabolic reprogramming is considered hallmarks of cancer. Aerobic glycolysis in tumors cells has been well-known for almost a century, but specific factors that regulate lactate generation and the effects of lactate in both cancer cells and stroma are not yet well understood. In the present study using breast cancer cell lines, human primary cultures of breast tumors, and immune deficient murine models, we demonstrate that the POU1F1 transcription factor is functionally and clinically related to both metabolic reprogramming in breast cancer cells and fibroblasts activation. Mechanistically, we demonstrate that POU1F1 transcriptionally regulates the lactate dehydrogenase A (LDHA) gene. LDHA catalyzes pyruvate into lactate instead of leading into the tricarboxylic acid cycle. Lactate increases breast cancer cell proliferation, migration, and invasion. In addition, it activates normal-associated fibroblasts (NAFs) into cancer-associated fibroblasts (CAFs). Conversely, LDHA knockdown in breast cancer cells that overexpress POU1F1 decreases tumor volume and [18F]FDG uptake in tumor xenografts of mice. Clinically, POU1F1 and LDHA expression correlate with relapse- and metastasis-free survival. Our data indicate that POU1F1 induces a metabolic reprogramming through LDHA regulation in human breast tumor cells, modifying the phenotype of both cancer cells and fibroblasts to promote cancer progression.
Similar content being viewed by others

Introduction
Cellular metabolism reprogramming is a hallmark of cancer cells [1, 2]. Metabolic plasticity includes aerobic glycolysis, also called the Warburg effect, a well-known feature of tumors early recognized by Otto Warburg [3]. The glycolysis occurring in cancer cells gives rise to a high-rate glucose uptake and the conversion of pyruvate into lactate, instead of converting into Acetyl CoA and leading to the tricarboxylic acid (TCA) cycle and mitochondrial oxidative phosphorylation (OXPHOS). Moreover, the lactate output to the extracellular space results in acidification of tumor microenvironment (TME) [4,5,6]. In addition to the key role of lactate in tumor cell maintenance [7, 8], lactate is known to affect stromal cells in the TME. For example, in immune cells, lactate induces M2-like polarization of tumor-associated macrophages (TAM) and TAM-induced angiogenesis [9]. It also impairs T and natural killer cell activation and compromises dendritic cell differentiation and maturation that suppresses anticancer immune responses [10, 11]. In endothelial cells, lactate has been demonstrated to increase angiogenesis [12]. However, the role of lactate on other stromal cell populations has not yet been fully elucidated. For example, administration of exogenous lactate in breast cancer cells has been shown to induce the cancer stem cell (CSC) phenotype [13], but it is not clear which source of lactate could contribute to inducing stemness: the cancer cell-derived lactate, the CSC-produced lactate, or both. Regarding fibroblasts, cancer cells reprogram the surrounding fibroblasts to induce aerobic glycolysis. In turn, fibroblasts produce lactate that can be taken up by cancer cells and oxidized in the mitochondria for energy production and tumor progression. This metabolic coupling is referred to as the “reverse Warburg effect” [14]. In addition, a recent study indicated that lactate secreted by pancreatic ductal adenocarcinoma cells reprogram mesenchymal stem cells epigenetically to differentiate into cancer-associated fibroblasts (CAFs), thus leading to tumor progression [15].
Lactate dehydrogenase A (LDHA) is the key enzyme generating lactate from pyruvate. HIF-1, c-Myc, and p53 are likely the most widely studied lactate regulators. Upregulation of HIF-1 and c-Myc and suppression of p53 are responsible for the metabolic switch to glycolysis in cancer cells [16]. LDHA overexpression promotes cell proliferation and invasion in pituitary adenomas [17], whereas reduction in LDHA activity compromises the ability of tumor cells to proliferate under hypoxia and severely diminishes tumorigenicity and tumor cell maintenance [7].
Transcriptional regulation of prolactin (PRL) and growth hormone (GH) in the pituitary gland by the POU1F1 transcription factor is well known [18, 19]. However, POU1F1 is also expressed in non-pituitary tissues, such as human breast, where it also regulates GH and PRL [20, 21]. High POU1F1 levels in breast cancer cells induce cell proliferation, reduce apoptosis, and increase migration and invasion [22]. In fact, high expression of POU1F1 in breast cancer correlates with poor clinical outcome [23]. Hypomethylation profiles in clusters of circulating breast cancer cells have recently been identified in binding sites for several transcription factors related to stemness and proliferation, including POU1F1. This epigenetic mechanism has been related to metastasis seeding [24].
The current study analyzes the role of POU1F1 in breast cancer cell metabolism. Human breast cancer cell lines and primary human breast tumors were used to evaluate the effect of POU1F1 overexpression and POU1F1 knockdown in the glycolysis pathway. Using immunodeficient mice, we studied how cancer cells with POU1F1 overexpression and LDHA blockade could affect tumor growth and glucose uptake. In patients, POU1F1 and LDHA mRNA expression was correlated with breast cancer clinical outcome. Finally, in primary cultures of human breast tumors, we studied the effect of both POU1F1 and LDHA on fibroblast activation.
Results
POU1F1 in breast cancer cells induces metabolic reprogramming
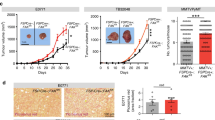
Bioinformatic analyses of human breast cancer datasets were carried out for glycolytic activity and other processes related with proliferation and metabolism (Fig. 1A–C). Glycolytic activity was found to be higher in breast tumors than in normal breast (GSE109169), in triple-negative breast cancer (TNBC) than in luminal A tumors (GSE45827), and in bone metastasis than in primary tumors (GSE103357). Interestingly, glycolytic activity was also found to be higher in the breast cancer subtypes Luminal B and HER2 as compared to luminal A (Supplementary Fig. S1A–B). Given that POU1F1 is related to pituitary gland development and cancer progression, we analyzed these biological processes in the context of breast cancer. We carried out an unbiased transcriptomic analysis by performing a microarray (GSE64101) in the control luminal A subtype of MCF7 cells with low endogenous POU1F1 expression (MCF7) and after transient transfection by a POU1F1 overexpression vector (MCF7-POU1F1). To analyze these data, a gene-set enrichment analyses was performed. We found that POU1F1 overexpression induced a clear enrichment of glycolysis signature (NES = 2.01, P < 0.001) (Fig. 1D). Next, we selected well-known enzymes and carriers involved in the glycolytic pathway (Fig. 1E) and a real-time PCR was carried out before and after POU1F1 overexpression. Our data confirmed significant upregulation in mRNA expression of eight glycolysis-related genes: HK2 (hexokinase 2), GAPDH (glyceraldehyde 3-phosphate dehydrogenase), PGK1 (phosphoglycerate kinase 1), PGAM1 (phosphoglycerate mutase), ENO1 (enolase 1), ENO2, PKM2 (pyruvate kinase M2), and LDHA (lactate dehydrogenase A). Downregulation of PFK1 (6-phosphofructokinase) and no changes of GPI (glucose-6-phosphate isomerase), or ALDOA (aldolase A) glycolysis-related genes (Fig. 1F) were observed. In addition, two key transporters in glucose metabolism, SLC2A1 (glucose transporter 1, GLUT1), and SLC2A4 (GLUT4), significantly decreased and increased, respectively, after POU1F1 overexpression. The mRNA expression of SLC16A3 (monocarboxylate transporter 4, MCT4), which is responsible for transporting lactate out of cells, also significantly increased after POU1F1 overexpression (Fig. 1F). Furthermore, the C12orf5 gene which codes for the TP53-inducible glycolysis and apoptosis regulator (TIGAR) protein, a well-known glycolysis p53-mediator [25, 26], decreases after POU1F1 overexpression, and this could suggest a possible role for TIGAR in POU1F1-induced glycolysis. To analyze possible changes in secreted metabolites, conditioned medium (CM) from MCF7 and MCF7-POU1F1 cells after 24 h of culture was analyzed by 1H-NMR assay (Supplementary Fig. S2, A-B, and Supplementary Table S1). Glycolytic- and TCA-derived metabolites showed dramatic changes after POU1F1 overexpression, with a significant decrease in glucose and a significant increase in lactate (both P < 0.001) (Fig. 1G). Several TCA-derived metabolites also presented significantly reduced levels in CM-MCF7-POU1F1 cells (Fig. 1G). Altogether, our data indicate a clear Warburg effect induced by POU1F1., i.e., pyruvate to lactate production increased, while pyruvate to TCA-cycle products decreased at least at the extracellular level. To confirm the functional relevance of our findings, glycolytic activity of breast cancer cells was assayed by measuring extracellular acidification rate (ECAR), quantification of basal glycolysis, compensatory glycolysis, and mitochondrial oxygen consumption rate (OCR)/proton extrusion rate (glycoPER) basal ratio in both MCF7 and MCF7-POU1F1 cells. The data obtained show that POU1F1 significantly increased the glycolytic profile as compared to control MCF7 cells (Fig. 2A, B). Conversely, knockout of POU1F1 (MDAsgPOU1F1) using the CRISPR methodology in the MDA-MB-231 cell line (triple-negative subtype, with high endogenous levels of POU1F1) presented a significant decrease in the glycolytic profile compared to control cells (MDAsgC) (Fig. 2C, D and Supplementary Fig. S3). Primary cultures of two human breast tumors with low POU1F1 protein expression (PDT 1) and high expression (PDT 6) (see below) were selected for glycolytic activity analyses. As shown in Fig. 2E, F, similar data were obtained for the human breast tumors and the breast cancer cell lines, which strongly suggests that POU1F1 regulates the glycolysis pathway in breast tumors.

A–C Dataset enrichment graphs related with metabolic and proliferation process categories from Hallmark collection (up) and GSEA plot of enrichment in Glycolysis geneset (down). Gene expression data obtained from GSE109169, GSE45827, and GSE103357 as indicated: A Tumor breast cancer patient samples (n = 25) vs normal breast (n = 25); B TNBC breast tumors (n = 41) vs Luminal A breast tumors (n = 29); C Breast cell line derived from bone metastasis patient (n = 3) vs primary tumor cells (n = 2). FDR false discovery rate, NES normalized enrichment score. D Dataset enrichment graphs related with metabolic, proliferation, and development process categories from Hallmark collection (left) and GSEA plot of enrichment in Glycolysis geneset (right) of microarray data from POU1F1-overexpressing MCF7 cells vs MCF7 cells (GSE64101). E Glycolysis pathway and catalytic enzymes (in red). F qPCR of glycolytic enzymes, carriers, and factors involved in the glycolytic pathway in control and POU1F1-overexpressing MCF7 cells. POU1F1, HK2 (hexokinase 2), GPI (glucose-6-phosphate isomerase), PFK1 (6-phosphofructokinase), ALDOA (aldolase A), GAPDH (glyceraldehyde 3-phosphate dehydrogenase), PGK1 (phosphoglycerate kinase 1), PGAM1 (phosphoglycerate mutase), ENO1 (enolase 1), ENO2, PKM2 (pyruvate kinase M2), LDHA (lactate dehydrogenase A), SLC2A1 (glucose transporter 1, GLUT1), SLC2A4 (GLUT4), SLC16A3 (monocarboxylate transporter 4, MCT4), and C12orf5 (TIGAR). G 1H-NMR assay of glycolytic- and TCA-derived metabolites in conditioned medium (CM) of MCF7 and MCF7-POU1F1 cells after 24 h culture. *FDR-adjusted p value (q-value) < 0.05; **< 0.01; ***0.001; ns not significant.

A Representative ECAR Glycolytic Rate Assay profile in MCF7 and MCF7-POU1F1 cells. cs cells. B Quantification of basal glycolysis, compensatory glycolysis and mitoOCR/glycoPER basal ratio in MCF7 and MCF7-POU1F1 cells. C Representative ECAR Glycolytic Rate Assay profile in control (MDAsgC) and POU1F1-knocked-out MDA-MB-231 (MDAsgPOU1F1) cells. D Quantification of basal glycolysis, compensatory glycolysis and mitoOCR/glycoPER basal ratio in MDAsgC and MDAsgPOU1F1 cells. E Representative ECAR Glycolytic Rate Assay profile in human primary breast tumor-derived cultures (see Fig. 6D) with low POU1F1 expression (PDT 1) and high POU1F1 expression (PDT 6). F Quantification of basal glycolysis, compensatory glycolysis and mitoOCR/glycoPER basal ratio in PDT 1 and PDT 6. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
POU1F1 regulates LDHA expression in breast cancer
LDHA is a key enzyme in the glycolysis pathway, transforming pyruvate into lactate. Aberrantly high expression of LDHA has been demonstrated in multiple cancers, including breast cancer, and it has been associated with malignant tumor progression. In fact, bioinformatic search of human breast cancer tumors and normal breast samples (GSE22820) revealed a significant (P < 0.001) increase of LDHA mRNA expression in human breast tumors as compared with normal mammary tissue (Fig. 3A). Using the same database, we also classified breast tumors and normal samples according to POU1F1 mRNA expression (Fig. 3B). Further analyses also showed a significant (P < 0.05) correlation between POU1F1 and LDHA mRNA expression (Fig. 3C, D).

A Dispersion plot of LDHA mRNA levels in human breast tumors (n = 176) and normal tissues (n = 10) (GSE22820). B, C POU1F1 and LDHA mRNA levels according to POU1F1 mRNA levels (high POU1F1: higher than 75th percentile, and lower POU1F1: with levels below 25th percentile). D Spearman correlation analysis of POU1F1 and LDHA mRNA expression (log2) (n = 186). E, F JASPAR analysis indicates two POU1F1 binding sites in the LDHA gene at positions −76/−62 bp and +276/+290 bp with respect to transcription start site. DEL1 and DEL2 indicate deletion of POU1F1 binding sites. G The wild LDHA promoter (−101 to +315 bp from the transcription start site; LDHApromoter-WT) and the LDHA promoter with specific deletions at the POU1F1 binding sites (LDHApromoter-DEL1 and LDHApromoter-DEL2) were subcloned into the pRP vector, transfected in HEK 293 cells, and co-transfected with the control (pDream) or the pDream-POU1F1 overexpression vector for 48 h, and luciferase activity was measured. Normalized relative luciferase units (RLU) were calculated as the ratio of luciferase activity in control and POU1F1-transfected cells. H H3K27Ac occupancy of LDHA promoter in human breast cancer cell lines (GSE69112), analyzed by ChIPseq (MCF7, ZR75 and T47D: Luminal A; CAL51, SUM159, and MDA-MB-231: Triple Negative Breast Cancer). I MCF7 cells were transiently transfected with the pDream-POU1F1 overexpression vector and 72 h later a qPCR was carried out to evaluate LDHA and POU1F1 mRNA expression. J Western blot of LDHA, POU1F1, and β-actin in MCF7 cells after transient POU1F1 overexpression (n = 2). K LDHA mRNA in MDA-MB-231 cells after POU1F1 knockdown for 72 h. L WB of POU1F1, LDHA, and β-actin in MDA-MB-231 cells after POU1F1 knockdown (n = 2). M Representative images of POU1F1 and LDHA after confocal microscopy in MCF7 and MDA-MB-231 cells after stable POU1F1 overexpression (MCF7-POU1F1) and POU1F1 knockout (MDAsgPOU1F1). DAPI was used as nuclei marker. Scale bar: 15 μm. Data are expressed as mean ± SEM. **P < 0.01, and ***P < 0.001.
In order to evaluate the hypothesis that POU1F1 could regulate the LDHA gene at transcriptional level, we searched for potential binding sites of POU1F1 in the LDHA promoter using the JASPAR database (http://jaspar.genereg.net/), and found two putative motifs for POU1F1 in the LDHA gene promoter region (Fig. 3E). These two consensus elements for POU1F1 were located at −76 to −62 bp upstream and +276 to +290 bp downstream from the start transcription site in the LDHA promoter (Fig. 3F). To evaluate the possible transcriptional regulation of LDHA by POU1F1, a luciferase reporter assay in the absence or presence of POU1F1 was carried out using three plasmids: (a) the wild LDHA promoter (LDHApromoter-WT, −101 to +315 bp), (b) a plasmid with deletion of the first POU1F1 binding site (LDHApromoter-DEL1, deletion of −76 to −62 bp), and (c) a plasmid with deletion of the second POU1F1 binding site (LDHApromoter-DEL2, deletion of +276 to +290 bp). HEK 293 cells co-transfected with the LDHApromoter-WT and the POU1F1 overexpression vector significantly increased luciferase activity as compared with control (P < 0.01), whereas deletion of both the first and the second POU1F1 binding sites significantly reduced luciferase activity (P < 0.01 and P < 0.001) (Fig. 3G). In fact, interrogation of the epigenetic status of LDHA gene promoter in breast cancer cell lines showed a peak enrichment in H3K27ac, an epigenetic mark associated with higher activation of transcription (Fig. 3H). Furthermore, transient POU1F1 overexpression in MCF7 cells significantly (P < 0.001) increased LDHA mRNA expression (Fig. 3I). Western blots also indicated a visible increase in LDHA protein expression after POU1F1 overexpression in both transient (Fig. 3J) and stable (Supplementary Fig. S4A) MCF7 cells. Conversely, transient knockdown of POU1F1 using a pLKO-shPOU1F1 vector in MDA-MB-231 cells significantly decreased LDHA mRNA (P < 0.001, Fig. 3K) and protein (Fig. 3L) levels. Confocal microscopy of MCF7 control cells transfected stably with the pTRE2 control vector (MCF7), MCF7 cells transfected stably with the pTRE2-POU1F1 vector (MCF7-POU1F1), MDA-MB-231 control cells transfected with the pLentiCRISPRv2 control (MDAsgC), and after POU1F1 knockout (MDAsgPOU1F1), indicated a correlation between POU1F1 (in red) and LDHA (in green) immunodetection (Fig. 3M). Supplementary Fig. S4B also shows a decrease in LDHA protein expression after knockout of POU1F1 in MDA-MB-231 cells using three different CRISPR clones.
POU1F1-induced lactate facilitates cancer progression in breast cancer cells
To evaluate the effect of lactate on breast cancer cells, glycolytic activity (ECAR and basal glycolysis) was assayed in stable MCF7 overexpressing POU1F1 cells before and after treatment with either a pharmacological LDHA inhibitor (GSK2837808A, referred as LDHAi, 10 μM for 24 h) or after genetic LDHA knockdown (shLDHA, Supplementary Fig. S4C). Figure 4A, B indicate a significant decrease in ECAR and basal glycolysis in MCF7-POU1F1 cells after both LDHA procedures. Previous studies have shown an LDHA-dependence on cancer cell proliferation under hypoxic environment conditions [27]. Our data are in line with these reports. Proliferation of MCF7 cells significantly increased after POU1F1 overexpression in both normoxic and hypoxic cultures, as we previously demonstrated [28], but only under hypoxia does the treatment of MCF7-POU1F1 cells with the LDHAi and knockdown of LDHA significantly (P < 0.05) reduce cell proliferation (Fig. 4C, D). Next, migration of MCF7-POU1F1 cells was evaluated using a trans-well assay. A significant increase in cell migration was observed in MCF7 cells after POU1F1 overexpression as compared with control cells (Fig. 4E). However, treatment either with LDHAi (P < 0.001) or shLDHA (P < 0.01) significantly reduced cell migration (Fig. 4E). Finally, to analyze cancer cell invasion, we carried out organotypic cultures using control MCF7-POU1F1 cells and MCF7-POU1F1 cells either treated with LDHAi or transfected with shLDHA. Our data indicate a significant decrease (P < 0.05) in cell invasion after both experimental manipulations (Fig. 4F). In summary, our data suggest that either blocking LDHA enzymatic activity or decreasing LDHA expression in MCF7-POU1F1 cells reduces proliferation, migration, and invasion of breast cancer cells.

A, B Administration of a LDHA inhibitor (10 μM, GSK2837808A, referred as LDHAi) or LDHA knockdown (shLDHA) in MCF7-POU1F1 cells for 24 h significantly decreases extracellular acidification rate (ECAR) and basal glycolysis. Data were obtained using an XFp seahorse cell analyzer. C, D Cell proliferation in control and POU1F1-overexpressing MCF7 cells treated with 10 μM LDHAi or transfected with the pKLO-shLDHA vector for 72 h under normoxic and hypoxic (1% O2) atmosphere. E Representative figure and quantitative analysis of trans-well migration assays in control cells (MCF7), POU1F1-overexpressing cells (MCF7-POU1F1), and MCF7-POU1F1 cells either treated with LDHAi (upper panel) or after LDHA knockdown (shLDHA, bottom panel). Scale bar: 100 μm. F Representative images of H&E-stained sections of MCF7-POU1F1 cells cultured in an organotypic system in the presence of a pharmacological LDHA inhibitor (LDHAi) or LDHA knockdown (shLDHA), and quantification of cell invasion. Data are expressed as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar: 50 μm.
POU1F1-regulated LDHA influences tumor growth and tumor glucose uptake in vivo, and both POU1F1 and LDHA expression are related to clinical outcome
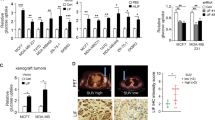
To evaluate in vivo the effect of LDHA knockdown in POU1F1-overexpressing cells, immunodeficient BALB/c-nu mice were injected with control MCF7 cells (stably transfected with the pTRE2 control vector, n = 7, MCF7), MCF7 cells with POU1F1 overexpression (pTRE2-POU1F1-overexpressing vector plus the pLKO control vector, n = 7, MCF7-POU1F1), and with MCF7 cells with POU1F1 overexpression and LDHA knockdown (pTRE2-POU1F1-overexpressing vector plus the pLKO-shLDHA plasmid, n = 7, MCF7-POU1F1-shLDHA). Xenografted tumors were monitored every 3 days, and mice were sacrificed at day 15 post injection. Body weight remained stable among all three groups (Fig. 5A). Tumor volume in MCF7-injected mice was negligible at day 15, but a continuous growth of tumor volume in both the MCF7-POU1F1 and the MCF7-POU1F1-shLDHA groups was observed during the study, being significatively higher at 9, 12, and 15 days in MCF7-POU1F1 mice comparing to LDHA knocked-down mice (Fig. 5B). POU1F1 and LDHA protein levels in three tumors from each group were assayed by western blot showing, as expected, reduced levels of LDHA in MCF7-POU1F1-shLDHA-injected mice with respect to MCF7-POU1F1 group (Fig. 5C). Immunohistochemistry analyses of tumor xenografts showed high cell proliferation (ki67 marker) in both MCF7-POU1F1 and MCF7-POU1F1-shLDHA groups with respect to MCF7-injected group, but ki67 staining was reduced in MCF7-POU1F1-shLDHA as compared to the MCF7-POU1F1 injected mice (Fig. 5D).

A Body weight of mice orthotopically injected with MCF7, MCF7-POU1F1, and MCF7-POU1F1-shLDHA human breast cancer cells (n = 7 mice per group). B Tumor volume of mice injected with MCF7, MCF7-POU1F1, and MCF7-POU1F1-shLDHA cells and representative images of tumors. Scale bar: 1000 μm. C qWB analysis of POU1F1, LDHA and β-actin in tumors from nine mice injected with MCF7 (n = 3), MCF7-POU1F1 (n = 3) and MCF7-POU1F1-shLDHA cells (n = 3). D ki67 immunostaining of mouse tumors. Scale bar: 50 μm. E On the left, SUVmax values for the three experimental groups (n = 4 mice per group). Significant differences can be observed between MCF7-POU1F1 and other groups (*P < 0.05, **P < 0.01). On the right, [18F]FDG PET/CT images from representative BALB/c-nu mice. Metabolic activity is coded on a color scale ranging from blue (low [18F]FDG uptake) to red (high [18F]FDG uptake). At the top, xenograft tumors indicated by arrows on CT images. At the bottom, tumors delineated using PET/CT images. Axial views are also shown. F Correlation between POU1F1 and LDHA mRNA expression and relapse-free survival (RFS) in human breast tumors (n = 3951). G Correlation between POU1F1 and LDHA mRNA expression and overall survival (OS) in human breast tumors (n = 42). Analysis of (F) and (G) were done using the KM plotter and the ProgGeneV2 online tools, respectively.
In addition, four mice per group were assayed for glucose uptake just before being sacrificed, using [18F]FDG PET/CT scans. SUVmax indicates a significant (P < 0.01) increase in glucose uptake in tumors from MCF7-POU1F1 injected mice as compared to control group (Fig. 5E). However, tumors in mice injected with MCF7-POU1F1-shLDHA cells had significantly reduced SUVmax (P < 0.05) as compared with MCF7-POU1F1 injected mice, but similar values to those found in control mice (Fig. 5E). Finally, to study the possible relationship between POU1F1/LDHA expression and clinical outcome, POU1F1 and LDHA mRNA was analyzed in a dataset of human breast cancer patients. We found a significant correlation between POU1F1/LDHA expression and both relapse-free survival (RFS) (P < 0.001) and overall survival (OS) (P < 0.001) (Fig. 5F, G, and Supplementary Fig. S5).
POU1F1 expression in breast cancer tumor samples correlates with CAF activation
Once we demonstrated that POU1F1 regulates LDHA and that pharmacological or genetic manipulation of LDHA induces phenotypic changes in breast cancer cells that modify cancer progression, we studied whether POU1F1 could have an impact on TME, specifically on fibroblasts. Numerous studies have shown increased NAF to CAF activation in breast cancer [29]. To study the role of POU1F1 in CAF activation, we first evaluated POU1F1 mRNA expression in 21 human breast tumors (Supplementary Table S2). Tumor samples were classified as high POU1F1 (POU1F1 higher than 75th percentile, n = 6) and low POU1F1 (with levels below 25th percentile, n = 5) mRNA expression (Fig. 6A). Based on this classification, actin alpha 2 smooth muscle mRNA (ACTA2) expression was evaluated as a key marker of CAFs. We found a significant (R = 0.597, P = 0.0043) correlation between POU1F1 and ACTA2 mRNA (Fig. 6B, C). POU1F1 and ACTA2 protein (α-SMA) expression were also evaluated in seven primary cultures of human breast tumors (Supplementary Table S3). Both western blot and immunohistochemistry (IHC) showed a positive relationship between POU1F1 and α-SMA protein expression (Fig. 6D–F). Immunohistochemistry shows a representative example of POU1F1 and α-SMA immunodetection in two of the above-mentioned human breast tumors with high (Fig. 6E) and low (Fig. 6F) POU1F1 protein levels. As noted, POU1F1 is expressed at nuclear level in cancer cells, whereas α-SMA immunoreactivity in CAFs is present at cytoplasmic level. In line with the results at the mRNA level, a significant (R = 0.764, P = 0.045) correlation between POU1F1 and α-SMA protein expression was found (Fig. 6G). Indeed, a significant (P = 0.048, HR = 1.13 (1–1.28)) relationship between POU1F1/ACTA2 mRNA levels and RFS was found in a human breast cancer dataset (n = 3951 samples) (Fig. 6H), which suggests a clinical prognostic value for both POU1F1 and α-SMA in breast tumors.

A–C POU1F1 and ACTA2 mRNA expression in 21 breast tumor samples were evaluated by qPCR and plotted by POU1F1 expression (high POU1F1: higher than 75th percentile, low POU1F1: below 25th percentile). Dispersion plot indicates a significant positive correlation between POU1F1 and ACTA2 mRNA. **P < 0.01. D Quantitative western blot (qWB) of POU1F1, α-SMA, and β-actin (as loading control) in seven patient-derived breast tumors (PDT 1–7). E Representative immunostaining of POU1F1 in tumor cells and α-SMA in fibroblasts in human breast invasive ductal carcinomas with high POU1F1 expression. Scale bar: 75 μm. F Representative immunostaining of POU1F1 in tumor cells and α-SMA in fibroblasts in human breast invasive ductal carcinomas with low POU1F1 expression. Scale bar: 75 μm. G Dispersion plot indicates a significant correlation between POU1F1/β-actin and α-SMA /β-actin protein levels in seven patient-derived breast tumors. H POU1F1 and ACTA2 mRNA expression significantly correlate with relapse-free survival (RFS) in human breast tumor samples (n = 3951) (http://kmplot.com). I–N Characterization of normal-associated fibroblasts (NAFs, in the breast tumor surrounding tissue) and cancer-associated fibroblasts (CAFs, in the breast tumor tissue) in three human breast tumor samples based on ACTA2 mRNA and α-SMA protein expression measured by qPCR and qWB. Data are expressed as mean ± SEM from three independent Western blots. **P < 0.01, ***P < 0.001.
To further evaluate the role of POU1F1 in NAF to CAF activation, we selected three human primary breast tumors: 035, 823, and 2920 (Supplementary Table S4). NAFs were obtained at least 2 cm away from the tumor. After tissue culture, both NAFs and CAFs were isolated and carefully characterized. Consistent with previous reports, fibroblasts were classified according to ACTA2/α-SMA mRNA/protein expression (NAFs have low ACTA2/α-SMA, and CAFs have high ACTA2/α-SMA) (Fig. 6I–N). In addition, other NAFs/CAFs markers, such as vimentin (VIM), fibroblast activation protein (FAP), vascular endothelial growth factor (VEGF), and C-X-C motif chemokine 12 (CXCL12) were evaluated, showing a significant increase in VIM, FAP, VEGF, and CXCL12 mRNA in CAFs with respect to NAFs (Supplementary Fig. S6A–C). MCF7 cells with and without POU1F1 overexpression were cultured for 24 h and CM from either MCF7 or MCF7-POU1F1 cultures was added to NAFs obtained from patient 823 (NAF823) to evaluate a possible NAF to CAF activation (Fig. 7A). Overexpression of POU1F1 (Fig. 7B) increased α-SMA protein levels in NAF823 (Fig. 7C), and conversely, treatment of NAFs with CM of MDA-MB-231 cells after knockout of POU1F1 (Fig. 7D, E) decreased α-SMA protein expression with respect to NAFs treated with CM-MDA-MB-231 control cells (Fig. 7F). Two similar experiments—treatment with CM from MCF7 cells before and after POU1F1 overexpression, and treatment with CM from MDA-MB-231 cells before and after POU1F1 knockout—were carried out in normal mammary fibroblasts (NMF) obtained from a normal patient after mammoplasty, confirming the results obtained with NAF823 (Supplementary Fig. S7A, B).

A Conditioned medium (CM) from 24 h culture of NAF823, MCF7, and MCF7-POU1F1 cells was added to NAF823 for 24 h and α-SMA and β-actin protein levels were evaluated by qWB. B qWB of POU1F1 and β-actin in MCF7 and MCF7-POU1F1 cells. C qWB of α-SMA and β-actin in NAF823 after administration of CM-NAF823, CM-MCF7 and CM-MCF7-POU1F1 for 24 h, and representative histograms from two independent qWB. D CM from 24 h culture of NAF823, control MDA-MB-231 (MDAsgC), and MDA-MB-231 cells after POU1F1 knockout (MDAsgPOU1F1) was added to NAF823 for 24 h and α-SMA and β-actin were evaluated by qWB. E qWB of POU1F1 and β-actin in MDAsgC and MDAsgPOU1F1 cells. F qWB of α-SMA and β-actin in NAF823 after CM-NAF823, CM-MDAsgC, and CM-MDAsgPOU1F1 treatments, and representative histograms from two independent qWB. G–H Western blot of α-SMA and β-actin in NAF823 and normal mammary fibroblasts (NMFs) treated with vehicle (0) and lactate (Lac, 20 mM) for 24 h. Data are expressed as mean ± SEM from three independent quantitative western blots. **P < 0.01, and ***P < 0.001. I LDHA and ACTA2 mRNA expression significantly correlate with relapse-free survival (RFS) in human breast tumor samples (http://kmplot.com). J Model of POU1F1-induced metabolic reprogramming and cancer progression. POU1F1 transcriptionally regulates LDHA leading pyruvate into lactate, which in turn increases proliferation, migration, and invasion of breast cancer cells and induces fibroblast activation.
POU1F1-induced lactate mediates CAF activation
To evaluate the effect of lactate on NAF to CAF activation, NAF823 and NMF were treated for 24 h with 20 mM lactate, and α-SMA protein levels were evaluated by western blot (Supplementary Fig. S8A). Both NAF823 and NMF showed a significant increase in α-SMA after lactate treatment with respect to untreated control fibroblasts (Fig. 7G, H). In fact, to correlate our in vitro data with clinical outcome, LDHA/α-SMA mRNA levels were analyzed in a breast cancer patient database (n = 3951) (http://kmplot.com). A significant (P < 0.001) correlation was found between LDHA/α-SMA and RFS (Fig. 7I). To analyze the effect of POU1F1 and lactate at protein level on CAF activation, CM was collected from a 24-h culture of stable MCF7-POU1F1 cells (control) treated either with 10 μM of an LDHA inhibitor (MCF7-POU1F1-LDHAi) or after LDHA knockdown (MCF7-POU1F1-shLDHA, see Supplementary Fig. S4C). Then, NAF823 and NMF were treated for 24 h with CM (Supplementary Fig. S8B, C), and α-SMA was immunoblotted to evaluate protein expression. The results were similar to those obtained after treatment with 20 mM lactate (Supplementary Fig. S8D–G).
Discussion
The current study demonstrates that the POU1F1 transcription factor induces metabolic reprogramming by enhancing aerobic glycolysis of human breast cancer cells through transcriptional regulation of the LDHA gene (Fig. 7J). Elevated expression of LDHA increases conversion of pyruvate to lactate. In turn, the LDHA-dependent lactate increases proliferation of hypoxic cancer cells as well as migration and invasion. In addition, cancer cell-secreted lactate modifies fibroblast phenotype, which activates the conversion of NAF into CAF. LDHA downregulation in tumors of mice with POU1F1 overexpression significantly reduces tumor growth and tumor [18F]FDG uptake. In summary, our data revealed POU1F1 as a new factor of glycolysis regulation and demonstrate an important role for lactate in both cancer cells and fibroblasts to mediate cancer progression. Both POU1F1/LDHA and POU1F1/α-SMA expression correlate with clinical outcome, suggesting POU1F1 as a prognostic factor in breast cancer.
High POU1F1 expression in breast cancer cells is related to high cell proliferation, migration, invasion, and low apoptotic rate [22]. These features together with induction of EMT indicate that POU1F1 induces breast cancer progression [22]. In the present study, our first objective was to evaluate possible metabolic changes in breast cancer mediated by POU1F1. Increased glucose uptake relative to the surrounding normal tissues has emerged as a hallmark of cancer metabolism [30]. High glucose metabolism is accompanied by increased aerobic glycolysis in breast cancer cells, which produce lactate from pyruvate instead of transforming pyruvate into Acetyl CoA, as occurs in normal cells. We found increased glycolysis in breast tumors vs. normal breast, in Luminal B vs. Luminal A tumors, in HER2 vs. Luminal A tumors, in TNBC vs. luminal A tumors, and in metastasis vs. primary breast tumors, corroborating previous reports that glycolysis is related to aggressiveness in breast tumors [31]. Although it has recently been reported that the main upregulated pathway in micrometastasis is OXPHOS [32], we found both OXPHOS and glycolysis to be a hallmark in metastasis. The availability of nutrients in metastatic niche is different than in primary tumors, and metabolism is not the same in all metastatic niches. After POU1F1 overexpression, we found a significant increase in the glycolysis signature. Microarray data were confirmed by qPCR, showing increased mRNA expression of multiple enzymes belonging to the glycolysis pathway, as well as elevated mRNA levels of glucose GLUT1 and lactate MCT4 transporters in MCF7 cells after POU1F1 overexpression. MCT4 lactate transporter is a critical determinant of disease prognosis and glycolytic metabolism. Targeting MCT4 suppresses tumor growth and yields vulnerability to metabolic stress [33]. In addition, high lactate levels are present in CM of MCF7-POU1F1 cells, demonstrating increased aerobic glycolysis and lactate secretion. Functionally, metabolic analyses in either MCF7 cells with POU1F1 overexpression or MDA-MB-231 cells after POU1F1 knockout indicate that POU1F1 reprograms cancer cell metabolism toward a glycolytic pattern. Importantly, primary cultures of human breast tumors with low and high POU1F1 expression showed similar metabolic results to those obtained in cell lines after POU1F1 knockout and overexpression.
The LDHA enzyme is a key component of the glycolysis pathway. However, only a few factors that transcriptionally regulate LDHA are currently known. Among them, the c-Myc oncogenic transcription factor transactivates LDHA gene and stimulates aerobic glycolysis [34], and the HIF-1α transcription factor upregulates the rate-limiting enzyme for glycolysis (GLUT1 transporter) and transcriptionally regulates LDHA [35, 36]. Our study showed that LDHA mRNA is highly expressed in tumors with respect to normal breast and we found a significant correlation between POU1F1 and LDHA mRNA expression. In fact, two POU1F1 binding sites were identified in the promoter region of the LDHA gene, and luciferase reporter assays demonstrated that POU1F1 transcriptionally regulates the LDHA gene. Interestingly, we found that the histone mark H3K27ac, which is related to active enhancer regions, is increased in TNBC as compared with luminal breast cancer cell lines. This suggests an increased transcriptional activity of POU1F1 and consequently increased expression of LDHA in highly aggressive breast cancer cell lines [37]. At functional level, the upregulation of LDHA by POU1F1 interferes with mitochondrial respiration, turning cancer cell metabolism into aerobic glycolysis instead of OXPHOS. Furthermore, pharmacological and genetic LDHA blockade reduces cell migration and invasion and, interestingly, decreases breast cancer cell proliferation under hypoxic environment. Because POU1F1 does not modify HIF-1α expression (data not shown), our data suggest that POU1F1 regulates LDHA in hypoxic cells independently of HIF-1α. Our results are in line with those previously obtained by Leder’s group, who demonstrated that LDHA knockdown resulted in a compromised ability of tumor cells to proliferate under hypoxia [7]. In vivo, LDHA knockdown in POU1F1-overexpressing tumors reduces cell proliferation and tumor growth, and, importantly, decreases [18F]FDG uptake. In fact, [18F]FDG PET/CT imaging is widely used in clinical practice for staging and follow-up after cancer treatment.
In addition to its role in breast tumors, POU1F1-mediated glycolytic activity could also have important physiological and pathological consequences. POU1F1 plays a key role in cell differentiation during anterior pituitary gland organogenesis in mammals [38], and the POU family of transcription factors (POU1F1, Oct, and Unc) are critical in key biological processes, such as cell proliferation, determination of cell lineage fate and regulation of cell migration, survival, and terminal differentiation [39]. Furthermore, as commented above, increased LDHA-mediated glycolysis is related to invasion and proliferation of pituitary adenomas [17]. Given that POU1F1 is mainly expressed in the pituitary gland, we could speculate that POU1F1-LDHA-mediated glycolysis plays a role in pituitary cell differentiation and proliferation.
TME consisting of tumor cells and host stromal cells has emerged as an important player in tumor progression. Among stromal cells, macrophages and fibroblasts are the major components of tumor mass. Recently, we have demonstrated that POU1F1 overexpression induces both recruitment of monocytes-macrophages to breast tumor area and polarization of macrophages into TAM [40]. In the present study, we evaluated the role of POU1F1 in fibroblast activation. CAFs are central elements of the TME, interacting with tumor cells and other stromal cells [41]. Although CAFs may have tumor suppressive properties, they mainly have tumor promoting capacities. In fact, high CAF presence in TME is related to cancer prognosis and response to therapy [42]. Our study demonstrates a clear relationship between POU1F1 and α-SMA in breast tumors as well as the clinical prognostic value of both POU1F1 and ACTA2 mRNA expression. In addition, POU1F1-derived lactate induces NAF into CAF activation, which is partially reverted by either POU1F1 knockdown or LDHA blockade. In some tumors, lactate has recently been reported to epigenetically reprogram mesenchymal stem cells to differentiate into CAFs, thus leading to tumor progression [15]. In addition, lactate binds to the GPR81 lactate receptor, which is highly expressed in cancer cells, including breast cancer cells [43, 44]. GPR81 is also expressed in stromal cells, such as antigen presenting dendritic cells, suggesting that tumoral lactate prevents dendritic stromal cells from presenting cancer cell-specific antigens to other immune cells, which may be of importance to cancer cell immune evasion [45]. The present study evaluated neither epigenetic reprogramming of NAFs through POU1F1-induced lactate nor GPR81 lactate receptors in NAFs, but we cannot exclude either possibility in lactate-mediated NAF to CAF activation.
Altogether, our data suggest that LDHA and POU1F1 could be important therapeutic targets in cancer. Interestingly, Sherman et al. [46] have demonstrated that pancreatic stellate cells, the predominant fibroblastic cell type in the TME of pancreas tumors, are reconverted to quiescent state by stromal remodeling after vitamin D treatment, indicating a molecular strategy to transcriptional reprogramming of tumor stroma. POU1F1 is transcriptionally repressed by vitamin D and its analogs [47, 48]. This led us to hypothesize that vitamin D treatment alone and/or in combination with LDHA inhibitors [49] might be a therapeutic strategy in breast cancer, acting on both cancer and stromal cells to potentiate the chemotherapeutic response in breast tumors. The effect of both treatments in glycolytic breast tumors should be further elucidated.
Material and methods
Cell culture, reagents, and CM
The human breast adenocarcinoma MCF7 and MDA-MB-231 cell lines were obtained from the European Collection of Cell Culture (ECACC; Porton Down, UK). MCF7 Tet-Off cells were purchased from Clontech-Takara (Kusatsu, Japan). All cells were negative and periodically tested for mycoplasm. Cell lines were cultured in DMEM supplemented with 10% FBS (Gibco, Paisley, UK) and grown in an air-CO2 (95:5) atmosphere at 37 °C. Hypoxic experiments were carried out at 1% O2. The LDHA inhibitor used at 10 μM in all experiments was purchased from Tocris (GSK 2837808 A, Minneapolis, USA). Lactate (Sigma-Aldrich, San Luis, USA) was used at 20 mM. CM was obtained after seeding the cells at 2 × 105 cs/cm2 in DMEM with 10% FBS and cultured overnight. Afterward, cells were washed in PBS and cultured again in DMEM/F12 without FBS for 24 h. Medium was centrifuged for 5 min at 300 g and the supernatant was collected and used immediately or stored at −80 °C.
Details of additional methodology can be found in Supplementary Data.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. 2019;20:436–50.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20.
Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–8.
McGuirk S, Audet-Delage Y, St-Pierre J. Metabolic fitness and plasticity in cancer progression. Trends Cancer. 2020;6:49–61.
Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–34.
Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–518.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumor-associated macrophages by tumour derived lactic acid. Nature. 2014;513:559–63.
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–9.
Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191:1486–95.
Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71:2550–60.
Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer. Cell Cycle. 2011;10:1271–86.
Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 2014;25:47–60.
Bhagat TD, Von Ahrens D, Dawlaty M, Zou Y, Baddour J, Achreja A, et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. Elife. 2019;8.pii:e50663.
Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65:3981–99.
An J, Zhang Y, He J, Zang Z, Zhou Z, Pei X, et al. Lactate dehydrogenase A promotes the invasion and proliferation of pituitary adenoma. Sci Rep. 2017;7:4734–46.
Lefevre C, Imagawa M, Dana S, Grindlay J, Bodner M, Karin M. Tissue specific expression of the human growth hormone gene is conferred in part by the binding of a specific trans-acting factor. EMBO J. 1987;6:971–81.
Nelson C, Albert VR, Elsholtz HP, Lu LI-W, Rosenfeld MG. Activation of cell specific expression of rat growth hormone and prolactin genes by a common transcription factor. Science. 1988;239:1400–5.
Gil-Puig C, Seoane S, Blanco M, Macia M, Garcia-Caballero T, Segura C, et al. Pit-1 is expressed in normal and tumoral human breast and regulates growth hormone secretion and cell proliferation. Eur J Endocrinol. 2005;153:335–44.
Ben-Batalla I, Seoane S, Macia M, Garcia-Caballero T, Gonzalez LO, Vizoso F, et al. The Pit-1/Pou1f1 transcription factor regulates and correlates with prolactin expression in human breast cell lines and tumors. Endocr Relat Cancer. 2010;17:73–85.
Ben-Batalla I, Seoane S, Garcia-Caballero T, Gallego R, Macia M, Gonzalez LO, et al. Deregulation of the Pit-1 transcription factor in human breast cancer cells promotes tumor growth and metastasis. J Clin Investig. 2010;120:4289–302.
Gao Z, Xue K, Zhang L, Wei M. Over-expression of POU class 1 homeobox 1 transcription factor (Pit-1) predicts poor prognosis for breast cancer patients. Med Sci Monit. 2016;22:4121–5.
Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. 2019;176:98–112.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20.
Ko YH, Domingo-Vidal M, Roche M, Lin Z, Whitaker-Menezes D, Seifert E, et al. TP53-inducible Glycolysis and Apoptosis Regulator (TIGAR) metabolically reprograms carcinoma and stromal cells in breast cancer. J Biol Chem. 2016;291:26291–303.
Firth JD, Ebert BL, Ratcliffe PJ. Hypoxic regulation of lactate dehydrogenase A. J Biol Chem. 1995;270:21021–7.
Martinez-Ordoñez A, Seoane S, Cabezas P, Eiro N, Sendon-Lago J, Macia M, et al. Breast cancer metastasis to liver and lung is facilitated by Pit-1-CXCL12-CXCR4 axis. Oncogene. 2018;37:1430–44.
Luo H, Tu G, Liu Z, Liu M. Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression. Cancer Lett. 2015;361:155–63.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47.
Lehuede C, Dupuy F, Rabinovitch R, Jones RG, Siegel PM. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. 2016;76:1–8.
Davis RT, Blake K, Ma D, Gabra MBI, Hernandez GA, Phung AT, et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat Cell Biol. 2020;22:310–20.
Baek G, Tse YF, Hu Z, Cox D, Buboltz N, McCue P, et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014;9:2233–49.
Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci USA. 1997;94:6658–63.
Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J Biol Chem. 1995;270:29083–9.
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–37.
Huang X, Li X, Xie X, Ye F, Chen B, Song C, et al. High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. Breast. 2016;30:39–46.
Dolle P, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M. Expression of GHH-1 protein in mouse pituitaries correlates both temporally and spatially with the onset of growth hormone gene activity. Cell. 1990;60:809–20.
Andersen B, Rosenfeld MG. POU domain factors in the neuroendocrine system: lessons from developmental biology provide insights into human disease. Endocr Rev. 2001;22:2–35.
Seoane S, Martinez-Ordoñez A, Eiro N, Cabezas-Sainz P, Garcia-Caballero L, Gonzalez LO, et al. POU1F1 transcription factor promotes breast cancer metastasis via recruitment and polarization of macrophages. J Pathol. 2019;249:381–94.
Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30:1002–19.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20:174–86.
Roland CL, Arumugam T, Deng D, Liu SH, Philip B, Gomez S, et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014;74:5301–10.
Lee YJ, Shin KJ, Park SA, Park KS, Park S, Heo K, et al. G-protein-coupled receptor 81 promotes a malignant phenotype in breast cancer through angiogenic factor secretion. Oncotarget. 2016;7:70898–911.
Brown TP, Bhattacharjee P, Ramachandran S, Sivaprakasam S, Ristic B, Sikder MOF, et al. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene. 2020;39:3292–304.
Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93.
Seoane S, Perez-Fernandez R. The vitamin D receptor represses transcription of the pituitary transcription factor Pit-1 gene without involvement of the retinoid X receptor. Mol Endocrinol. 2006;20:735–48.
Seoane S, Arias E, Sigueiro R, Sendon-Lago J, Martinez-Ordoñez A, Castelao E, et al. Pit-1 inhibits BRCA1 and sensitizes human breast tumors to cisplatin and vitamin D treatment. Oncotarget. 2015;6:14456–71.
Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, et al. Targeting lactate dehydrogenase−a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014;19:795–809.
Acknowledgements
This study was supported by FEDER/Ministerio de Ciencia, Innovación y Universidades- Agencia Estatal de Investigación-PGC2018-100776-B-I00 and from Conselleria de Cultura, Educación e Ordenacion Universitaria (GPC2014/001), AM-O was supported by an FPU grant (Ministerio de Educación—FPU14/00548). We are grateful to Dr. M. Fidalgo and Dr. D. Guallar for helping us to design some experiments, to MJ Otero Fraga for IHC analysis, and the BioBank Complejo Hospitalario Universitario de Santiago (CHUS)(PT17/0015/0002) for their collaboration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martínez-Ordoñez, A., Seoane, S., Avila, L. et al. POU1F1 transcription factor induces metabolic reprogramming and breast cancer progression via LDHA regulation. Oncogene 40, 2725–2740 (2021). https://doi.org/10.1038/s41388-021-01740-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-021-01740-6
This article is cited by
-
Identification of POU4F1 as a novel prognostic biomarker and therapeutic target in esophageal squamous cell carcinoma
Cancer Cell International (2024)
-
The hsa_circ_0000276-ceRNA regulatory network and immune infiltration in cervical cancer
BMC Cancer (2023)
-
Transcription factors in fibroblast plasticity and CAF heterogeneity
Journal of Experimental & Clinical Cancer Research (2023)
-
Glycolysis-related lncRNA TMEM105 upregulates LDHA to facilitate breast cancer liver metastasis via sponging miR-1208
Cell Death & Disease (2023)
-
Cancer-associated fibroblasts: from basic science to anticancer therapy
Experimental & Molecular Medicine (2023)
