
Abstract
In epithelial ovarian cancer (EOC), response to platinum (PT)-based chemotherapy dictates subsequent treatments and predicts patients’ prognosis. Alternative splicing is often deregulated in human cancers and can be altered by chemotherapy. Whether and how changes in alternative splicing regulation could impact on the response of EOC to PT-based chemotherapy is still not clarified. We identified the splicing factor proline and glutamine rich (SFPQ) as a critical mediator of response to PT in an unbiased functional genomic screening in EOC cells and, using a large cohort of primary and recurrent EOC samples, we observed that it is frequently overexpressed in recurrent PT-treated samples and that its overexpression correlates with PT resistance. At mechanistic level, we show that, under PT treatment, SFPQ, in complex with p54nrb, binds and regulates the activity of the splicing factor SRSF2. SFPQ/p54nrb complex decreases SRSF2 binding to caspase-9 RNA, favoring the expression of its alternative spliced antiapoptotic form. As a consequence, SFPQ/p54nrb protects cells from PT-induced death, eventually contributing to chemoresistance. Overall, our work unveils a previously unreported SFPQ/p54nrb/SRSF2 pathway that in EOC cells plays a central role in regulating alternative splicing and PT-induced apoptosis and that could result in the design of new possible ways of intervention to overcome PT resistance.
Similar content being viewed by others


Introduction
Epithelial ovarian cancer (EOC) is a relatively rare but highly lethal disease, representing the fifth leading cause of cancer death in women in the western world. The high death-to-incidence rate is mainly due to the late diagnosis, the peculiar way of metastatic dissemination and the appearance of chemoresistance [1, 2]. Gold standard therapy for EOC patients is cytoreductive surgery followed by platinum (PT) plus taxane-based chemotherapy. In particular, PT compounds (i.e., carboplatin and cisplatin) represent the backbone of first-line chemotherapy in EOC and also occupy an integral role in the management of relapsed ovarian cancer. The appearance of PT resistance dictates the choice of subsequent treatments and is predictive of poor prognosis [1, 2]. In EOC patients, de novo PT resistance is quite infrequent while acquired PT resistance is very common and has been linked to different mechanisms, including alteration in drug influx/efflux, increased DNA repair or tolerance to DNA lesions, defective activation, and/or execution of cell senescence/cell death [3, 4].
To identify new molecular alterations that could modify the sensitivity of EOC to PT, we performed a loss-of-function genomic approach for genes belonging to DNA repair, apoptosis, and TP53 pathways and identified SFPQ (splicing factor proline and glutamine rich) as a gene functionally involved in the response to PT.
SFPQ, also known as PSF (PTB-associated splicing factor), is a multifunctional nuclear protein first identified as a splicing factor, that participates to several cellular activities, including RNA transport, apoptosis, and DNA repair. Together with p54nrb and PSPC1, it belongs to the well-conserved multifunctional DBHS (Drosophila behavior/human splicing) protein family that forms obligated homo- and heterodimers [5]. DBHS proteins are found in the nucleoplasm and, under various conditions, within sub-nuclear bodies termed paraspeckles, localized close to chromatin, or DNA damage foci [6]. SFPQ has both DNA- and RNA-binding domains and shares with its preferential binding partner p54nrb most, but not all of its activities. Accordingly, SFPQ but not p54nrb or PSPC1 seems to be essential for cell viability [7]. The precise mechanism by which SFPQ is required for cell growth and survival is not completely clarified, although some evidences indicate its involvement in the control of apoptosis [8,9,10]. A remarkable number of transcripts encoding for proteins involved in the apoptotic pathway are subjected to alternative splicing, eventually leading to the expression of proteins with opposite functions, either pro- or antiapoptotic [11, 12]. Changes in alternative splicing patterns were observed in association with acquired chemoresistance or under the selective pressure of PT treatment. For instance, it has been shown in breast cancer cells that cisplatin treatment induces the alternative splicing of more than 600 transcripts [13, 14]. Among those, several transcripts involved in apoptosis regulation, such as caspase-8, BCL2L1, Diablo, P73, Fas, were identified [14], suggesting that alternative transcripts could participate to the response of cancer cells to PT. In particular, it has been reported that basal and cisplatin-induced caspase-8 and -9 activation is regulated by the splicing factor SRSF2 (also known as SC35) [11, 15]. Accordingly, SRSF2 knock-down increased EOC cell survival under cisplatin exposure [16].
Here, we report that SFPQ is critically involved in EOC cell sensitivity to PT treatment and we identify a previously unknown activity of the SFPQ/p54nrb complex that, via SRSF2 modulation, regulates caspase-9 alternative splicing and, eventually, leads to reduced PT-induced apoptosis.
Results
SFPQ modulates platinum (PT) response in epithelial ovarian cancer (EOC) cells
To identify new putative mediators of PT response in EOC cells, we performed an shRNA-based loss-of-function screening to target 680 genes, belonging to apoptosis, TP53 and DNA repair pathways, which play key functions in EOC tumor progression and response to PT-based chemotherapy [17]. One TP53 mutated (MDAH-2774, hereafter MDAH) and one TP53-null (SKOV3) cell line were used, allowing us to identify genes involved in PT response both in the absence of p53 and in the presence of TP53 gain of function mutations, which represent the most common scenarios in EOC. Cells were transduced with the shRNA library and treated with sub-optimal doses of carboplatin (CBDCA) to obtain a ~20% reduction in cell viability (Fig. 1a). By this approach, we identified 50 candidate genes able to increase CBDCA activity when silenced, in at least one cell line. Validation screening by using five-specific shRNA/gene on four different EOC cell lines (i.e., MDAH, SKOV3, TOV112D, and OV-90), demonstrated that silencing of five genes, namely ATR, BCL2L1 (also known as Bcl-XL), SGK2, USP1, and SFPQ, were able to significantly increase CBDCA response, in at least 3/4 tested cell lines. Since ATR and Bcl-XL involvement in PT response has been already well characterized, we decided to study the role of USP1 [17], SGK2 (manuscript in preparation) and SFPQ. Here, we report the data on the role of SFPQ in PT response. First, we validated the screening results using two-specific shRNAs against SFPQ. SFPQ knock-down significantly decreased the IC50, in MDAH and SKOV3 cells, to both CBDCA and Cisplatin (CDDP) (hereafter referred to as PT) (Fig. 1b, c). The similar results were obtained using two other independent EOC cell lines, OVCAR-3 and KURAMOCHI (Supplementary Fig. 1a, b), overall confirming that SFPQ played a role in the response to PT and that this role was independent from BRCA1/2 status and the type of TP53 alteration (Supplementary Fig. 1c).

a Experimental design of the loss-of-function screening. Briefly MDAH and SKOV3 cells were transduced with 2040 shRNAs targeting 680 genes in 96-well plates in duplicate and treated or not with CBDCA for 16 h. Cell viability was evaluated by MTS assay, 24 h after the end of the treatment. Statistical analyses (Z-score and adjusted p value for multiple testing) identified 50 potential targets (22 in MDAH, 24 in SKOV3, and 4 in both cell lines). The 50 genes were then validated in a subsequent screening using five shRNAs/gene in four different cell lines. Validated genes were considered genes for which at least three shRNAs increased the sensitivity to CBDCA in at least three cell lines. Using these parameters we identified SFPQ (and other four genes). Nonlinear regression analyses of cell viability in MDAH (b) and SKOV3 cells (c). Cells were transduced with control shRNA (sh-ctrl) or two different SFPQ shRNAs (sh-SFPQ#2 and #3) and treated with increasing doses of CBDCA and CDDP for 16 h. The table shows the IC50 and the confidence interval (CI) of each condition. Data are expressed as percentage of viable cells compared with untreated cells and represent the mean (±SD) of three biological replicates. Fisher’s exact test was used to calculate the global p value reported in the graph. On the right western blot (WB) analysis reporting SFPQ expression in corresponding silenced cells. Tubulin and Vinculin were used as loading control.
SFPQ expression increases after exposure to PT
SFPQ was generally more expressed in EOC cell lines compared with normal epithelial ovarian cells (i.e., HuNoEC). It was also widely expressed in a panel of primary EOC available in our Institute (Fig. 2a–c and Supplementary Table 1) [18, 19]. SFPQ expression (both at mRNA and protein levels) was significantly higher in EOC samples from patients that had previously received a PT treatment (samples collected after neo-adjuvant chemotherapy or recurrent EOC) (Fig. 2b, c) and in samples from PT-resistant (PT-Res) versus PT-sensitive EOC (Fig. 2d and Supplementary Table 2). Finally, the expression of SFPQ and of its preferential binding partner p54nrb was higher in MDAH isogenic PT-Res cells, recently generated in our lab [20], compared with their parental counterpart (Fig. 2e). Next, using these cells in time course analysis of PT response [19,20,21], we evaluated the expression of SFPQ protein during the phases of PT-induced DNA damage and repair. The results showed that higher SFPQ expression in PT-Res cells correlated with decreased and delayed apoptosis and DNA damage response, as evidenced by PARP-1 and caspase-3 cleavage and γ-H2AX expression, respectively (Fig. 2e). Overall, these data suggest a possible role for SFPQ in the regulation of PT response.

a WB analysis of SFPQ expression in normal human epithelial ovarian cells (HuNoEC) and in the indicated EOC cell lines. qRT-PCR (b) and WB analyses (c) reporting the normalized SFPQ expression in samples derived from untreated and PT-exposed (after neo-adjuvant chemotherapy or recurrent) EOC patients. A representative WB analysis of SFPQ expression is shown in (c, bottom panel). d qRT-PCR analysis reporting the normalized SFPQ mRNA expression in samples from PT-sensitive and PT-resistant EOC patients. e WB evaluating the expression of SFPQ and p54nrb and of DNA damage (γ-H2AX) and apoptotic markers (PARP-1 and cleaved caspase-3) in parental and isogenic PT-resistant (MIres) MDAH cells treated or not with CDDP for 16 h (t0) and then released for 24 h (t24). In the figure statistical significance was determined by a two-tailed, unpaired Student’s t test. (*p < 0.05; **p < 0.01; ***p < 0.001). Vinculin or GAPDH expression was used as loading control.
SFPQ knock-down increases basal and PT-induced apoptosis
Long-term SFPQ knock-down (i.e., more than 1 week after antibiotic selection) induced massive cell death (Fig. 3a). As already reported by others [7], it was not feasible to generate stable SFPQ-silenced clones. This massive cell death was accompanied by increased ratio of cleaved/total PARP-1 and decreased expression of p54nrb, total caspase-8, and Bcl-XL (Fig. 3b), suggesting that the mechanism of apoptosis was involved. However, in transiently transduced MDAH and SKOV3 cells SFPQ knock-down decreased cell growth starting 3–4 days after transduction (Fig. 3c), leaving an opportunity for mechanistic investigation. Using this approach and FACS analysis we observed that SFPQ knock-down slightly but consistently increased the sub-G1 population both in basal condition and after PT treatment, accompanied by increased cell accumulation in S-phase, 24 h after PT removal (Fig. 3d and Supplementary Fig. 2a). At molecular level, the combination of SFPQ knock-down and PT treatment, in two different cell lines (i.e., MDAH and OVCAR-3), anticipated the activation of caspase-dependent cell death, compared with control cells. This was evidenced by the precocious expression of cleaved PARP-1, cleaved caspase-8, -9, and -3 (Fig. 3e and Supplementary Fig. 2b–d). Overall, these data supported the possibility that SFPQ directly participates to the regulation of basal and PT-induced apoptosis.

a Typical phase contrast images (10× objective) of MDAH cells stable transduced with sh-ctrl or sh-SFPQ#2, after few days in culture. b WB analysis evaluating the expression of SFPQ and of DNA damage (γ-H2AX) and apoptotic markers (PARP-1, caspase-8, Bcl-XL) in cells silenced for SFPQ as in (a). c Growth curve of MDAH and SKOV3 cells transiently transduced with sh-ctrl and sh-SFPQ#2 and #3 as indicated. Cells growth was monitored by Trypan Blue Exclusion test. Data are the mean (±SD) of three biological replicates. d Cell cycle distribution of MDAH cells transiently transduced with the indicated shRNAs (sh-ctrl and sh-SFPQ#3) and analyzed by FACS 72 h after transduction in untreated and CDDP-treated conditions (CDDP 50 µM, T0 = 16 h treatment; T24 = 16 h treatment + 24 h release). The percentages of cells in each phase of the cell cycle are indicated. Data are the mean (±SD) of three biological replicates. e WB analysis evaluating the expression of PARP-1, caspase-8, -9, -3, γ-H2AX and Bcl-XL in MDAH cells transduced with sh-ctrl and sh-SFPQ#2 and #3 and treated as in (d). f Representative RT-PCR analyses of pro (+) and anti (−) apoptotic isoforms of caspase-9 in MDAH cells transduced as indicated and treated with CDDP 50 µM for the indicated times. g Graphs reporting the quantification of the ratio between pro-/antiapoptotic isoforms of caspase-9, normalized on Gapdh expression and folded on untreated (UNT) condition. Data are the mean (±SD) of three independent experiments. h WB analysis evaluating PARP-1 cleavage in MDAH cells treated with CDDP 25 µM for 16 h, alone or in combination with Z-LEHD-FMK (caspase-9 inhibitor). i Dose-response curves evaluating cell viability of MDAH cells transiently transduced with sh-ctrl or SFPQ#3 shRNAs and treated with increasing doses of CDDP alone or in combination with 20 µM Z-LEHD-FMK. Data are expressed as percentage of viable cells compared with untreated cells and represent the mean (±SD) of three biological replicates. In the figure GAPDH was used as loading control and statistical significance was determined by a two-tailed, unpaired Student’s t test. (*p < 0.05; **p < 0.01; ***p < 0.001).
SFPQ regulates the alternative splicing of caspase-9 mRNA in PT-treated cells
To get more insights on the molecular pathway(s) that could drive apoptosis in SFPQ-silenced cells, we carried out a gene expression profiling (GEP) of MDAH and SKOV3 cells, silenced or not for SFPQ. Two KEGG-pathways were commonly enriched in both cell lines, namely the spliceosome and the ubiquitin-mediated proteolysis (Supplementary Fig. 3a, b and Supplementary Tables 3 and 4) and, given the involvement of SFPQ in splicing regulation, we decided to focus on the spliceosome. We observed that long-term SFPQ knock-down significantly increased the ratio of pro-/antiapoptotic forms of caspase-9 RNAs, both in MDAH and SKOV3 cells (Supplementary Fig. 3c–e), supporting the possibility that SFPQ participates to the regulation of caspase-9 alternative splicing, favoring the expression of antiapoptotic spliced forms.
We next verified if PT treatment induced changes in the splicing of apoptotic genes and how these changes preceded or accompanied cell death. In PT-treated MDAH cells, apoptosis was massively observed 24 h after PT removal, while it was minimal under PT treatment for up to 16 h (Fig. 3d, e and Supplementary Fig. 2a–d), as previously observed [18, 19]. We tested if PT treatment could alter the expression ratio of RNA of proapoptotic (marked with +) and antiapoptotic (marked with −) forms of caspases and BCL2L1, before the appearance of massive apoptosis. Time course analyses in MDAH cells showed that, starting from 16 h of PT treatment, a decrease of the antiapoptotic (−) forms of caspase-8 and -9 and a slight increase of the proapoptotic (+) ones, paralleled the induction of DNA damage (Supplementary Fig. 4a–c). In accord to its established role in mediating the intrinsic apoptotic pathway, these changes were particularly evident for caspase-9 (Supplementary Fig. 4a, b). Consistent observations were made also for the expression of BCL2L1, with an increase of the Bcl-XS (proapoptotic) and decrease of the Bcl-XL (antiapoptotic) form (Supplementary Fig. 4a, b), demonstrating that PT treatment altered the alternative splicing of apoptotic genes in EOC cells.
More importantly, under PT treatment, knock-down of SFPQ in MDAH cells resulted in an increased expression of caspase-9 (+) proapoptotic form, already visible after 1.5 h, while it had minor effects on caspase-8 and BCL2L1 splicing (Fig. 3f, g and Supplementary Fig. 5a–c). Accordingly, both the caspase-9-specific inhibitor Z-LEHD-FMK and the pan-caspase inhibitor Z-VAD-FMK, effectively prevented PARP-1 cleavage in PT-treated MDAH cells and increased cell survival of SFPQ-silenced MDAH cells (Fig. 3h, i and Supplementary Fig. 5d, e), confirming a central role for caspase-9 activation in PT-induced cell death when SFPQ was silenced.
SFPQ regulates caspase-9 splicing via SRSF2 regulation
Basal and PT-induced caspase-8 and -9 alternative splicing is mainly regulated by the splicing factor SRSF2 [11, 15], which has been reported to play an opposite effect compared with SFPQ in PT-treated EOC cells [16]. Accordingly, knock-down of SRSF2 in MDAH cells delayed the increase of the proapoptotic form of caspase-9 (+) induced by PT, having minor effects on BCL2L1 (Supplementary Fig. 6a, b). Based on these evidences, we better dissected the mechanism whereby SFPQ and SRSF2 controlled caspase-9 splicing, in basal and PT-treated EOC cells.
First, we co-expressed SRSF2 and SFPQ, as full length or deleted isoforms (Fig. 4a) [22] in 293T17 cells then treated or not with PT. By co-immunoprecipitation assay we observed that SFPQ and SRSF2 co-precipitated under basal conditions and their binding was increased by PT treatment (Fig. 4b). The SFPQ-ΔN mutant lost the ability to bind SRSF2, indicating that this interaction requires the protein–protein interaction domain of SFPQ (Fig. 4b). SFPQ–SRSF2 interaction and its increase under PT treatment were confirmed using endogenous proteins from basal and PT-treated HeLa cells (Fig. 4c). Notably, also p54nrb, the preferential binding partner of SFPQ, co-precipitated with SRSF2 (Fig. 4c).

a Schematic representation of SFPQ WT protein. RRM1 and RRM2, in light-blue indicate the RNA recognition motifs, NOPS (green) the NonA/paraspeckle domain and coiled coil (orange) the coiled coil domain. GPQ and G-rich (yellow) mark the low complexity regions and DBD (pink) the uncharacterized DNA-binding domain. SFPQ deletion mutants (ΔN and ΔRRM) used are indicated in the upper part of the scheme, reporting the correspondent amino-acidic deletion (SFPQ ΔN lacks 9–264 residues; SFPQ ΔRRM lacks 298–429 residues). b Co-IP analyses in 293T17 cells, transfected with the indicated vectors, treated or not with CDDP 50 µM for 3 h. Input reports the expression of the indicated protein in the lysates used for IPs experiments. Tubulin was used as loading control. Densitometric analysis of the IP ratio is reported under the blot. Anti-MYC Ab was used to reveal SFPQ expression. c Endogenous co-IP analyses of SFPQ, p54nrb, and SRSF2 in nuclear fraction of HeLa cells, in untreated (UNT) and CDDP-treated conditions (3 and 6 h of 50 µM CDDP). Lysates were IP with anti-SRSF2 antibody. Densitometric analysis of normalized SFPQ IP/SRSF2 IP and p54nrb IP/SRSF2 IP expression is reported. RNA Immunoprecipitation analysis (RIP) in MDAH cells transfected with GFP-SRSF2 (d) or MYC-SFPQ WT (e) vectors and treated or not with CDDP 50 µM for 3 h. Samples were IP with the indicated antibodies. Densitometric analysis of normalized RIPs is reported under the blot. Overexpression of GFP-SRSF2 and MYC-SFPQ WT was confirmed by WB analysis. Fibrillarin was used as loading control. f RIP assay in OVCAR8 cells transfected with MYC-SFPQ WT and mutants constructs. Cells were treated with CDDP 50 µM for 3 h before harvesting. Samples were IP with anti-MYC antibody and amplified for caspase-9 as described in (e).
Using RNA immunoprecipitation (RIP) in MDAH cells treated or not with PT, we next demonstrated that both SRSF2 and SFPQ bound caspase-9 RNA (Fig. 4d, e). Interestingly, PT treatment had opposite effects, on one side increasing SFPQ and the other decreasing SRSF2 binding to caspase-9 RNA. (Fig. 4d, e). Using SFPQ-ΔN and -ΔRRM mutants or the SFPQ WT proteins we observed that SFPQ needs both domains to efficiently bind caspase-9 RNA under PT treatment (Fig. 4f). These data support the possibility that this RNA-binding activity of SFPQ required the presence of other partners in the RNA/protein complex.
At biological level, we observed that SFPQ overexpression enhanced and SRSF2 overexpression reduced cell survival, in both basal and PT-treated OVCAR8 cells (Fig. 5a), used as a model of low SFPQ expression. When SFPQ and SRSF2 were co-transfected the effect of SRSF2 prevailed and cells were more prone to cell death (Fig. 5a). Accordingly, the increased PT-induced apoptosis observed in SFPQ-silenced cells was prevented by concomitant SRSF2 knock-down in MDAH cell (Fig. 5b and Supplementary Fig. 7a, b), indicating that SRSF2 was necessary in this process.

a Dose-response curve of OVCAR8 cells overexpressing MYC-SFPQ WT and GFP-SRSF2 alone or in combination treated with increasing doses of CDDP. Data are expressed as absorbance of viable cells (λ = 492 nm) and represent the mean (±SD) of three biological replicates. On the right, the WB analysis reports MYC-SFPQ and GFP-SRSF2 expression in the used samples. b Dose-response analysis of MDAH cells transduced with the indicated shRNAs alone or in combination and then treated with increasing doses of CDDP. Caspase-3/7 activity was used as readout of apoptotic pathway activation. Data are expressed as luminescence (×106) and represent the mean (±SD) of three biological replicates. c IF analysis evaluating SRSF2 (green) localization and expression in MDAH cells transduced with the indicated shRNAs left untreated or with CDDP 50 µM for 16 h. Nuclei were stained with propidium iodide (pseudocolored in blue); scale bar = 7 µm. d RIP analysis in OVCAR8 cells transfected with empty vector, GFP-SRSF2 and MYC-SFPQ WT alone or in combination and then treated with CDDP 50 µM for 3 h. Samples were IP with anti-GFP antibody, and the RNAs purified from the IP samples evaluated for caspase-9 expression by RT-PCR. Overexpression of MYC-SFPQ WT and GFP-SRSF2 was confirmed by WB analysis. e Expression of caspase-9 pro (+) and anti (−) apoptotic isoforms in OVCAR8 cells (left panel), transfected with decreasing (6–0 µg) and increasing (6–0 µg) doses of MYC-SFPQ WT and GFP-SRSF2, respectively. Cells were treated for 3 h with 50 µM CDDP. Gapdh was used as loading control. In the left panel WB confirming MYC-SFPQ and GFP-SRSF2 overexpression and caspase-9 expression and cleavage. In the figure Vinculin and Fibrillarin were used as loading control. Statistical significance was determined by a two-tailed, unpaired Student’s t test. (*p < 0.05; **p < 0.01; ***p < 0.001).
Using immunofluorescence analyses, we observed that SFPQ and SRSF2 co-localized in PT-treated MDAH cells in nuclear speckles (Supplementary Fig. 7c, d) and that SFPQ knock-down altered the PT-induced re-localization of SRSF2 (Fig. 5c). Moreover, RIP analyses in PT-treated OVCAR8 cells demonstrated that SFPQ reduced the ability of SRSF2 to bind caspase-9 mRNA (Fig. 5d) and competition experiments, using increasing/decreasing concentrations of SFPQ and SRSF2 expressing vectors in OVCAR8 cells, showed that the antiapoptotic variant of caspase-9 mRNA progressively decreased with the increased expression of SRSF2 and the reduced expression of SFPQ (Fig. 5e). Accordingly, the expression of caspase-9 proapoptotic form paralleled the increased expression of SRSF2 protein (Fig. 5e).
p54nrb loss impairs SFPQ/SRSF2 interaction and influences caspase-9 splicing
Our data indicated that SFPQ binding to caspase-9 RNA could be mediated by another protein (Fig. 4f). The most common binding partner of SFPQ is p54nrb, which co-precipitated with both SFPQ and SRSF2, especially in PT-treated cells (Fig. 4c). Thus, we tested if p54nrb also played a role in caspase-9 mRNA splicing. First, we assessed that p54nrb was generally well expressed in EOC cells and its expression slightly increased in PT-Res MDAH clones (Fig. 2e and Supplementary Fig. 8a). As expected, SFPQ WT and mutant proteins readily bound to endogenous p54nrb and PT treatment did not modify the extent of their interaction in 293T17 cells (Fig. 6a). Differently to what observed in SFPQ-silenced cells (Fig. 3), MDAH cells stably silenced for p54nrb displayed a growth rate that was only slightly reduced compared with their control counterpart (Fig. 6b). However, the downregulation of p54nrb expression significantly increased PT-induced cell death (Fig. 6c). FACS analyses confirmed that in PT-treated MDAH cells, p54nrb loss led to increased fraction of apoptotic cells (sub-G1 fraction) and reduced cell accumulation in G2/M-phase, particularly evident 24 h after PT removal, during the DNA repair phase (Fig. 6d and Supplementary Fig. 8b).

a SFPQ-p54nrb co-IP analyses in cells, transfected with the indicated vectors treated or not with CDDP 50 µM for 3 h. Input reports the expression of the indicated protein in the lysates used for IPs experiments. An anti-MYC Ab was used for the IP and to evaluate SFPQ expression. GAPDH was used as loading control. Densitometric analysis of normalized p54nrb/SFPQ interaction is reported under the blot. b Representative phase contrast images (10× objective) of MDAH cells stable transduced with sh-ctrl or sh-p54nrb after puromycin selection. In the upper western blot the expression of p54nrb in control and silenced pools is shown. c Nonlinear regression analyses evaluating cell viability of MDAH cells transiently transduced with sh-p54nrb #2 and #5, as indicated, and treated with increasing doses of CDDP. The table reports the IC50 and the confidence interval (CI) of each condition. Data are expressed as percentage of viable cells compared with untreated cells and represent the mean (±SD) of three biological replicates. Fisher’s exact test was used to calculate the global p value reported in the graph. On the left, WB showing p54nrb and levels in transduced cells. d Cell cycle distribution of MDAH cells transduced with sh-ctrl and sh-p54nrb#5, subjected to FACS analysis, 72 h after transduction in untreated and CDDP-treated conditions (CDDP 50 µM, t0 = 16 h treatment; t24 = 16 h treatment + 24 h release). The percentages of cells in each cycle phase are indicated. Data represent the mean (±SD) of three biological replicates. e Co-IP analysis of endogenous SFPQ and GFP-SRSF2 in MDAH cells described in (b) (pool sh-p54nrb#1), transiently transfected with GFP-SRSF2. Cells were treated with 50 µM CDDP for 3 h, and subjected to cytoplasmic/nuclear fractionation. IP was performed with anti-SFPQ antibody on nuclear extracts. Under the blot is reported the quantification of SFPQ–SRSF2 interaction in the different conditions that represents the mean (±SD) of three biological replicates. f RIP analysis in MDAH cells transfected with Flag-p54nrb vector in untreated and treated conditions (CDDP 50 µM 3 h). Samples were IP with the indicated antibodies and the RNAs purified from the IP samples evaluated for caspase-9 expression by RT-PCR. g RIP analysis in MDAH cells (described in (b)), transiently transfected with GFP-SRSF2 and treated with CDDP 50 µM for 3 h. Samples were IP with the indicated antibodies, and RNAs purified from the IP samples evaluated for caspase-9 expression by RT-PCR (left panel). The right graph shows the densitometric analysis of the RIP and represents the expression of caspase-9 mRNA normalized on sh-ctrl set at 1 as reference. Data represent the mean (±SD) of three biological replicates. In the figure, GAPDH, Vinculin or Fibrillarin were used as loading control. In (d) and (g) statistical significance was determined by a two-tailed, unpaired Student’s t test (*p < 0.05; **p < 0.01).
Interestingly, in p54nrb silenced MDAH cells SFPQ binding to SRSF2 was reduced (Fig. 6e and Supplementary Fig. 8c, d), indicating that, in this context, p54nrb regulated the reciprocal interaction between SFPQ and SRSF2. Further, the RIP assay demonstrated that p54nrb bound the caspase-9 RNA and that this binding was increased by PT treatment (Fig. 6f). Moreover, in MDAH cells stably silenced for p54nrb, SRSF2 bound with higher affinity caspase-9 RNA (Fig. 6g), while its binding to caspase-8 was unaltered compared with control cells (Supplementary Fig. 8e), again confirming that the expression of SFPQ/p54nrb complex specifically regulated the splicing activity of SRSF2 on caspase-9 RNA, in response to PT.
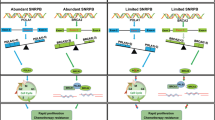
Overall, our work unveils a novel role for the SFPQ/p54nrb complex in the regulation of caspase-9 splicing. First, we observed that SFPQ/p54nrb binds to SRSF2 and their relative expression levels are critical to determine whether exons 4–7 of caspase-9 are retained or excluded from the mRNA, leading to different expression ratios of caspase-9 pro- or antiapoptotic variants. The inhibition SFPQ/p54nrb complex, combined with PT treatment, can increase the ability of SRSF2 to bind caspase-9 RNA, thus enhancing its proapoptotic form and consequently cell death (Fig. 7).

SFPQ, in complex with p54nrb, binds and regulates the activity of the splicing factor SRSF2, already identified as a key factor in caspase-9 alternative splicing regulation. In basal conditions, SFPQ/p54nrb complex prevents/decreases SRSF2 binding to caspase-9 RNA, favoring the expression of its antiapoptotic alternative spliced form and promoting cell survival. SFPQ/p54nrb downregulation augment the binding of SRSF2 to caspase-9 RNA and leads to increased expression of caspase-9 proapoptotic isoform, inducing cell death, in particular under platinum treatment. In this manner SFPQ/p54nrb complex protect EOC cells from PT-induced death, eventually contributing to chemoresistance.
Discussion
Here, we describe a previously unknown pathway that links the SFPQ/p54nrb complex to the regulation of PT-induced apoptosis, via the modulation of SRSF2 activity. Our data indicate that SFPQ binds to SRSF2 and counteracts its splicing activity on caspase RNAs, favoring the expression of antiapoptotic variants.
Several reports suggest that alternative splicing of specific genes, such as BRCA1, BRCA2, BARD1, or ERCC1, can impact on the response of cancer cells to PT and some studies also suggest that this correlates with PT resistance in human cancer [23,24,25,26,27,28]. Accordingly, it has been proposed that evaluation of alternative splicing of selected genes could serve as prognostic marker in ovarian cancer [29]. Our data demonstrate that the expression levels of SRSF2, SFPQ or p54nrb impact on the sensitivity of ovarian cancer cells to PT-induced death and support the possibility that targeting alternative splicing, could also represent a promising therapeutic option to overcome PT resistance.
RNA sequencing and proteomic screenings have already demonstrated that treatment with PT impacts on the expression of genes involved in splicing regulation, suggesting again that this pathway represents an effective and dynamic way for cancer cells to respond to treatment-induced DNA damage [13, 14, 30, 31]. Indeed, our data suggest that PT treatment induced alternative splicing of apoptotic genes well before the appearance of cell death, supporting the possibility that it represents a rapid adaptive modification to survive.
The observations that SFPQ is highly expressed in primary EOC human samples and that its expression increased in samples from PT-exposed and PT-Res patients, support the possibility that SFPQ plays a role during cancer progression. Therefore, its evaluation as prognostic or predictive biomarker of PT response might merit further investigation in additional EOC cohorts. In line with our findings in EOC, very recent data have proposed a role for SFPQ in prostate cancer, where it promotes the expression of the androgen receptor variant 7, associated with castration resistance [32], supporting a role for SFPQ in promoting cancer-specific progression and resistance to therapy via the regulation of the alternative splicing.
We have previously reported a role for the splicing factor SRSF2 in the regulation of PT-induced alternative splicing in ovarian cancer and other groups also supported this possibility [15, 16]. Since SRSF2 is often overexpressed in ovarian cancer [33] and mutated in PT-treated ovarian cancer [16], we have focused on the interaction between SFPQ, p54nrb, and SRSF2. Our findings indicate that PT finely regulates the extent and the duration of this binding. Further studies will be necessary to precisely identify if and which PT-induced modification(s), either transcriptional or posttranscriptional, could alter this newly described interaction.
Overall, SFPQ plays pleiotropic roles in cells and acts differently when cells are under basal conditions or PT treated. Here, guided by the results of an unbiased GEP of SFPQ-silenced EOC cells (Supplementary Fig. 3), we focused on the role of SFPQ in the regulation of the spliceosome. We are aware that SFPQ could regulate other pathways like the ubiquitin-mediated proteolysis (enriched in two different EOC cell lines) that will need to be investigated further and this certainly represents a limitation of our study. Future studies will likely clarify if SFPQ exerts additional functions that also contribute to the survival of EOC, under basal and PT-treated conditions.
Indeed, several evidences suggest that SFPQ plays additional roles beyond the control of SRSF2-mediated regulation of caspase-9 splicing. For instance, our data on the evaluation of PT-induced DNA damage (evaluated as γ-H2AX expression) suggested that SFPQ also participated to the control of DNA damage and repair. Further, we observed that caspase-9 inhibition reduced but did not completely reverted the PT-induced cell death observed in SFPQ-silenced cells (Fig. 3h, i), implying that other SFPQ-regulated mechanisms likely contributed to the response to PT in EOC cells. Accordingly, we observed an increased and anticipated expression of γ-H2AX in PT-treated cells that, conversely, was not changed in cells silenced for SRSF2 (Supplementary Fig. 9), suggesting that while SFPQ participated to the PT-induced DNA damage response, probably SRSF2 did not. This possibility is also supported by other data showing how the SFPQ/p54nrb complex regulates the DNA damage response in vitro and in vivo in other models [7, 34, 35].
In our screening, we used shRNA libraries targeting genes involved in the apoptosis/DNA damage and TP53 pathways and our experimental condition preferentially selected genes able to protect rather than sensitize to PT-induced death. It is therefore possible that we have missed the possibility to identify other targets or pathways that could be relevant for the regulation of PT response in EOC, or that some of the targets that have been identified could represent the consequence rather than the cause of EOC PT resistance. Also, it would be interesting to verify if the specific overexpression of caspase-9 antiapoptotic form could rescue the higher apoptosis induced by SFPQ/p54nrb silencing. Despite these limitations, our work has unveiled a critical role for the SFPQ/p54nrb complex in the response to PT in EOC cells. Our findings open the way for many new interesting investigations into the predictive and therapeutic implications of the alternative splicing in the processes of PT resistance and DNA damage response in ovarian cancer. Targeting the alternative splicing pathway is increasingly becoming a feasible opportunity to treat human diseases, including cancer [36,37,38]. Due to its crucial role in cell survival, the direct targeting of SFPQ seems not a pursuable strategy in patients. However, our data support the possibility that targeting p54nrb could represent an alternative and more specific way to improve PT activity, especially in PT-Resistant EOC patients, for whom new therapeutic options are urgently needed.
Materials and methods
Primary EOC collection
Human EOC samples were collected by CRO institutional Biobank, immediately frozen and stored in liquid nitrogen until needed. Informed consent was obtained from all patients. The CRO Internal Review Board approved this study with the number CRO-IRB #05-2014.
Loss-of-function screening
Loss-of-function screening was performed as described [17]. Briefly, the shRNAs library obtained by Sigma-Aldrich was used to perform the loss-of-function screening. On day one MDAH-2774 and SKOV3 cells were seeded in 96-well plates and the day after transduced with the specific shRNA or the control sh-ctrl. Seventy-two hours posttransduction cells were treated with CBDCA 140 µg/ml for 16 h. Cell viability was evaluated 24 h after the end of treatment using CellTiter 96 AQueous cell proliferation assay (MTS) kit (Promega).
RNA immunoprecipitation (RIP)
EOC cells (~1 × 107 for each condition), transfected with the indicated constructs, were lysed in complete RIP lysis buffer (nuclear isolation buffer, added with protease inhibitors cocktail (Roche) and RNAse (Promega)). After nuclear isolation and lysis of nuclear pellets, chromatin was sheared mechanically using sonication. The indicated (GFP/MYC/Flag or control IgG) antibodies were added to nuclear supernatant and incubated O/N at 4 °C. Protein A/G beads were added and incubated for 1 h 30 min at 4 °C. The immunoprecipitated samples were then centrifuged and washed with ice-cold RIP wash buffer, and the co-precipitated RNA was isolated by resuspending beads in TriZol (Invitrogen).
Gene expression profiling
GEP were performed essentially as described [20, 39]. Briefly, 150 ng of total RNA were labeled with Cyanine (Cy)-3 dye andCy3-labeled RNA was hybridized using the Affymetrix Human Whole Genome (8 × 60 k) oligo-microarray platform (Agilent Technologies) and analyzed with an Agilent Microarray Scanner (Agilent Technologies) using the Agilent Feature Extraction Software 10.7.3 (Agilent Technologies). Microarray data have been deposited in NCBI Gene Expression Omnibus repository (#GSE131539) and could be accessed through the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131539.
Statistical analyses
All statistical analyses and graphs were performed using GraphPad PRISM software (version 6.0) using the most appropriate test, as specified in each figure. Independent groups are subjected to the comparison under the assumptions of normal distribution and equal variances. Data are presented as the mean ± SD of at least three biological replicates unless otherwise specified. Sample size for each experiment is indicated in the figure legend; no a priori criteria were established to predetermine sample size. Significant differences between means were determined with two-tailed Student’s t tests. For nonlinear regression analyses, Fisher’s exact test was used to calculate the global p value reported in the graph. SFPQ/SRSF2 co-localization (immunofluorescence analysis) was analyzed by Pearson correlation. The differences were defined as statistically significant (*p < 0.05), highly significant (**p < 0.01), and extremely significant (***p < 0.001). No samples were excluded from the analyses and no randomization or blinding was used.
“Materials and methods” are better described in Supporting Materials.
References
Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384:1376–88.
Lheureux S, Gourley C, Vergote I, Oza AM. Epithelial ovarian cancer. Lancet. 2019;393:1240–53.
Konstantinopoulos PA, Matulonis UA. Targeting DNA damage response and repair as a therapeutic strategy for ovarian cancer. Hematol/Oncol Clin N Am. 2018;32:997–1010.
Freimund AE, Beach JA, Christie EL, Bowtell DDL. Mechanisms of drug resistance in high-grade serous ovarian cancer. Hematol/Oncol Clin N Am. 2018;32:983–96.
Knott GJ, Bond CS, Fox AH. The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 2016;44:3989–4004.
Shav-Tal Y, Zipori D. PSF and p54(nrb)/NonO—multi-functional nuclear proteins. FEBS Lett. 2002;531:109–14.
Yarosh CA, Iacona JR, Lutz CS, Lynch KW. PSF: nuclear busy-body or nuclear facilitator? Wiley Interdiscip Rev RNA. 2015;6:351–67.
Shav-Tal Y, Cohen M, Lapter S, Dye B, Patton JG, Vandekerckhove J, et al. Nuclear relocalization of the pre-mRNA splicing factor PSF during apoptosis involves hyperphosphorylation, masking of antigenic epitopes, and changes in protein interactions. Mol Biol Cell. 2001;12:2328–40.
Heyd F, Lynch KW. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol Cell. 2010;40:126–37.
Tsukahara T, Matsuda Y, Haniu H. PSF knockdown enhances apoptosis via downregulation of LC3B in human colon cancer cells. Biomed Res Int. 2013;2013:204973.
Merdzhanova G, Edmond V, De Seranno S, Van den Broeck A, Corcos L, Brambilla C, et al. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008;15:1815–23.
Kędzierska H, Piekiełko-Witkowska A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017;396:53–65.
Gabriel M, Delforge Y, Deward A, Habraken Y, Hennuy B, Piette J, et al. Role of the splicing factor SRSF4 in cisplatin-induced modifications of pre-mRNA splicing and apoptosis. BMC Cancer. 2015;15:227.
Lambert CA, Garbacki N, Colige AC. Chemotherapy induces alternative transcription and splicing: facts and hopes for cancer treatment. Int J Biochem Cell Biol. 2017;91:84–97.
Edmond V, Moysan E, Khochbin S, Matthias P, Brambilla C, Brambilla E, et al. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J. 2011;30:510–23.
Nicoloso MS, Schiappacassi M, Dall’Acqua A, D’Andrea S, Benevol S, Sorio R, et al. SRSF2 mutations in epithelial ovarian cancer. Cancer Break News. 2017;5:25–9.
Sonego M, Pellarin I, Costa A, Vinciguerra GLR, Coan M, Kraut A, et al. USP1 links platinum resistance to cancer cell dissemination by regulating Snail stability. Sci Adv. 2019;5:eaav3235.
Sonego M, Schiappacassi M, Lovisa S, Dall’Acqua A, Bagnoli M, Lovat F, et al. Stathmin regulates mutant p53 stability and transcriptional activity in ovarian cancer. EMBO Mol Med. 2013;5:707–22.
Dall’Acqua A, Sonego M, Pellizzari I, Pellarin I, Canzonieri V, D’Andrea S, et al. CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol Med. 2017;9:1415–33.
Sonego M, Pellizzari I, Dall’Acqua A, Pivetta E, Lorenzon I, Benevol S, et al. Common biological phenotypes characterize the acquisition of platinum-resistance in epithelial ovarian cancer cells. Sci Rep. 2017;7:7104.
Lorenzon I, Pellarin I, Pellizzari I, D’Andrea S, Belletti B, Sonego M, et al. Identification and characterization of a new platinum-induced TP53 mutation in MDAH ovarian cancer cells. Cells. 2019;9. https://doi.org/10.3390/cells9010036.
Rosonina E, Ip JYY, Calarco JA, Bakowski MA, Emili A, McCracken S, et al. Role for PSF in mediating transcriptional activator-dependent stimulation of pre-mRNA processing in vivo. Mol Cell Biol. 2005;25:6734–46.
Yu JJ, Mu C, Dabholkar M, Guo Y, Bostick-Bruton F, Reed E. Alternative splicing of ERCC1 and cisplatin-DNA adduct repair in human tumor cell lines. Int J Mol Med. 1998;1:617–20.
Sun Y, Li T, Ma K, Tian Z, Zhu Y, Chen F, et al. The impacts of ERCC1 gene exon VIII alternative splicing on cisplatin-resistance in ovarian cancer cells. Cancer Investig. 2009;27:891–7.
Sevcik J, Falk M, Kleiblova P, Lhota F, Stefancikova L, Janatova M, et al. The BRCA1 alternative splicing variant Δ14-15 with an in-frame deletion of part of the regulatory serine-containing domain (SCD) impairs the DNA repair capacity in MCF-7 cells. Cell Signal. 2012;24:1023–30.
Sevcik J, Falk M, Macurek L, Kleiblova P, Lhota F, Hojny J, et al. Expression of human BRCA1Δ17-19 alternative splicing variant with a truncated BRCT domain in MCF-7 cells results in impaired assembly of DNA repair complexes and aberrant DNA damage response. Cell Signal. 2013;25:1186–93.
Meyer S, Stevens A, Paredes R, Schneider M, Walker MJ, Williamson AJK, et al. Acquired cross-linker resistance associated with a novel spliced BRCA2 protein variant for molecular phenotyping of BRCA2 disruption. Cell Death Dis. 2017;8:e2875.
Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). PNAS. 2008;105:20852–7.
Zhu J, Chen Z, Yong L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol Oncol. 2018;148:368–74.
Wu W, Yan C, Gan T, Chen Z, Lu X, Duerksen-Hughes PJ, et al. Nuclear proteome analysis of cisplatin-treated HeLa cells. Mutat Res/Fundam Mol Mech Mutagen. 2010;691:1–8.
Zhang G, Sun L, Lu X, Chen Z, Duerksen-Hughes PJ, Hu H, et al. Cisplatin treatment leads to changes in nuclear protein and microRNA expression. Mutat Res. 2012;746:66–77.
Takayama K, Suzuki T, Fujimura T, Yamada Y, Takahashi S, Homma Y, et al. Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA-binding protein PSF. PNAS. 2017;114:10461–6.
Fischer D-C, Noack K, Runnebaum IB, Watermann DO, Kieback DG, Stamm S, et al. Expression of splicing factors in human ovarian cancer. Oncol Rep. 2004;11:1085–90.
Jaafar L, Li Z, Li S, Dynan WS. SFPQ•NONO and XLF function separately and together to promote DNA double-strand break repair via canonical nonhomologous end joining. Nucleic Acids Res. 2017;45:1848–59.
Li S, Shu F-J, Li Z, Jaafar L, Zhao S, Dynan WS. Cell-type specific role of the RNA-binding protein, NONO, in the DNA double-strand break response in the mouse testes. DNA Repair. 2017;51:70–8.
Lee SC-W, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22:976–86.
Lee SC-W, Dvinge H, Kim E, Cho H, Micol J-B, Chung YR, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22:672–8.
Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24:497–504.
Segatto I, Zompit MDM, Citron F, D’Andrea S, Vinciguerra GLR, Perin T, et al. Stathmin is required for normal mouse mammary gland development and Δ16HER2-driven tumorigenesis. Cancer Res. 2019;79:397–409.
Acknowledgements
We are grateful to the patients who consented to donating their samples. We thank Dr Benjamin Blencowe for providing the p54nrb and the SFPQ wild type and mutant expression vectors (through Addgene) and Dr Beatrice Eymin for providing the SRSF2 vector. We like to thank all members of the SCICC lab for fruitful scientific discussion, Dr Riccardo Bomben for helping us with the analysis of gene expression profiles and Dr Francesca Rossi for FACS analyses. This work was supported by grants from Ministero della Salute (RCR-2019-23669115 and RF-2016-02361040 to GB and GR-2016-02361041 to MS); Ministero degli Affari Esteri e Cooperazione Internazionale (PGR01036 to GB), CRO-Ricerca Corrente core grant (linea 1); 5 × 1000 CRO Intramural to GB and Regione Friuli Venezia Giulia (POR-FERS TICHEP grant to GB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pellarin, I., Dall’Acqua, A., Gambelli, A. et al. Splicing factor proline- and glutamine-rich (SFPQ) protein regulates platinum response in ovarian cancer-modulating SRSF2 activity. Oncogene 39, 4390–4403 (2020). https://doi.org/10.1038/s41388-020-1292-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-1292-6
This article is cited by
-
The pleiotropic nature of NONO, a master regulator of essential biological pathways in cancers
Cancer Gene Therapy (2024)
-
Single-cell RNA binding protein regulatory network analyses reveal oncogenic HNRNPK-MYC signalling pathway in cancer
Communications Biology (2023)
-
RNA-binding proteins in ovarian cancer: a novel avenue of their roles in diagnosis and treatment
Journal of Translational Medicine (2022)
-
SF3B4 promotes ovarian cancer progression by regulating alternative splicing of RAD52
Cell Death & Disease (2022)
-
Transcriptome analysis of clock disrupted cancer cells reveals differential alternative splicing of cancer hallmarks genes
npj Systems Biology and Applications (2022)
