
Abstract
Radiation is a significant treatment for patients with head and neck cancer. Despite advances to improve treatment, many tumors acquire radiation resistance resulting in poor survival. Radiation kills cancer cells by inducing DNA double-strand breaks. Therefore, radiation resistance is enhanced by efficient repair of damaged DNA. Head and neck cancers overexpress EGFR and have a high frequency of p53 mutations, both of which enhance DNA repair. This review discusses the clinical criteria for radiation resistance in patients with head and neck cancer and summarizes how cancer cells evade radiation-mediated apoptosis by p53- and epidermal growth factor receptor (EGFR)-mediated DNA repair. In addition, we explore the role of cancer stem cells in promoting radiation resistance, and how the abscopal effect provides rationale for combination strategies with immunotherapy.
Similar content being viewed by others
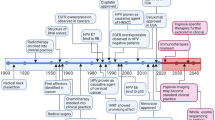

Introduction
Globally, over 650,000 head and neck cancers are diagnosed each year, and ~50% of patients succumb to their disease [1]. More than 90% of head and neck cancers are squamous cell carcinomas (HNSCC). In the United States, HNSCC account for 4% of all cancers. It is projected that in 2020 alone over 53,000 people in the United States will develop the disease and nearly 11,000 people will die within the year [2]. Some of the major risk factors include tobacco use, alcohol consumption, and human papilloma virus 16 (HPV16) infection. Treatment decisions are determined by taking into consideration factors such as the primary site, stage, surgical accessibility, associated morbidity, and the patient’s general health. Radiation is one of the chief treatment modalities for the management of HNSCC. Early stage disease is typically treated with radiation or surgery; locoregionally advanced disease is usually tackled with combined approaches including surgery followed by adjuvant therapy, definitive chemoradiation, or bioselection [3]. Therefore, a critical determinant of local control of HNSCC is dependent on the tumor’s sensitivity to radiation (Fig. 1). Overexpression of epidermal growth factor receptor (EGFR) and p53 mutations have been linked to treatment resistance in HNSCC. EGFR is overexpressed in 90% of HNSCCs and p53 is the most common somatic mutation [4,5,]. Both EGFR and p53 are implicated in repairing radiation-induced DNA damage. This review discusses the clinical criteria for radiation resistance in patients with HNSCC and summarizes how cancer cells evade radiation-mediated apoptosis by p53- and EGFR-mediated DNA repair. Additional entities such as the presence of cancer stem cells (CSCs) as a cause of resistance are also addressed.

Early stage disease is treated with a unimodality approach while later stage disease requires a multimodality approach.
Radiation resistance
The significant role of ionizing radiation for the management of HNSCC is irrefutable. Currently, a large number of patients with HNSCC undergo some form of external beam radiotherapy (EBRT). Intensity-modulated radiation therapy (IMRT) is a form of EBRT that delivers very precise radiation doses to the entire tumor or specific areas within the tumor, utilizing computer-controlled X-ray accelerators that conform to the three-dimensional structure of the tumor. This helps to significantly reduce damage to neighboring normal tissue during treatment. Alternatively, radiation may be internally administered utilizing radioactive implants (brachytherapy) that are removed when the appropriate dose has been administered.
Proton therapy is a recent advance in radiotherapy. Bragg peak refers to the physical properties of a proton beam that allow rapid decrease in radiation dose beyond the tumor, leading to lower normal tissue doses. Phase II data demonstrated efficacy, safety, and favorable toxicity profiles in base of skull malignancies, periorbital tumors, nasopharynx cancer, and re-irradiation while studies in other head and neck malignancies are ongoing [5, 6]. Furthermore, the Patel et al.’s meta-analysis of multiple single institution series has shown very favorable 5-year overall survival of patients treated with charged particles compared with those treated with photon radiotherapy [7]. However, there is not yet Phase III data in HNSCC to suggest improved tumor control or decreased toxicity associated with the use of proton-beam therapy. NCT01893307 is an ongoing Phase II/III randomized trial in oropharyngeal cancer that compares outcomes after chemoradiation given by proton versus photon therapy. The results from this trial should provide insight after its completion.
Patients with HNSCC typically receive ionizing radiation over several weeks in daily sessions on weekdays. A standard dose of 70 Gy in 35 fractions is usually given over 7 weeks. The doses may be administered via accelerated fractionation, hyperfractionation, or hypofractionation. Accelerated fractionation involves dividing the total dose into small doses that may be given more than once per day with reduction in total treatment time. Accelerated fractionation and particularly hyperfractionation (twice daily treatment) are more efficacious in maintaining local tumor control and improving disease-free survival over standard fractionation radiation alone, with inferior outcomes compared with concurrent chemoradiation [8]. Hypofractionation involves administering larger doses once per day or less frequently but spans a shorter period than the standard therapy. Hypofractionation is not commonly used in the definitive management of HNSCC due to concerns of late side effects including dysphagia and fibrosis.
Locoregional relapse is a significant cause of patient mortality and morbidity, and is an indicator of treatment failure [9]. Within 5 years from the end of treatment, approximately a third of patients with HNSCC develop locoregional failure and their prognosis is significantly attenuated [10]. p16-negative disease is a risk factor for locoregional failure following definitive treatment [11]. Locoregional control is also critically important to quality of life as tumor progression can lead to deficits in speech, eating, social interactions, physical deformities, and painful nonhealing wounds.
Although radiation eradicates a large fraction of tumor cells, selected groups of tumor cells (clonogens) are able to survive and repopulate irradiated areas. While 5-year overall survival ranges from 40 to 60% in patients treated exclusively with radiation or chemoradiation, ~25% of patients treated with radiation suffer from local relapse [12]. Tumors are labeled as “radiation-resistant” if recurrences are observed within 6 months following the first course of radiation. Treatment options are significantly diminished for patients with recurrent disease or second primary tumors in the previously irradiated field. The time a patient remains disease-free prior to tumor recurrence is a significant factor when deciding the therapeutic benefit of re-irradiation. Surgical salvage is often the primary treatment modality following failure of radiation [13] with reported 5-year survival rates up to 30%. Patients with positive margins, lymphovascular invasion, perineural invasion, and extra nodal extension are deemed high risk for recurrence subsequent to surgical salvage. In addition, some patients may benefit from post-operative re-irradiation, which was reported to improve progression-free survival in combination with chemotherapy in a randomized Phase III trial conducted in France [14].
Radiation-induced DNA damage and repair
Upon cellular exposure to radiation, charged particles either directly pass through and ionize DNA or produce reactive oxygen species by ionizing nearby water molecules that in turn react with DNA. These interactions elicit lethal DNA damage. Whereas reactive free radicals can induce a wide gamut of lesions, DNA double-strand breaks (DSBs) are credited to be the most cytotoxic lesions generated from radiation. DSBs involve breaks in the phosphodiester backbone of both strands of DNA separated by ~10 bases or less [15]. The production of DSBs increases linearly with radiation dose [16]. In fact, radiation induces approximately 850 pyrimidine lesions, 450 purine lesions, 1000 single-strand breaks, and on average 30 DSBs/cell/Gy of low linear energy transfer γ-radiation [17, 18]. The standard fractionating dose of 2 Gy/cycle is capable of inducing roughly 3000 DNA lesions per exposed cell, and merely 40 DSBs are required to induce cell death [17, 19]. Generation of free radicals that may lead to DNA damage also occurs during normal cellular metabolic processes, recombination during meiosis, and DNA replication. Evolutionarily, eukaryotes have developed the innate ability to repair DNA. Homologous recombination (HR) and nonhomologous end joining (NHEJ) are the major pathways responsible for repairing DSBs. HR requires homologous DNA sequences from sister chromatids to repair DSBs. HR is therefore restricted to phases of the cell cycle where sister chromatids are available (late S to G2 phases) [20]. In contrast, NHEJ is a promiscuous repair mechanism that directly ligates two broken ends and does not require sequence homology. Hence, NHEJ occurs at all stages of the cell cycle. NHEJ is more error-prone than HR and often includes base deletions and insertions [21]. NHEJ is initiated by Ku proteins. Ku is a heterodimeric protein consisting of Ku70 and Ku80 subunits. It has a high affinity for the ends of DNA and once bound, forms a holoenzyme with DNA-dependent protein kinase catalytic subunit (DNA-PKcs). The DSB termini are processed via activation of artemis, polynucleotide kinase/phosphatase (PNKP), and aprataxin-PNKP-like factor (APLF) if necessary. Subsequently, other proteins involved in the repair pathway get phosphorylated including autophosphorylation of DNA-PKcs. Then gaps are filled by polymerase μ and γ followed by ligation by ligase IV, X-ray repair complementing 4 (XRCC4), and XRCC4-like factor (XLF) (Fig. 2). It should be noted that while Ku is drafted to the site of all DSBs, DNA-PKcs only participates in repair of complex DSBs [22].

Broken DNA ends are protected by Ku70/80 before the recruitment of DNA/PKcs and processing by artemis. Damaged bases are replaced and broken ends are ligated.
Radiation-induced DNA damage and proliferation
The cell cycle involves a highly regulated set of events that allows cells to proliferate. During the cycle, cells pass through the G0 (resting), G1 (first growth phase), S (DNA replication), G2 (growth), and M (mitosis) phases. Progression through the cell cycle is regulated by cyclin-dependent kinases (CDKs) and cyclins. CDKs remain inactive until they are bound to their associated cyclin subunits. The availability of cyclins is controlled by regulating their synthesis and proteolysis. When cyclins bind CDKs, the conformation of the CDKs is switched to an active state, thereby phosphorylating downstream molecules necessary for cell-cycle progression. Checkpoints are responsible for sensing defects during DNA synthesis and chromosome segregation to maintain cell-cycle integrity. Checkpoint pathways maintain CDKs in inactive states until a DNA lesion is resolved. It is crucial that cells maintain tightly regulated mechanisms to direct cell-cycle progression, especially when cells confront cellular stressors such as radiation. If checkpoints are too tightly regulated, while there would be less occurrences of chromosomal instability, cell proliferation would be significantly attenuated, and survival of the organism would be compromised. In contrast, if checkpoints are bypassed easily, cells would proliferate regardless of the presence of DNA damage and ultimately increase risks of chromosomal instability and cancer development.
Activation of ataxia telangiectasia mutated (ATM) subsequent to recognition of radiation-induced DSBs is a key event in activating checkpoints. Sensing DNA damage in the G1 phase postirradiation results in phosphorylation of H2AX adjacent to the DSB predominantly by ATM [23]. ATM and Chk2 stabilize p53 by dissociating it from MDM2, thereby activating downstream effectors such as p21. Accumulation of p21 in G1 inhibits cyclin/CDK complexes and deters cell-cycle progression. p21 binds to cylin E/CDK2 and cyclin D/CDK4/6 complexes. Deng et al. reported that complete knockout of p53 or p21 results in total loss of the G1 checkpoint [24]. In addition, ATM stabilizes p21 encoding mRNA by activating p38 MAPK [25]. Since p53 activation and effector functions involve transcriptional activation subsequent to posttranscriptional modifications, cell-cycle arrest may require 4–6 h post irradiation. Consequently, cells may progress to the S phase even with high doses of radiation administered in the late G1 phase [26]. DNA damage during the S phase may disturb replication fork progression, and damage to bases may result in point mutations from base mispairings. In addition, formation of DNA–DNA or DNA–protein crosslinks could impede replication fork progression. If DSBs remain unrepaired, further DSBs and chromosomal breaks may occur. Hence, following DSB formation, cell-cycle progression is halted to allow repair. Radiation may also induce cell-cycle arrest in the G2 phase of the cell cycle. The G2/M checkpoint prevents cell division until DSBs are efficiently repaired. Entry into mitosis is induced by the cyclin B1/CDK1 complex. DNA-damaging agents such as radiation disrupt feedback loops that are responsible for further activation of cyclin B1/CDK1. DSBs activate ATM, which phosphorylates Chk2 which phosphorylates Cdc25 resulting in inactivation of cyclin B1/CDK1. The consequence of this chain of events is rapid G2 arrest. The role of p53 in G2/M arrest needs further investigation [27]. There is a caveat to G2/M arrest; increasing evidence suggests that cells are released from G2 arrest after reduction of DSBs below a 10–20 DSB threshold [28]. Regulation of both G1/S and G2/M cell-cycle checkpoints maintain proper cell proliferation.
Immunomodulatory effects of radiation
In addition to cytotoxic effects, radiation plays a role in immune modulation in the tumor and the tumor microenvironment. Radiation augments antigen-specific antitumor immune responses through various proposed mechanisms. These mechanisms include: (a) increasing activation and proliferation of tumor-infiltrating lymphocytes, (b) altering chemokines that preferentially recruit cytotoxic T lymphocytes and lead to MHC I upregulation, (c) fostering the release of tumor neoantigens via inflammatory cell death, and (d) activation and migration of dendritic cells. Furthermore, an abscopal effect has been noted where irradiation in one area results in tumor regression outside the field of radiation. Interestingly, one of the first reports of this effect was in head and neck cancer in the early 1900s; it is a rare phenomenon [29]. Radiation in combination with immunotherapy has therefore gained traction for HNSCC evidenced by the numerous ongoing clinical trials (Table 1). Keynote 048 showed that pembrolizumab/chemotherapy had a superior overall survival with comparable safety in the overall population versus cetuximab/chemotherapy leading to FDA approval of pembrolizumab as a first-line standard-of-care treatment for recurrent or metastatic HNSCC. Reports of abscopal effects induced by radiation have been noted with higher doses of radiation. For this reason, clinical trials investigating immune checkpoint blockade (ICB) and stereotactic body radiation therapy (SBRT) where precise intense doses can be administered in a shorter period of time as compared with conventional fractionation schemes, are being explored. High ablative doses of RT may superiorly potentiate the abscopal effect compared with fractionated RT [30]. The timing and dose schedules of RT and ICB are still being optimized, but concurrent administration with hypofractionated SBRT seems reasonable.
Mechanism of radiation resistance in HNSCC
Radiation sensitivity is dependent on the amount of DNA damage induced within the cell and the cell’s ability to activate repair mechanisms through DNA-damage response (DDR) pathways [31]. Subsequent to failure of DDR activation and repair of cellular DNA, cells cannot divide, and die via mechanisms that include apoptosis, necrosis, senescence, mitotic catastrophe, or autophagy [32]. Radioresistant cancer cells have an increased propensity to augment the DDR rate. As mentioned earlier, the major radiation-induced DSB repair mechanism is NHEJ. Multiple proteins involved in NHEJ are associated with radioresistance in HNSCC. For example, overexpression of TRIP13 promotes NHEJ repair and treatment resistance in vitro [33]. In addition, Ku80 expression is correlated with radiation resistance in vitro, and abrogating Ku80 restores sensitivity [34]. In fact, Ku80 protein expression is associated with locoregional failure and death, post radiotherapy [35]. ATM is a key player in DSB repair, and its kinase activity is responsible for the activation of key proteins such as CHK2. In addition, ATM is crucial for phosphorylation of DNA-PKcs at Thr-2609 in response to radiation, thereby playing a fundamental role in NHEJ repair [36]. Therefore, not surprisingly, disrupting ATM function permits radiosensitization of tumors [37].
There has been increased emphasis on elucidating the role of HPV16 in response to radiation. While HPV16 accounts for between 60 and 80% of HPV-related HNSCC, HPV18 accounts for ~2.5% and other subtypes (HPV33, 35, 31, 52, 39, and 45) have been reported to account for between 11 and 16% [38]. HPV16 is a DNA virus with oncogenic properties. Many studies have made etiological associations between HPV16 and oropharyngeal squamous cell carcinomas originating from the base of tongue and tonsils, and to a lesser extent in laryngeal and oral cavity cancers [39, 40]. Expression of HPV16 E6 and E7 oncoproteins allows cells to bypass normal antiproliferative control mechanisms and supports tumorigenesis. E6 protein can bind to p53 resulting in ATP-dependent degradation of p53, whereas E7 targets and binds to the retinoblastoma tumor suppressor protein pRb, impairing function. Since HPV16-associated HNSCC is relatively radiosensitive and given the role of HPV16-associated oncoproteins in inactivating wild-type (WT) p53 function, it is possible that failure to respond to radiation in the same subset of patients can be overcome by restoring p53 function through mechanisms such as gene therapy.
P53 activation/inactivation
A crucial element of the DDR machinery is the activation of tumor suppressor protein p53 by kinases such as ATM and DNA-PKcs. The major outcomes associated with p53 induction are cell-cycle arrest and DNA repair or apoptosis [41]. Tissue type, extent of damage, duration of stress, and the cell’s environment determine these outcomes. Details of this decision-making process are especially relevant in clinical settings where p53 status may be important for response to treatment with DNA-damaging agents such as radiation. Low levels of transient stress could trigger repairable DNA damage; then a survival response is elicited, and p53 acts as a protector. In this case, activation of p53 mediates cell-cycle arrest and DNA repair. In contrast, high levels of sustained stress, which lead to irreparable damage, activate the killer functions of p53 [42]. When the killer function is activated, p53 eliminates damaged cells via apoptosis or cellular senescence [43].
Mutations in TP53 have been associated with high rates of locoregional recurrence, increased resistance to therapy, and reduced survival. TP53 mutations may be classified as disruptive or nondisruptive. Disruptive mutations involve aberrations within the DNA binding domains or a truncated p53 protein due to the presence of an early stop codon; these alterations result in a complete loss of function. In contrast, nondisruptive mutations partially affect the normal functionality of p53 [44]. Cellular exposure to radiation induces DSBs that activate p53. In cancer cells with mutated p53, repair of radiation-induced DNA damage is substandard yet proficient enough to generate clones with an accumulation of genetic mutations that confer resistance. Furthermore, loss-of-function mutations in p53 disable cell-cycle arrest and apoptosis resulting in cell survival and radiation failure [45]. Dominant negative mutations may also inactivate the function of the remaining WT allele. Similarly, gain-of-function mutations may lead to resistance to DNA damage-induced cell death via downregulation of proapoptotic genes and upregulation of prosurvival genes [46]. Gain-of-function mutations were identified as p53 aberrations that conferred additional oncogenic properties to tumor cells. The cell type, stimuli, position, and nature of the substitution of p53 determines its gain-of-function potential [4]. Gain-of-function mutations can also increase DNA repair, induce replicative stress and genomic instability, inactivate ATM, promote proliferation, migration, invasion, and deregulate metabolism, all of which may contribute to resistance [47].
Induction of ATM by radiation inhibits MDM2, the negative regulator of p53. DNA repair at the G1/S and G2/M checkpoints prevents propagation of errors to daughter cells. Activated p53 binds to and transactivates the p21 promoter [48]. The predominant consequence of p21 activation is the inhibition of Cdk2 and Cdk4 allowing a hypophosphorylated form of pRb to remain bound to E2F1 resulting in G1 arrest (Fig. 3) [49]. E2F1 is a transcription factor with the pivotal function of regulating genes responsible for cell proliferation in a cell-cycle dependent manner.

Activation of ATM activates p53, which depending on severity of the damage executes one of the two functions: cell-cycle arrest or apoptosis. Cells undergo cell-cycle arrest if damage is repairable and apoptose if irreparable.
Although HPV16-positive oropharyngeal HNSCCs are relatively radiosensitive the mechanism is unclear. Speculations include an association with p53 status. TP53 mutations are present in 75–85% non-HPV16-related HNSCC [50]. Most HPV16-positive tumors have WT p53 and are more sensitive to radiation than HPV-negative tumors. Radiation-sensitive cells generally have higher basal levels of p53 mRNA while suppression of p53 in tumors results in increased resistance to radiation. With functional p53, apoptotic pathways can be initiated upon exposure to radiation. In contrast, HPV16 E6 protein ubiquitinates p53, targeting it for degradation. Hence, cell-cycle arrest is inhibited in response to DNA damage and makes cells more susceptible to genomic instability [51]. This may be a mode of escape for radiation-resistant HPV16-associated tumors. The differential response to treatment between HPV16-positive and -negative HNSCC provides a valid reason to consider treatment de-escalation for HPV16-positive patients. However, deeper knowledge of the biology is necessary to justify this approach. Clinical trials have been designed to assess the clinical outcomes associated with reduced radiation treatment in HPV16-driven HNSCC (Table 2).
Tumors of lymphoid or myeloid lineage are usually more sensitive to radiation than epithelial-derived tumors like HNSCC, which undergo significantly less apoptosis [52,53,]. The desired outcome for patients treated with radiation is to activate the killer functions of p53. With sufficient DNA damage, p53 induces expression of a number of genes involved in apoptosis. Radiation induces apoptosis by activating caspases via intrinsic or extrinsic pathways. Through the intrinsic pathway, p53 promotes transcription of proapoptotic proteins primarily belonging to the BCl-2 and BH3-only family. Some of these proteins include Bax, Puma, Noxa, and Bim [53, 54]. p53 may also upregulate genes that lower the apoptotic threshold and mediate repression of antiapoptotic genes [55,56,57]. The extrinsic apoptotic pathway also called the death receptor pathway requires activation of TNF receptor family members by specific ligands. Radiation increases many of these receptors leading to activation of caspases. Several studies in mice have highlighted p53’s ability to induce apoptosis thereby inhibiting spontaneous tumors, delaying tumor progression in mice with activated oncogenes, and inducing regression of fully developed tumors. p53 null mice were reported to be susceptible to spontaneous thymic lymphomas, and apoptosis of thymocytes post irradiation was dependent on p53 [58]. In addition, tumors with mutated T antigen in the p53 binding domain showed rapid growth and reduced apoptosis [59]. This suggests that p53 restricts tumor expansion by inducing apoptosis. Furthermore, restoring normal p53 in hepatocellular carcinomas and sarcomas resulted in tumor regression via p53-mediated senescence [60, 61].
Epidermal growth factor receptor
EGFR is a tyrosine kinase transmembrane receptor that is activated by ligands including EGF and transforming growth factor (TGFα and β). Ligand-mediated EGFR activation results in autophosphorylation of the intracellular domain with downstream activation of PI3K and Ras pathways to elicit survival and proliferation. EGFR is overexpressed in over 90% of HNSCC and is associated with poor clinical outcome [62]. Patients with high EGFR show higher rates of postradiation locoregional failure, suggesting that EGFR contributes to radiation resistance [63]. Radiation mimics ligand–receptor interaction by inducing autophosphorylation of EGFR [64], which leads to hyperactivation of Ras and PI3K pathways that support proliferation and survival of tumor cells, and ultimately radiation failure. Another caveat of radiation is that after autophosphorylation and Ras activation, the downstream effector MAPK mediates synthesis of EGF, amphiregulin, and TGF monomers that induce an autocrine loop, leading to overactive proliferative pathways.
EGFR may modulate repair of radiation-induced DSBs via formation of an EGFR–DNA-PK complex [65]. There is uncertainty about the intracellular site of the initial EGFR and DNA-PK interaction. These proteins may associate in the nucleus following translocation of EGFR, which results in increased repair of radiation-induced DNA damage [66]. The EGFR–DNA-PK complex is formed within 5 min of radiation with optimal activity of DNA-PK at ~10 min [65, 67]. EGFR is present on the plasma membrane, and upon radiation, evades internalization and degradation, and translocates to the nucleus [68]. In addition, EGFR in the perinuclear space translocates into the nucleoplasm in a ligand-independent manner when subjected to radiation [65]. DNA-PKcs and Ku are generally nuclear proteins, but may localize in lipid rafts and interact with membrane proteins, and can translocate from the nucleus to cytosol [69, 70]. Regardless of where the interaction occurs, it undoubtedly constitutes a critical component of EGFR-mediated radioprotection.
Cells with WT EGFR display diminished radiosensitivity compared with mutant counterparts. However, the radioprotective effect of WT EGFR is lost in DNA-PKcs-deficient cells [66, 71]. EGFR also regulates transcription of ATM. ATM plays a pivotal role in phosphorylation of DNA-PKcs, which mediates DSB repair [36]. When cancer cells are irradiated and undergo cell-cycle arrest, EGFR contributes to successful repair, allowing cells to exit the arrested phase.
For these reasons, EGFR inhibition and radiation has been an attractive treatment combination. Clinical trial data suggested that cetuximab plus radiation was superior to radiation alone for patients with locoregional disease [72]. However, the RTOG 0522 study demonstrated no benefit of cetuximab-radiation over cisplatin-radiation for treatment of locoregionally advanced oropharyngeal HNSCC [73]. Furthermore, in two randomized trials of definitive chemoradiation with concurrent cisplatin versus cetuximab, cetuximab resulted in inferior outcomes to platinum with similar overall toxicity [74, 75]. RTOG 1016 reaffirmed concurrent cisplatin as the standard of care for platinum eligible patients. Cetuximab (versus altered fractionated RT alone) is an option for nonplatinum eligible patients, both HPV16-positive and HPV16-negative. To this end, NRG HN 004 is currently testing RT/cetuximab versus RT/anti-PD1 in noncisplatin eligible patients. Results of these trials highlight the need to predict patient populations who will respond to the combination treatment and/or identify other targetable molecular markers that may improve patient outcome. The future of EGFR inhibition in HNSCC likely depends on predictive biomarkers.
Additional players in radiation resistance
Phenotypic and functional heterogeneity of HNSCC contributes to the challenges faced when treating the disease. Genomic instability results in an accumulation of mutations that contribute to tumor heterogeneity and developmental diversity of cells within the tumor mass. CSCs have been identified within tumors as self-renewing cells with the potential to differentiate into heterogenous lineages in the tumor. Although these cells account for only 1–5% of the HNSCC population, they have been implicated in radiation resistance, recurrence, and metastases. The mechanism of CSC-mediated radioresistance is unclear, but these slow cycling cells are believed to exhibit increased efficiency in DNA repair (via regulation of DNA repair genes and activation of DNA-damage checkpoint responses) and an upregulation of antiapoptotic proteins. Paradoxically, there are reports highlighting that radiation itself may induce non-CSCs to become CSCs [76].
Radiation sensitizers in HNSCC: targeting DNA repair pathways
Since radiation-induced DSBs are primarily repaired via NHEJ, targeting NHEJ has the potential to radiosensitize tumor cells. NHEJ repair consists of four stages including termini recognition, bridging, processing, and ligation of DNA. In the initial step, the Ku70/Ku80 complex is recruited to the DNA terminus of a DSB, and then recruits DNA-PKcs leading to formation of a heterotrimeric complex. Ku localizes within seconds of DSB formation due to high affinity for DNA termini and high concentration within the cell. There are ~0.5 × 106 Ku molecules per cell with a DNA binding affinity between 2.4 × 10−9 and 5 × 10−10 M−1 [77, 78]. Ku forms a ring structure and slides onto the DNA ends where it binds to the sugar backbone of the DNA rather than to the bases [79]. Binding of Ku protects DNA ends from nonspecific processing thereby maintaining DNA stability. When Ku-deficient cells are irradiated in S phase, severe chromosome instability is observed [80]. Therefore, inhibition of Ku proteins with concurrent radiation offers an attractive treatment option in cancer. Li et al. showed that depletion of Ku70 or Ku80 sensitizes pancreatic cancer cells to radiation [81]. DNA-PK is responsible for phosphorylation of other key proteins needed in subsequent steps of the repair pathway and without it, repair would be compromised. Therefore, several DNA-PK inhibitors have been designed, including wortmannin and LY294002. Wortmannin is a potent inhibitor of DNA-PK by making an irreversible covalent modification on Lys802 in the active site of DNA-PK, which is crucial for a necessary phosphate transfer reaction. This compound is a general noncompetitive inhibitor of PI-3 kinases. LY294002 on the other hand is a nonspecific competitive inhibitor that binds irreversibly to the DNA-PK kinase domain [82]. Both these compounds have failed to make it into the clinic due to lack of specificity, poor solubility, and severe toxicity [83]. NU7026 and NU7441, more selective DNA-PK inhibitors, have shown efficacy in vitro but failed to make a clinical impact. One of the major issues with NU7026 was the metabolic instability of the morpholino ring. This challenge made it difficult to deliver appropriate concentrations of the drug in the time frame required for synergy with radiation [84]. More recently, a selective inhibitor of NHEJ, VX-984, that preferentially affects transformed cells was described [85]. Studies in glioblastoma cell lines and tumor-bearing mice showed that VX-984 enhances radiosensitivity of brain tumors [86]. Notably, VX-984 dose dependently inhibited radiation-induced DNA-PKcs phosphorylation and attenuated clonogenic survival [86]. The safety and tolerability of VXλ84 in combination with chemotherapy was assessed in participants with solid tumors in clinical trial NCT02644278. The study was discontinued due to business-related reasons limiting the conclusions [87].
Additional attempts have been made to target DNA end-processing to disrupt NHEJ repair subsequent to radiation-induced DSBs. DNA ends are processed to facilitate ligation since very rarely are blunt ends formed during radiation damage [88]. DSB ends may contain 5′ hydroxyls or 3′ phosphates that cannot be ligated. PNKP phosphorylates the 5′ hydroxyl termini and dephosphorylates the 3′ phosphate termini that often result from radiation treatment [89]. A12B4C3, a noncompetitive inhibitor, has been designed to dysregulate PNKP’s phosphatase activity by disrupting protein secondary structure and leading to radiation sensitivity. The inhibitor sensitizes breast and lung cancer to radiation in vitro and needs further assessment in vivo [90]. Artemis is another protein involved in processing the DNA ends in preparation for repair. Phosphorylation of artemis by DNA-PK permits its endonuclease activity, which nicks 5’ overhangs on a duplex end and displays its hairpin-opening activity [91]. Likewise, artemis has single-strand specific 5′-3′ exonuclease activity. Optimizing a drug to inhibit artemis may show efficacy in treating cancer.
The final step of NHEJ repair is ligation of broken ends and disintegration of the NHEJ complex. XRCC4 stabilizes DNA ligase IV, enhancing its activity by promoting adenylation, which ligates discordant DNA ends across gaps [92, 93]. While phosphorylation of XRCC4 disrupts its ability to bind DNA, it maintains the capacity to bind DNA ligase IV [94]. Therefore, phosphorylation of XRCC4 may dissociate the XRCC4/ligase IV complex from DNA after repair. Additional proteins including XLF and APLF stimulate ligation by the XRCC4–DNA ligase complex [95, 96]. XLF encourages ligation of mismatched and noncohesive DNA ends [95].
There is uncertainty about whether DNA-PKcs is released from the repair site before ligation. DNA-PKcs is released from DNA after conformational changes resulting from autophosphorylation events [97]. However, Ku may remain on the DNA molecule after ligation until it is ubiquitylated and degraded [98]. Inhibitors of DNA ligase IV have been developed to inhibit the ligation step of NHEJ repair. L189 displayed very poor specificity; it could inhibit all three mammalian ligases [99]. SCR7 was a more specific inhibitor of ligase IV. Treatment with SCR7 resulted in accumulation of DSBs and subsequent activation of apoptotic pathways in vitro and in vivo. In fact, tumor-bearing mice treated with SCR7 and radiation displayed significant reduction in tumor burden [100]. Further investigation of specific NHEJ inhibitors is necessary. Treatment with a NHEJ inhibitor has the potential to substantially improve radiosensitivity (Table 3).
Conclusion
Radiation is a potentially curative treatment for HNSCC if used within the appropriate patient population. Radiation induces significant DSBs, which if unrepaired, lead to cell death. Unfortunately, cancer cells have cleverly devised mechanisms to repair radiation-induced DNA damage predominantly by NHEJ. Identifying new targetable players in NHEJ repair could lead to combination therapies to improve disease control and patient survival. With knowledge of p53 and EGFR involvement in the repair process, identification of binding partners may highlight additional avenues to target in order to improve response. Moreover, combination approaches with immunotherapy may be efficacious.
References
Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2018;144:ijc.31937.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. Cancer J Clin. 2020;70:7–30.
Furness S, Glenny A-M, Worthington HV, Pavitt S, Oliver R, Clarkson JE, et al. Interventions for the treatment of oral cavity and oropharyngeal cancer: chemotherapy. Cochrane Database Syst Rev. 2011;2011:1–209.
Zhou G, Liu Z, Myers JN. TP53 mutations in head and neck squamous cell carcinoma and their impact on disease progression and treatment response. Prospect J Cell Biochem. 2016;117:2682–92.
Gunn GB, Blanchard P, Garden AS, Zhu XR, Fuller CD, Mohamed AS, et al. Clinical outcomes and patterns of disease recurrence after intensity modulated proton therapy for oropharyngeal squamous carcinoma. Int J Radiat Oncol Biol Phys. 2016;95:360–7.
Lee N. Study of Proton Versus Photon Beam Radiotherapy in the Treatment of Head and Neck Cancer [Internet]. 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT02923570.
Patel SH, Wang Z, Wong WW, Murad MH, Buckey CR, Mohammed K, et al. Charged particle therapy versus photon therapy for paranasal sinus and nasal cavity malignant diseases: a systematic review and meta-analysis. Lancet Oncol. 2014;15:1027–38.
Fu KK, Pajak TF, Trotti A, Jones CU, Spencer SA, Phillips TL, et al. A radiation therapy oncology group (RTOG) phase III randomized study to compare hyperfractionation and two variants of accelerated fractionation to standard fractionation radiotherapy for head and neck squamous cell carcinomas: first report of RTOG 9003. Int J Radiat Oncol. 2000;48:7–16.
Oksuz DC, Prestwich RJ, Carey B, Wilson S, Senocak MS, Choudhury A, et al. Recurrence patterns of locally advanced head and neck squamous cell carcinoma after 3D conformal (chemo)-radiotherapy. Radiat Oncol. 2011;6:54.
De Felice F, Thomas C, Barrington S, Pathmanathan A, Lei M, Urbano TG. Analysis of loco-regional failures in head and neck cancer after radical radiation therapy. Oral Oncol. 2015;51:1051–5.
Fakhry C, Zhang Q, Gillison ML, Nguyen-Tân PF, Rosenthal DI, Weber RS, et al. Validation of NRG oncology/RTOG-0129 risk groups for HPV-positive and HPV-negative oropharyngeal squamous cell cancer: implications for risk-based therapeutic intensity trials. Cancer. 2019;125:2027–38.
Haimovitz-Friedman A, Kolesnick RN, Fuks Z. Modulation of the apoptotic response: potential for improving the outcome in clinical radiotherapy. Semin Radiat Oncol. 1996;6:273–83.
De Crevoisier R, Domenge C, Wibault P, Koscielny S, Lusinchi A, Janot F, et al. Full dose reirradiation combined with chemotherapy after salvage surgery in head and neck carcinoma. Cancer. 2001;91:2071–6.
Janot F, De Raucourt D, Benhamou E, Ferron C, Dolivet G, Bensadoun RJ, et al. Randomized trial of postoperative reirradiation combined with chemotherapy after salvage surgery compared with salvage surgery alone in head and neck carcinoma. J Clin Oncol. 2008;26:5518–23.
Hanai Yazu K, Hieda RM. On the experimental distinction between ssbs and dsbs in circular DNA. Int J Radiat Biol. 1998;73:475–9.
Rothkamm K, Lö M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 2003;100:5057–62.
Lomax ME, Folkes LK, O’Neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol. 2013;25:578–85.
Cadet J, Douki T, Ravanat J-L. Oxidatively generated damage to the guanine moiety of DNA: mechanistic aspects and formation in cells. Acc Chem Res. 2008;41:1075–83.
Dynan W, Takeda Y, Roth D, Bao G. Understanding and re-engineering nucleoprotein machines to cure human disease. Nanomedicine. 2008;3:93–105.
Zhao X, Wei C, Li J, Xing P, Li J, Zheng S, et al. Cell cycle-dependent control of homologous recombination. Acta Biochim Biophys Sin. 2017;49:655–68.
Emerson CH, Bertuch AA. Consider the workhorse: nonhomologous end-joining in budding yeast 1. Biochem Cell Biol. 2016;94:396–406.
Reynolds P, Anderson JA, Harper JV, Hill MA, Botchway SW, Parker AW, et al. The dynamics of Ku70/80 and DNA-PKcs at DSBs induced by ionizing radiation is dependent on the complexity of damage. Nucleic Acids Res 2012;40:10821–31.
Stucki M, Jackson SP. γH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair. 2006;5:534–43.
Deng C, Pumin Zhang T, Wade Harper J, Elledge SJ, Leder P, McLean M. Mice Lacking p21 c~P7/wAF7 undergo normal development, but are defective in Gl checkpoint control. Cell. 1995;82:675–84.
Lafarga V, Cuadrado A, Lopez de Silanes I, Bengoechea R, Fernandez-Capetillo O, Nebreda AR. p38 mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol. 2009;29:4341–51.
Deckbar D, Stiff T, Koch B, Reis C, Löbrich M, Jeggo PA. The limitations of the G 1 -S checkpoint. Cancer Res. 2010;70:4412–21.
Chan TA, Hwang PM, Hermeking H, Kinzler KW, Vogelstein B. Cooperative effects of genes controlling the G(2)/M checkpoint. Genes Dev. 2000;14:1584–8.
Löbrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer. 2007;7:861–9.
McCulloch HD. On the analogy between spontaneous recoveries from cancer and the specific immunity induced by X ray irradiations of the lymphatic glands involved. Br Med J. 1908;2:1146–8.
Ngwa W, Credit Irabor O, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18:313–22.
Jackson StephenP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8.
Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumor Biol. 2010;31:363–72.
Banerjee R, Russo N, Liu M, Basrur V, Bellile E, Palanisamy N, et al. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nat Commun. 2014;5:4527.
Chang HW, Kim SY, Yi S-L, Son S-H, Song DY, Moon SY, et al. Expression of Ku80 correlates with sensitivities to radiation in cancer cell lines of the head and neck. Oral Oncol. 2006;42:979–86.
Moeller BJ, Yordy JS, Williams MD, Giri U, Raju U, Molkentine DP, et al. DNA repair biomarker profiling of head and neck cancer: Ku80 expression predicts locoregional failure and death following radiotherapy. Clin Cancer Res. 2011;17:2035–43.
Chen BPC, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582–7.
Mansour WY, Bogdanova NV, Kasten-Pisula U, Rieckmann T, Köcher S, Borgmann K, et al. Aberrant overexpression of miR-421 downregulates ATM and leads to a pronounced DSB repair defect and clinical hypersensitivity in SKX squamous cell carcinoma. Radiother Oncol. 2013;106:147–54.
Spence T, Bruce J, Yip KW, Liu FF. HPV associated head and neck cancer. Cancers (Basel). 2016;8:75.
Chung CH, Zhang Q, Kong CS, Harris J, Fertig EJ, Harari PM, et al. P16 protein expression and human papillomavirus status as prognostic biomarkers of nonoropharyngeal head and neck squamous cell carcinoma. J Clin Oncol. 2014;32:3930–8.
Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tân PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl J Med. 2010;363:24–35.
Gatz SA, Wiesmüller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–16.
Bensaad K, Vousden KH. p53: new roles in metabolism. Trends Cell Biol. 2007;17:286–91.
Helton ES, Chen X. p53 modulation of the DNA damage response. J Cell Biochem. 2007;100:883–96.
Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012;18:290–300.
Valente LJ, Gray DHD, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, et al. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013;3:1339–45.
Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53. Oncogene. 2007;26:2202–11.
Zhou X, Hao Q, Lu H. Mutant p53 in cancer therapy-the barrier or the path. J Mol Cell Biol. 2019;11:293–305.
Weinberg RL, Veprintsev DB, Bycroft M, Fersht AR. Comparative binding of p53 to its promoter and DNA recognition elements. J Mol Biol. 2005;348:589–96.
Wade Harper J, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60.
Korzeniewski N, Spardy N, Duensing A, Duensing S. Genomic instability and cancer: lessons learned from human papillomaviruses. Cancer Lett. 2011;305:113–22.
Radford IR, Murphy TK, Radley JM, Ellis SL. Radiation response of mouse lymphoid and myeloid cell lines. Part II. Apoptotic death is shown by all lines examined. Int J Radiat Biol. 1994;65:217–27.
Findley HarryW, Gu Lubing, Yeager AndrewM, Z M. Expression of Bcl-xL can confer a multidrug resistance phenotype. Blood. 1995;86:1903–10.
Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15:1019–29.
Fei P, El-Deiry WS. P53 and radiation responses. Oncogene. 2003;22:5774–83.
Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53*. J Biol Chem. 2002;277:3247–57.
Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM. Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer Res. 1994;54:2095–7.
Clarke AR, Purdie CA. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849.
Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, et al. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–11.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60.
Ventura A, Kirsch DG, Mclaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5.
Harari PM, Wheeler DL, Professor P, Grandis JR. Molecular target approaches in head and neck cancer: EGFR and beyond. Semin Radiat Oncol. 2009;19:63–8.
Kian Ang K, Berkey BA, Tu X, Zhang H-Z, Katz R, Hammond EH, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–6.
Schmidt-Ullrich RK, Mikkelsen RB, Dent P, Todd DG, Valerie K, Kavanagh BD, et al. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene. 1997;15:1191–7.
Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, et al. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280:31182–9.
Liccardi G, Hartley JA, Hochhauser D. EGFR Nuclear Translocation Modulates DNA Repair following Cisplatin and Ionizing Radiation Treatment. Cancer Res. 2011;71:1103–14.
Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem. 1998;273:1568–73.
Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–8.
Lucero H, Gae D, Taccioli GE. Novel localization of the DNA-PK complex in lipid rafts: a putative role in the signal transduction pathway of the ionizing radiation response. J Biol Chem. 2003;278:22136–43.
Huang S-M, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis 1. Clin Cancer Res. 2000;6:2166–74.
Das AK, Chen BP, Story MD, Sato M, Minna JD, Chen DJ, et al. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res. 2007;67:5267–74.
Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78.
Kian Ang K, Zhang Q, Rosenthal DI, Felix Nguyen-Tan P, Sherman EJ, Weber RS, et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J Clin Oncol. 2014;32:2940–50.
Gillison ML, Trotti AM, Harris J, Eisbruch A, Harari PM, Adelstein DJ, et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): a randomised, multicentre, non-inferiority trial. Lancet. 2019;393:40–50.
Mehanna H, Robinson M, Hartley A, Kong A, Foran B, Fulton-Lieuw T, et al. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): an open-label randomised controlled phase 3 trial. Lancet. 2019;393:51–60.
Ghisolfi L, Keates AC, Hu X, Lee D, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS ONE. 2012;7:e43628.
Downs JA, Jackson SP. A means to a DNA end: the many roles of Ku. Nat Rev Mol Cell Biol. 2004;5:367–78.
Mari P-O, Florea BI, Persengiev SP, Verkaik NS, Brü HT, Modesti M, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci USA. 2006;103:18597–602.
Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–14.
Nagasawa H, Brogan JR, Peng Y, Little JB, Bedford JS. Some unsolved problems and unresolved issues in radiation cytogenetics: a review and new data on roles of homologous recombination and non-homologous end joining. Mutat Res Toxicol Environ Mutagen. 2010;701:12–22.
Li Y-H, Wang X, Pan Y, Lee D-H, Chowdhury D, Kimmelman AC, et al. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS ONE. 2012;7:e39588.
Davidson D, Amrein L, Panasci L, Aloyz R. Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front Pharmacol. 2013;4:5.
Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene. 2005;24:949–61.
Nutley BP, Smith NF, Hayes A, Kelland LR, Brunton L, Golding BT, et al. Preclinical pharmacokinetics and metabolism of a novel prototype DNA-PK inhibitor NU7026. Br J Cancer. 2005;93:1011–8.
Khan AJ, Misenko SM, Thandoni A, Schiff D, Jhawar SR, Bunting SF, et al. VX-984 is a selective inhibitor of non-homologous end joining, with possible preferential activity in transformed cells. Oncotarget. 2018;9:25833–41.
Timme CR, Rath BH, O’neill JW, Camphausen K, Tofilon PJ. The DNA-PK inhibitor VX-984 enhances the radiosensitivity of glioblastoma cells grown in vitro and as orthotopic xenografts. Mol Cancer Ther. 2018;17:1207–16.
EMD Serono Research & Development Institute. First-in-Human Study of the Safety, Tolerability, and Pharmacokinetic/Pharmacodynamic Profile of VX-984 in Combination With Chemotherapy [Internet]. 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT02644278.
Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013;2:130–43.
Karimi-Busheri F, Rasouli-Nia A, Allalunis-Turner J, Weinfeld M. Human polynucleotide kinase participates in repair of DNA double-strand breaks by nonhomologous end joining but not homologous recombination. Cancer Res. 2007;67:6619–25.
Freschauf GK, Karimi-Busheri F, Ulaczyk-Lesanko A, Mereniuk TR, Ahrens A, Koshy JM, et al. Identification of a small molecule inhibitor of the human DNA repair enzyme polynucleotide kinase/phosphatase. Cancer Res. 2009;69:7739–85.
Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–94.
Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, Mann M, et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388:492–5.
Gu J, Lu H, Tippin B, Shimazaki N, Goodman MF, Lieber MR. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. EMBO J. 2007;26:1010–23.
Modesti M, Hesse JE, Gellert M. DNA binding of Xrcc4 protein is associated with V(D)J recombination but not with stimulation of DNA ligase IV activity. EMBO J. 1999;18:2008–18.
Tsai CJ, Kim SA, Chu G, Lehman IR. Cernunnos XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc Natl Acad Sci USA. 2007;104:7851–6.
Grundy GJ, Rulten SL, Zeng Z, Arribas-Bosacoma R, Iles N, Manley K, et al. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J. 2013;32:112–25.
Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285:1414–23.
Postow L, Ghenoiu C, Woo EM, Krutchinsky AN, Chait BT, Funabiki H. Ku80 removal from DNA through double strand break-induced ubiquitylation. J Cell Biol. 2008;182:467–79.
Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM, et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68:3169–77.
Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151:1474–87.
Acknowledgements
This work was supported by grants from NIH/NIDCR DE027551 and DE007057-43.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hutchinson, MK.N.D., Mierzwa, M. & D’Silva, N.J. Radiation resistance in head and neck squamous cell carcinoma: dire need for an appropriate sensitizer. Oncogene 39, 3638–3649 (2020). https://doi.org/10.1038/s41388-020-1250-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-1250-3
This article is cited by
-
NSUN6-mediated 5-methylcytosine modification of NDRG1 mRNA promotes radioresistance in cervical cancer
Molecular Cancer (2024)
-
CHD4 acts as a prognostic factor and drives radioresistance in HPV negative HNSCC
Scientific Reports (2024)
-
Architecting lymphoma fusion: PROMETHEE-II guided optimization of combination therapeutic synergy
International Journal of Information Technology (2024)
-
Salivary microbiome changes distinguish response to chemoradiotherapy in patients with oral cancer
Microbiome (2023)
-
Identify and validate circadian regulators as potential prognostic markers and immune infiltrates in head and neck squamous cell carcinoma
Scientific Reports (2023)
