
Abstract
Transcription factor PAX8 expression is upregulated in several types of cancers. However, little is known about the function of PAX8 in the progression of hepatoma and its regulatory mechanisms. Here, we show that PAX8 silencing inhibits the proliferation and clonogenicity of hepatoma cells and its growth in vivo. The HBV X protein (HBx) does not directly interacts, but stabilizes PAX8 by inhibiting proteasome-dependent ubiquitination and degradation. Furthermore, the E3 ubiquitin ligase complex component Skp2 through its LRR domain directly interacts with the Prd domain of PAX8 and targets PAX8 by recognizing its lysine 275 for ubiquitination and degradation in hepatoma cells. In addition, HBx directly interacts and is colocalized with Skp2 to inhibit its recognition and subsequent ubiquitination and degradation of PAX8 in hepatoma cells. Moreover, HBx upregulates the expression and phosphorylation of Aurora A, a serine–threonine kinase, which interacts with and phosphorylates PAX8 at S209 and T277, compromising the Skp2-recognized PAX8 ubiquitination and destabilization. Thus, HBx stabilizes PAX8 protein by inhibiting the Skp2 targeted PAX8 ubiquitination and enhancing the Aurora A-mediated its phosphorylation, contributing to the progression of hepatoma. Our findings suggest that PAX8 may a new target for design of therapies and uncover new insights into the pathogenesis of hepatoma.
Similar content being viewed by others


Introduction
Liver cancer now is the fourth leading cause of cancer-related death globally. Hepatocellular carcinoma (HCC) accounts for 90% of primary liver cancers and is attributed to chronic infection with hepatitis B virus (HBV) or hepatitis C virus, alcohol abuse and metabolic syndrome [1, 2]. Among all the etiology, chronic hepatitis B (CHB) is the common pathogenic factor for the development of HCC [3]. The HBV X protein (HBx), a multifunctional regulator, can facilitate hepatocytes to oncogenic transformation [4]. Although extensive investigations have advanced our understanding of HBx roles in the pathogenesis of HCC, the molecular mechanisms by which HBx regulates the progression of HCC have not been clarified.
Protein stability and turnover is a key process to regulate the cellular homeostasis. The ubiquitin-proteasome pathway (UPP) is crucial for the degradation of misfolded and aggregated proteins [5]. During the UPP process, the ubiquitin ligase E3 recognizes specific lysine residue in a specific protein by recruiting ubiquitin from ubiquitin-conjugating enzyme E2 to promote polyubiquitin chain formation and subsequent proteasome-dependent degradation. Hence, dysfunction of E3 and UPP processes are important for cell cycle, survival and apoptosis, and associated with the development of many types of diseases, including malignancies [6]. Viral proteins, such as the V protein of SV5, Vpx protein of human immunodeficiency virus, can target the UPP to support their replication and oncogenicity [7, 8]. Previous studies have shown that HBx can interact with some proteins in the proteasome complex to facilitate the viral replication [9, 10]. A recent study reveals that HBx modulates Skp2, a component of the E3 ubiquitin-protein ligase complex, and MSL2 E3 ligase, to promote hepatocarcinogenesis [11, 12]. However, how HBx promotes hepatocarcinogenesis is still unclear.
PAX8 is a member of the paired box (PAX) transcription factor family. PAX8, like other members in the same family, has a highly conserved DNA-binding paired domain (Prd) of 128 amino acids [13]. PAX8 is important for embryogenesis, and morphogenesis of the thyroid and kidney [14]. Furthermore, PAX8 expression is upregulated in several types of cancers and is a valuable biomarker for diagnosis of glioma, ovarian carcinoma, gastric cancer and others [15,16,17]. A recent study has reported that single nuclear polymorphism of the PAX8 gene is associated with prognosis of Chinese patients with HCC [18]. However, little is known on the expression profile and role of PAX8 during the progression of HCC. There is also no information on how PAX8 metabolizes, and whether and how HBx regulates PAX8 turnover in HCC cells.
In present study, we examined the expression of PAX8 and explored the effect of PAX8 silencing on the proliferation, cell cycle and clonogenicity of HCC cells and HCC growth in vivo. Furthermore, we explored the potential mechanisms by which HBx regulated PAX8 stability. Our data indicated that higher levels of PAX8 expression were associated with poor prognosis of HCC and PAX8 silencing inhibited the proliferation and clonogenicity of HepG2 cells by inducing cell cycle arrest in S phase. Although HBx did not interact with PAX8, but stabilized its protein by inhibiting the Skp2-dependent ubiquitination and enhancing the Aurora A-mediated phosphorylation of PAX8 in HCC cells. Our findings uncover new mechanisms by which HBx stabilizes PAX8, contributing to the progression of HBV-related HCC.
Results
PAX8 silencing inhibits the growth of HCC in vitro and in vivo
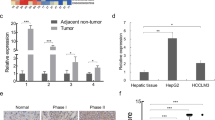
PAX8 is a biomarker for diagnosis of several types of malignant tumors [19, 20]. To understand the potential role of PAX8 in the progression of HCC, the expression of PAX8 in HCC and nontumor liver tissues in the Oncomine and TCGA was analyzed. As expected, the relative ratios of mRNA transcript of PAX8 in HCC in both Oncomine and TCGA databases were significantly higher than that in the nontumor tissues (p < 0.01 for both, Fig. 1a). Kaplan–Meier analysis indicated that higher levels of PAX8 expression were associated with poor survival of patients with HCC (p = 0.032, Fig. 1b). To further understand the role of PAX8, HepG2 cells were transfected with lentivirus for expression of PAX8-specific shRNA to silence its expression effectively (Fig. 1c). PAX8 silencing significantly inhibited the proliferation and clonogenicity of HepG2 cells (Fig. 1d, e) and led to S phase arrest (Fig. 1f). Consistently, PAX8 silencing obviously decreased E2F1, CDK2, and p53 expression and Rb phosphorylation as well as p21 expression in HepG2 cells (Fig. 1g). Similarly, PAX8 silencing also inhibited the proliferation and clonogenicity of MHCC97H and MHCC97L cells (Fig. S1). More importantly, PAX8 silencing significantly inhibited the growth of implanted HCC and reduced the tumor size, accompanied by reducing KI67 staining in the tumor tissues (Fig. 1h–j). Such data suggest that PAX8 may act as an oncogenic factor to promote the progression of HCC.

PAX8 silencing inhibits the growth of HCC in vitro and in vivo. a The expression levels of PAX8 mRNA transcripts in 102 HCC and 86 nontumor tissues in the Oncomine database and 367 HCC and 50 nontumor tissues in the TCGA (the data was download from the database on Oct 18, 2018). b Kaplan–Meier analysis of overall survival in 179 patients with low PAX8 HCC and 183 those with high PAX8 expressing HCC in the TCGA. c Western blot and qRT-PCR analysis of PAX8 expression in HepG2 cells following stably PAX8 silencing. d PAX8 silencing inhibits the proliferation of HepG2 cells, determined by CCK-8 assay. e PAX8 silencing reduces the clonogenicity of HepG2 cells. f PAX8 silencing induces cell cycle arrest in S phase in HepG2 cells. g Western blot analysis of the expression of cell cycling regulators in HepG2 cells. h, i PAX8 silencing inhibits the growth of implanted HepG2 tumors and reduces their weights in BALB/c node mice (n = 6 per group). j Hematoxylin and eosin (HE) staining and immunohistochemistry analysis of Ki67+ tumor cells in xenograft tumor tissues. Data are representative images or expressed as the mean ± SD of each group from three separate experiments. *P < 0.05, **P < 0.01
HBx stabilizes the PAX8 protein
Chronic HBV infection is a common risk factor for the development of HCC. HBx is crucial for HBV replication and the pathogenesis of HCC. Our protein/DNA Array results have shown that HBx upregulated the transcriptional activity of PAX8 (Fig. S2). Next, we examined how HBx regulated PAX8. We detected significantly higher levels of PAX8 protein expression, but not PAX8 mRNA transcripts in HepG2-HBx and HepG2.2.15 cells (Fig. 2a, b). The positively regulatory effects of HBx on PAX8 protein appeared to be dose and time-dependent in HepG2 cells (Fig. S3A–C). Furthermore, while treatment with CHX (50 µM) for 8–16 h significantly decreased the levels of PAX8 in HepG2-Ctrl cells the same treatment did not significantly affect the levels of PAX8 in HepG2-HBx and HepG2.2.15 cells, suggesting that HBx may prolong the half-life of PAX8 by inhibiting its degradation in HepG2 cells (Fig. 2c). The proteasome pathway is important for degradation of many proteins. Actually, treatment with MG132 prevented the degradation of PAX8 in all cells tested (Fig. 2d, Fig. S3D). A previous study has reported that sumoylation is critical for the steady state of PAX8 protein [21], but it is still unknown whether it can be regulated by ubiquitination. To address it, HEK293T cells were transfected with plasmids for expression of Flag-PAX8, Myc-HBx, and HA-Ub (K48 or K63, respectively), Flag-PAX8, Myc-HBx for 48 h. It was found that HBx inhibited the Ub-K48-mediated polyubiquitination of PAX8, but not the K63-linked pathway (Fig. 2e, f). Such data indicated that HBx stabilized PAX8 by inhibiting the Skp2-dependent proteasomal degradation.

HBx stabilizes PAX8 protein by inhibiting the proteasomal degradation of PAX8. a, b HBx increases PAX8 protein, but does not alter PAX8 mRNA transcripts in HBx+ and HBx- HepG2 cells. c HBx preserves PAX8 protein in HBx+, but not HBx-, HepG2 cells after treatment with cycloheximide (50 µmol/ml). d HBx and MG132, prevent PAX8 proteasomal degradation in HepG2 cells. e, f HBx inhibits the Ub-K48, but not Ub-K63, mediated polyubiquitination of PAX8 in HEK293T cells after cotransfection with the indicated plasmids. Data are representative images or expressed as the mean or mean ± SD of each group from three separate experiments. *P < 0.05, **P < 0.01
Skp2 interacts with PAX8
To further identify the PAX8-associated proteins, immunoprecipitation and Mass spectrometry indicated that PAX8 interacted with several proteins in HepG2 cells (Fig. S4). Given that the strongest interaction of PAX8 with one component of the ubiquitin complex was Skp2, we implied that Skp2 would interact with PAX8. To test the hypothesis, we confirmed the endogenous Skp2-PAX8 interaction by coimmunoprecipitation in HepG2 (Fig. 3a). The cotransfection experiment in HEK293T revealed Flag-PAX8 interacted with His-Skp2 by coimmunoprecipitation (Fig. 3b). Furthermore, the purified GST-PAX8 effectively pulled down Skp2 (Fig. 3c). Confocal microscopy revealed that PAX8 was colocalized with Skp2 in the perinuclear of HeG2 cells (Fig. 3d). These indicate that PAX8 directly interacts with Skp2 in HepG2 cells. To determine the specific regions for interaction between Skp2 and PAX8, several mutants of Skp2 and PAX8 were constructed, respectively. We found that the T1 fragment (1-138aa, containing the Prd domain), but not T2 (139-187aa, containing octapeptide), T3 (188-363aa, containing the homeodomain) and T4 (364-450aa, including the DNA activation domain) effectively pulled down skp2, indicating that the Prd domain was responsible for the interaction of PAX8 with Skp2 in HepG2 cells [22] (Fig. 3e). The Skp2 is a substrate recognition component of the E3 ubiquitin–protein ligase complex, which is crucial for the ubiquitination and subsequent proteasomal degradation of target proteins. Skp2 is one member of the SCF complex family, which contains a consensus protein motif of F-box [23, 24]. To determine the importance, we generated two Skp2 mutations of the F-box depletion (94-140aa, Skp2ΔF) and Leucine-rich Repeat (LRR) domain depletion (151-401aa, Skp2ΔLRR). Following cotransfection, we found that Flag-PAX8 effectively precipitated in wild-type Skp2 and Skp2-ΔF, but not Skp2-ΔLRR (Fig. 3f), indicating that PAX8 interacted with the LRR domain of Skp2. Such data demonstrated that PAX8 directly interacted with Skp2 through the interaction of the Prd domain in PAX8 with the LRR domain in Skp2 in HepG2 cells.

Skp2 interacts with PAX8. a, b Immunoprecipitation and western blot indicate that Skp2 directly interacts with PAX8 in HepG2 and HEK293T cells following cotransfection with the plasmids. c The purified GST-PAX8 pulls down Skp2 from HEK293T after transfection with the plasmid. The purified GST and GST-PAX8 proteins are resolved by Coomassie blue staining. * One probably degraded smaller molecule of PAX8. d Immunofluorescent confocal microscopy analysis of the colocalization of PAX8 and Skp2 proteins in HepG2. e The purified GST-PAX8 or GST-PAX8-T1 protein pulls down Skp2 from HEK293T cells. The purified truncated GST-PAX8 proteins are resolved by Coomassie blue staining. The PAX8 was truncated into T1 (containing the paired domain (Prd)), T2 (octapeptide), T3 (homeodomain) and T4 (DNA activation domain). f Skp2 interacts with PAX8, dependent on the LRR domain. HEK293T cells were cotransfected with the indicated plasmids for 48 h and precipitated with anti-Flag, followed by western blot using anti-His and anti-Flag. His-Skp2ΔF: depletion of 40 amino acids (F-box domain) of the Skp2; His-Skp2ΔLRR: depletion of 239 amino acids (Leucine-rich repeat domain) of the Skp2. Data are representative images of each group of cells from three separate experiments
Skp2 is crucial for PAX8 protein ubiquitination and degradation
Skp2 is required for the substrate recognition component of an E3 ubiquitin–protein ligase complex, which mediates the ubiquitination and subsequent proteasomal degradation of target proteins to regulate the pathogenic process of cancer. Previous studies have shown that c-Myc, E2F1, p27, and p21 are substrates of Skp2 [25, 26]. Given that Skp2 directly interacted with PAX8, we hypothesized that Skp2 might be required to target PAX8 for ubiquitination and subsequent degradation. To test the hypothesis, we investigated the effect of altered Skp2 expression on PAX8 expression in HepG2 cells. Skp2 silencing increased the levels of PAX8 protein in a trend of dose-dependent while Skp2 overexpression dramatically decreased the levels of PAX8 protein in HepG2 cells (Fig. 4a–d). Similar patterns of c-Myc, p27, and p21 proteins were detected in Skp2 silencing or overexpressing HepG2 cells (Fig. S5A and B). Moreover, MG132 treatment rescued the decreased PAX8 protein mediated by forced expression of His-Skp2 (Fig. 4e). Next, we examined the half-life of PAX8 in HepG2 cells following CHX treatment. While there was little reduction in the levels of PAX8 protein in the CHX-treated control cells, induction of His-Skp2 expression significantly reduced the levels of PAX8 protein in the CHX-treated HepG2 cells throughout the observation period (Fig. 4f). To determine Skp2-dependent ubiquitination of PAX8, HEK293T cells were cotransfected with plasmids for expression of HA-Ub and Flag-PAX8, together either His-Skp2 or Skp2-specific siRNA. Anti-Flag immunoprecipitation revealed that Skp2 silencing significantly decreased the PAX8 polyubiquitination while wild-type Skp2, but not Skp2ΔF, overexpression, remarkably increased its polyubiquitination (Fig. 4g–i). Skp2ΔF mutant overexpression failed to destabilize PAX8 and promote its ubiquitination (Fig. 4j). A similar pattern of p21 protein was observed in different groups of cells (Fig. S5C). Together, such data clearly indicated that Skp2 was important for PAX8 ubiquitination and degradation.

Skp2 mediate PAX8 protein ubiquitination and degradation. a, b Western blot and qRT-PCR analysis of PAX8 expression in HepG2 cells following transfected with the indicated dose of Skp2-specific siRNA or control siRNA. c, d Western blot analysis of PAX8 and His-Skp2 in HepG2 or HEK293T cells following inducing variable levels of His-Skp2. +:2 ug, ++:3 ug, +++:4 ug. e Treatment with MG132 mitigates the Skp2-decreased PAX8 protein in HepG2 cells. f Induction of His-Skp2 expression reduces PAX8 protein ubiquitination and degradation in HepG2 cells after treatment with CHX. g Skp2 silencing mitigates PAX8 protein ubiquitination and degradation in HEK293T cells following cotransfection with the indicated plasmid and siRNA. h Induction of His-Skp2 expression enhances PAX8 protein ubiquitination and degradation in HEK293T cells. i, j Skp2, but not its Skp2ΔF mutant promotes PAX8 protein ubiquitination and degradation in HepG2 and HEK293T cells. Data are representative images or expressed as the mean or mean ± SD of each group from three separate experiments. *P < 0.05, **P < 0.01
Lysine 275 in PAX8 is essential for Skp2-dependent PAX8 ubiquitination and degradation
To determine which lysine of PAX8 is essential for Skp2 recognition-related proteasome-dependent degradation, we divided 14 lysine residues in two groups of the N-terminus and C-terminus (seven per group, Fig. 5a). After point mutation of each lysine residue to arginine in each terminus to generate Flag-PAX8NT or Flag-PAX8CT, we found that while both wild-type Flag-PAX8WT and Flag-PAX8NT exhibited similar reduction following treatment with CHX, the levels of Flag-PAX8CT were slightly reduced (Fig. 5b), indicating that the C-terminus of PAX8 contained a lysine residue for Skp2-dependent ubiquitination of PAX8. Further K/R mutations of individual lysine residues in the C-terminus of PAX8 revealed that there was no obvious reduction in the levels of Flag-PAX8 K197R and Flag-PAX8K275R, but not other mutants, in HepG2 cells following CHX treatment (Fig. 5c). Both Flag-PAX8K197R and Flag-PAX8K275R exhibited much longer half-life than that of the wild-type of PAX8 (Fig. S6A). To determine which lysine was an ubiquitin acceptor for Skp2-dependent proteasomal degradation of PAX8, we found that His-skp2 overexpression decreased the levels of wild-type Flag-PAX8 and Flag-PAX8K197R, but not Flag-PAX8K275R in HepG2 cells (Fig. 5d). Further ubiquitination assay demonstrated that Skp2 failed to induce the Flag-PAX8K275R ubiquitination in HepG2 cells (Fig. 5e). Collectively, such data clearly uncovered that Skp2 recognized the K275 of PAX8 to induce the ubiquitination and degradation of PAX8 in HepG2 cells (Fig. 5f).

Skp2 recognizes Lysine 275 in PAX8 to promote its proteasomal degradation. a Schematic illustration of lysine residues in PAX8. All lysine residues were divided to the PAX8 N-terminal and PAX8 C-terminal parts (each with seven lysine residues). b Western blot analysis of the sensitivity of Flag-PAX8WT, Flag-PAX8NT, or Flag-PAX8CT mutants to proteasomal degradation in HepG2 cells after treatment with CHX. Flag-PAX8NT or Flag-PAX8CT: all K residues in each part were replaced with R residues. c Western blot reveals that the Flag-PAX8 K197R and K275R mutants are resistant to proteasomal degradation in HepG2 cells following transfection with individual plasmids for each mutant. d Increased Skp2 expression promotes the proteasomal degradation of Flag-PAX8 and Flag-PAX8K197R, but not Flag-PAX8K275R mutant in HepG2 cells. e Increased Skp2 expression promotes the ubiquitination and degradation of Flag-PAX8, but not Flag-PAX8K197R in HEK293T cells. Data are representative images of each group of cells from three separate experiments. f The schematic diagram of that SCF-Skp2 complex targets PAX8 protein for proteasomal degradation
HBx inhibits the Skp2-mediated PAX8 ubiquitination
Given that HBx inhibited the ubiquitination and degradation of PAX8, but did not directly interact with PAX8 (Fig. S7A), we explored whether HBx inhibited the PAX8 ubiquitination by directly interacting with Skp2 to affect its activity in HepG2 cells [27, 28]. Actually, we found that HBx overexpression did not significantly affect the levels of Skp2 expression in HepG2 cells (Fig. S7B). Immunoprecipitation indicated that HBx directly interacted with endogenous and inducible Skp2 in both HepG2 and HEK293T cells (Fig. 6a, b). Immunofluorescent confocal microscopy revealed that HBX and Skp2 were colocalized in the perinuclear of HepG2 cells (Fig. 6c). Immunoprecipitation displayed that anti-Myc antibody precipitated Myc-HBx with the wild-type and Skp2-ΔF, but not Skp2-ΔLRR, indicating that the LRR domain of Skp2 was necessary for direct interaction of Skp2 with HBx (Fig. 6d and Fig. S7C). Given that the LRR domain was important for the interaction of Skp2 with PAX8, we examined the effect of HBx on the Skp2-PAX8 interaction in HepG2 cells. We found that Myc-HBx overexpression impaired the interaction of Skp2 with PAX8 and decreased the Skp2-dependent ubiquitination and degradation of PAX8 in HepG2 cells (Fig. 6e–g). Because both PAX8 and HBx interacted with and colocalized with Skp2 in the perinuclear region, we detected the accumulated levels of PAX8 protein in the nuclear of HBx-expressing cells by western blot and immunofluorescence (Fig. 6h and Fig. S7D). Similar results were obtained in HEK293T cells following HBx overexpression (Fig. S7E). Such results suggest that HBx may stabilize PAX8 by impairing the Skp2-PAX8 interaction and subsequent ubiquitination and degradation of PAX8.

HBx inhibits the Skp2-mediated PAX8 ubiquitination. a, b Immunoprecipitation indicates that HBx directly interacts with Skp2 in HepG2 and HEK293T cells. c Immunofluorescent confocal microscopy reveals that HBx and Skp2 are colocalized in the perinuclear of HepG2 cells. d HBx interacts with His-Skp2, but not His-Skp2ΔLRR, in HEK293T cells. e, f HBx inhibits the Skp2-reduced PAX8 in HEK293T and HepG2 cells. g HBx mitigates the Skp2-mediated PAX* ubiquitination and degradation in HEK293T cells. h Western blot analysis of cytosol and nuclear PAX8 in HBx+ HepG2-HBx and HepG2.2.15 cells. Data are representative images of each group of cells from three separate experiments
Aurora A phosphorylates PAX8 and attenuates the Skp2-dependent ubiquitination and degradation of PAX8
The intracellular levels and activity of multiple proteins are also determined by their site-specific phosphorylation by specific protein kinase. Given that mass spectrometry indicated that PAX8 was possible to interact with Aurora A, a serine/threonine kinase, we further explored the role of Aurora A in the Skp2-dependent PAX8 ubiquitination. First, immunoprecipitation indicated that PAX8 directly interacted with Aurora A in HepG2 and HEK293T cells (Fig. 7a and S8A) and the GST-PAX8 effectively pulled down Aurora A (Fig. S8B). Induction of Aurora A overexpression increased the levels of PAX8 Thr/Ser phosphorylation in both HepG2 and HEK293T cells (Fig. 7b). It was notable that two sequences of amino acids (206-212aa/273-279aa) in PAX8 contained the consensus motif (R/K-X-S/T-I/V/L) recognized by Aurora A (Fig. 7C1). We found that Aurora A phosphorylated recombinant wild-type PAX8, but did not effectively phosphorylate the mutant GST-PAX8S209A and GST-PAX8T277A (Fig. 7C2). Similarly, treatment with MLN8237, an inhibitor of Aurora A [29], also impaired the PAX8 phosphorylation (Fig. S8C). Accordingly, Aurora A phosphorylated PAX8 at S209 and T277 to modulate its stability. Because different types of posttranslational modifications can cross talk and the phosphorylation of a substrate can affect its ubiquitination [30], we examined the impact of Aurora A-phosphorylated PAX8 on its stability and the Skp2-dependent ubiquitination and degradation of PAX8. Treatment with MLN8237 or Aurora A silencing decreased the levels of PAX8 protein even in HBx overexpressing cells, but did not affect the relative levels of PAX8 mRNA transcripts (Fig. 7d, S8D, E). Inhibition of Aurora A reduced the steady-state of endogenous PAX8 following treatment with CHX (Fig. 7e), which was abrogated by treatment with the MG132 (Fig. 7f). Induction of Aurora A overexpression significantly increased the levels of wild-type PAX8 protein than that of PAX8S209A/PAX8T277A (Fig. S9A). Such data clearly indicated that PAX8 phosphorylation by Aurora A prolonged its stability, which may be attributed its low sensitivity to the Skp2-dependent ubiquitination and degradation. Actually, induction of Aurora A overexpression mitigated the Skp2 overexpression reduced PAX8 protein in HepG2 cells (Fig. 7g). Aurora A overexpression abrogated Skp2-dependent ubiquitination of PAX8 (Fig. 7h), but failed significantly to modulate the Skp2-dependent ubiquitination of PAX8S209A and PAX8T277A mutants (Fig. S9B). Therefore, Aurora A phosphorylated PAX8 and inhibited the Skp2-recruited PAX8 for its ubiquitination and degradation in hepatoma cells.

Aurora A phosphorylates PAX8 and attenuates the Skp2-mediated PAX8 ubiquitination and degradation. a Immunoprecipitation exhibits that Aurora A directly interacts with PAX8 in HepG2 cells. b Induction of Aurora A overexpression enhances PAX Thr and Ser phosphorylation in HepG2 and HEK293T cells. c1 The consensus recognition motifs of Aurora A ;c2 Aurora A phosphorylates GST-FAX8, but not GST-PAX8S209A and GST-PAX8T277A mutants in HEK293T cells. d Inhibition of Aurora A by MLN8237 reduces its phosphorylation and PAX8 protein in HBx+ HepG2-HBx and HepG2.2.15 cells. e Treatment with MLN8237 decreases Aurora A phosphorylation, HBx expression, and PAX8 protein in HepG2-HBx cells after treatment with CHX. f Treatment with MG132 and/or MLN8237 modulates Aurora A phosphorylation and PAX8 in HepG2-HBx cells. g Induction of Myc-Aurora A expression mitigates the increased Skp2-mediated PAX reduction in HepG2 cells. h Induction of Myc-Aurora A expression prevents the Skp2-medaited PAX8 ubiquitination in HEK293T cells. i HBx Enhances Aurora A phosphorylation in HepG2-HBx and HepG2.2.15 cells. Data are representative images of each group of cells from three separate experiments
A previous study has suggested that Aurora A may act as an oncogenic factor for the tumorigenesis of HCC [31]. However, it is currently unclear whether HBx can regulate the activity of Aurora A. Given that HBx and Aurora A positively regulated PAX8 stability, we hypothesized that HBx might stabilize PAX8 protein by activating Aurora A. In fact, we found that HBx overexpression enhanced the Aurora A expression and phosphorylation, and promoted the accumulation of wild-type PAX8, but not PAX8S209A/PAX8T277A mutants, in HepG2 and HepG2.2.15 cells (Fig. 7i and S9C). Together, such novel findings demonstrated that PAX8 phosphorylation by Aurora A mitigated its sensitivity to the Skp2-dependent PAX8 ubiquitination and degradation, which was enhanced by HBx.
Discussions
CHB is a major cause of advanced liver disease and HCC in the world. It is well known that several factors contribute to the pathogenesis of HBV-mediated HCC. PAX8 is a transcription factor and implicated in progression of cancer [32, 33]. PAX8 expression is upregulated in most types of tumors, and it can promote the progression of cancer by inhibiting cancer cell apoptosis and/or promoting their proliferation. In this study, we found that PAX8 mRNA transcripts increased in HCC tissues and higher levels of PAX8 transcription was associated with poor survival of HCC patients. These suggest that PAX8 may act as an oncogenic factor for the progression of HCC. More importantly, we found that PAX8 silencing inhibited the proliferation and clonogencity of HCC cells in vitro and the growth of implanted HCC xenografts in vivo. Furthermore, PAX8 silencing induced HCC cell cycle arrest in S phase by downregulating E2F1, RB and P53 expression in HCC cells, consistent with previous reports [16, 33]. Therefore, PAX8 may be a new biomarker and therapeutic target for prognosis and intervention of HCC.
Precise and decisive control of protein expression, degradation and turnover is crucial for cellular homeostasis [34]. Previous studies have shown that HBx, a protein of the HBV, contributes to the development of HBV-related HCC, and participates into the process of cellular functions [35, 36]. Here, we show that although HBx does not directly interact with PAX8, but it stabilizes PAX8 by inhibiting its ubiquitination. Evidently, we found that the Skp2, a component of the SCF type ubiquitin ligase complex and a well-studied member of the F-box protein family, directly interacted with PAX8 and promoted its ubiquitination and degradation through its LRR domain [37]. Previous studies have shown that the sumoylation of PAX8 affects its stability, the mechanisms responsible for recognizing and targeting of PAX8 for ubiquitination and proteasomal degradation have remained unidentified [21] and HBx can regulate proteasomal functions [38]. Furthermore, HBx can interact directly with Skp2 although Skp2 binding domain with HBx remains controversial [27, 28, 39]. In this study, we found that HBx directly interacted with Skp2, dependent on its LRR domain and induction of HBx overexpression mitigated the Skp2-dependent PAX8 ubiquitination and degradation. It is possible that HBx through interacting with Skp2 to reduce/prevent the interaction of Skp2 with PAX8, inhibiting the PAX8 polyubiquitination to stabilize PAX8. Therefore, our findings may uncover a new mechanism by which HBx regulates the PAX8 stabilization and promotes the progression of HCC.
The E3 ubiquitin ligase usually recognizes a specific lysine acceptor in its substrate to promote proteasomal degradation [40]. A previous study has shown that the lysine 309 (K309) is a major acceptor in PAX8 for its sumolytion [21]. In this study, we identified that the lysine 197 and 275 residues (K197and K275) within PAX8 served as acceptors for PAX8 polyubiquitination and the K275 was essential for the Skp2-dependent proteasomal degradation. Thus, sumolytion and the Skp2-dependent PAX8 polyubiquitination appear to be through different acceptors in PAX8 to regulate its degradation in HCC cells.
Protein phosphorylation can inhibit or promote ubiquitination to prevent or enhance proteasomal degradation [41]. Aurora A, a member of the serine–threonine kinase family, is commonly elevated in epithelial malignancies [42, 43]. In this study, we found that Aurora A interacted with and phosphorylated PAX8 at S209 and T277, impairing the Skp2-mediated ubiquitination and degradation of PAX8. However, we did not detect the direct interaction between Skp2 and Aurora A (data not show), suggesting that alternative mechanisms underlying the action of Aurora A in regulating the Skp2-dependent PAX8 ubiquitination and degradation. A previous study has shown that Aurora A can stabilize N-Myc by inhibiting the Fbxw7 mediated degradation [44]. They argue that K63-linked or K11-linked ubiquitination plays a partial role in Aurora A-mediated stabilization of N-Myc. This suggests that Aurora A may recruit a S82 phosphatase or a de-ubiquitinating (DUB) enzyme to block the access of degradation [45, 46]. Furthermore, Aurora A can phosphorylate p53 to promote the Mdm2-mediated p53 destabilization and inhibition [47], which explains its oncogenic transformation in mammalian cells. As Aurora A is amplified and overexpressed in human cancers and correlates with poor prognosis of HCC patients, we examined whether HBx upregulated the expression and activity of Aurora A. We found that Aurora A phosphorylated PAX8 and HBx upregulated Aurora A expression to stabilize PAX8 protein, but not the phosphorylation site mutated PAX8. These data indicated that HBx indirectly enhanced PAX8 stability by up-regulating Aurora A expression and subsequent PAX8 phosphorylation-related stability. Therefore, our novel findings show that Aurora kinase A as a PAX8-activated molecule makes it an important factor in the pathogenesis of HCC.
Here, our data indicate that PAX8 is a major regulator of the progression of HCC and uncover new mechanisms by which HBx contributes to the pathogenesis of HCC. HBx stabilizes PAX8 protein by inhibiting the skp2-dependent ubiquitination and enhancing the Aurora A-mediated PAX8 phosphorylation. Potentially, our findings may aid in design of new therapies for intervention of HBV-related HCC.
Materials and methods
Cell culture
Human hepatoma HepG2 (passage 9), HepG2.2.15 (passage 11), and embryonic kidney HEK293T cells were from ATCC and maintained in our laboratory. MHCC97H (passage 6), MHCC97L (passage 6) cells were purchased from Guangzhou Cellcook Biotech and maintained in our laboratory. The cells were cultured in Dulbecco’s-modified Eagle’s medium (Hyclone) supplemented with 10% fetal bovine serum and 100 units/ml of penicillin and 100 µg/ml of streptomycin (Gbico). HepG2 cells were transduced with lentivirus for expression of control and PAX8-specific shRNA1 or shRNA2 to generate control HepG2-shNC, PAX8-stably silencing HepG2-shPAX8-1 and HepG2-shPAX8-2 cells, respectively. The cells were cultured in the presence or absence of cycloheximide (CHX, 50 µM, Cell Signal Technology), MG132 (25 µM Selleck), and/or MLN8237 (1 or 2 µM, Selleck). The cells were subjected to subsequent experiments.
Western blot assay
Individual groups of cells were lyzed in lysis buffer containing proteinase and phosphatase inhibitors (Roche) and centrifuged. After quantification of protein concentrations by BCA assay (ThermoFisher Scientific, 23235), individual cell lysate samples (30 µg/lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels and transferred onto polyvinylidene difluoride membranes (Millipore, IPVH00010). After being blocked with 5% fat-free dry milk in TBST, the membranes were incubated with antibodies against PAX8 (Novus, NBP1-32440), HBx (XIAMEN INNOVAX BIOTECH), Rb (#9313), phosphor-Rb (#D59B7), P21 (#2947), E2F1 (sc-251), p53 (sc-126), CDK2 (#78B2), and GAPDH (60004-1-Ig, Santa Cruz Biotechnology or Proteintech). The bound antibodies were detected with horseradish peroxidase (HRP)-conjugated second antibodies and visualized using the enhanced chemiluminescent reagents. The relative levels of target protein to the control GAPDH were determined by densitometric analysis using the ImageJ software.
Immunoprecipitation
The potential direct interaction between PAX8 and HBx was determined by immunoprecipitation, as previously reported [48]. Briefly, HEK293T cells were transfected with the control pcDNA/HA-Ub alone or together with pcDNA/Myc-HBx and/or pcDNA/Flag-PAX8 for 48 h using lipofectamine 2000 (ThermoFisher Scientific). After lyzed and centrifuged, the cell lysate samples were incubated with anti-Flag M2 (Sigma) 4 °C overnight and the immunocomplex was precipitated with the protein A/G agarose beads (Santa Cruz Biotechnology, sc-2003) at 4 °C for 2–3 h. After intensively washed, the bound immunocomplex was eluted and separated by SDS-PAGE, followed by probing with anti-HA. The input samples were simultaneously analyzed using anti-Flag, anti-Myc, and anti-HA.
In addition, the potential direct interaction between PAX8, HBx, Skp2, and Aurora A in HepG2 cells was determined by immunoprecipitation using anti-PAX8, anti-HBx, or anti-Skp2 and visualized by western blot. Similarly, HEK293T cells were cotransfected with pcDNA/HA-Ub alone or together with pcDNA/Flag-PAX8 and pcDNA/His-Skp2 or its mutants of Skp2ΔF, Skp2ΔLRR or Skp2-specific siRNA for 48 h. Some cells were transfected with pcDNA/Flag-HBx and pcDNA/His-Skp2 or pcDNA/Flag-PAX8 and pcDNA/Myc-HBx for 48 h. The cells were treated with vehicle DMSO or 25 mM MG132 for 6 h. The potential interaction of PAX8, HBx, and Skp2 was examined by immunoprecipitation using anti-Flag, anti-His (GenScript 66005-1-Ig), anti-HA (sc-57592), anti-Myc or isotype control, and visualized by immunoblot. Moreover, HepG2 and HEK293T cells were transfected with pcDNA3.1 or pcDNA/Aurora A for 48 h and immunoprecipitated with anti-PAX8 and Aurora A (#14474), followed by immunoblot with anti-phosphoseine (P-Ser, sc-81516) and anti-phosphothreonine (P-Thr, sc-81526), phosphor-Aurora Athr288 (#3079), C-Myc (#13987), and Myc-Tag (#2276, Santa Cruz Biotechnology).
Immunofluorescence
HepG2 cells were cultured on glass coverslips in six-well plates up to 30–40% confluency. After being washed, the cells were fixed with 4% paraformaldehyde and blocked with 5% BSA (including 0.1% Triton-100). The cells were incubated with rabbit anti-PAX8 and mouse anti-Skp2 at 4 °C overnight. After being washed, the cells were further stained with Alexa Fluor 568-conjugated anti-rabbit IgG or Alxa Fluor 488-conjugated anti-mouse IgG or control (Invitrogen) and counterstained with DAPI (ThermoFisher Scientific). The fluorescent signals were examined under a confocal microscope (Olympus, Tokyo, Japan). In addition, HepG2 cells were cotransfected with pcDNA/Flag-HBx and pcDNA/His-Skp2 for 48 h and stained with anti-HBx and anti-Skp2, followed by fluorescent second antibodies (green for HBx and red for Skp2).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from individual groups of cells using Trizol Reagent (Life Technology, 15596-26) and reversely transcribed into cDNA using PrimeScript RT Reagent Kit (Takara, RR047A), according to the manufacturer’s instruction. The relative levels of target mRNA transcripts to the control GAPDH were determined by qRT-PCR using the SYBR green kit and specific primers. The sequences of primers were PAX8-F: 5′-CAATGCCTTTCCCCATGCTG-3′ PAX8-R: 5′-ATGGCAGAGGAGGCATAGC-3′; Skp2-F: 5′-ATGCCCCAATCTTGTCCATCT-3′; Skp2-R: 5′-CACCGACTGAGTGATAGGTGT-3′. The PCR reactions were performed in duplicate, as previously reported [49].
GST pull down
GST-PAX8 and its truncated mutations were purified using GST Sefinose Resin (Sangon Biotech). The purified proteins were characterized by Coomassie blue staining. Appropriate amounts of purified GST-tagged protein were mixed with HEK293T cell lysates, incubated at 4 °C overnight. The resins were washed and bound proteins were analyzed by immunoblot using anti-GST (sc-53909).
In vivo ubiquitination assay
HEK293T cells were transfected with specific plasmids for 48 h and treated with 50 µM MG132 for additional 6 h. The cells were lyzed in RIPA buffer containing protease inhibitors at 4 °C for 30 min and centrifuged at 20,000 g for 15 min. Equal amounts of cellular extracts were immunoprecipitated with anti-Flag at 4 °C overnight. The generated immunocomplex was precipitated by protein A/G agarose beads. After being washed, the immunocomplex was resolved by SDS-PAGE and characterized by immunoblot.
In vitro phosphorylation assay
The DNA fragments for mutant PAX8 at Ser209 or Thr277 were amplified by PCR and cloned into the plasmid of pGEX-4T-1 for expression of control GST-PAX8, GST-PAX8S209A, and GST-PAX8T277A mutants. After being transfected, the generated GST-PAX8, GST-PAX8S209A, and GST-PAX8T277A were purified by GST Sefinose Resin. Subsequently, the sensitivity of individual GST-PAX8, GST-PAX8S209A, and GST-PAX8T277A samples to phosphorylation induced by Aurora A was determined in vitro. Briefly, a mixture of 1 mM ATP (Invitrogen, AM8110G), 3 µg GST-PAX8, PAX8S209, or PAX8T277, 50 µl 1 × Kinase Buffer (Cell Signal Technology, #9802) and the Flag-Aurora A protein immunoprecipitated from 293T lysates was reacted at 30 °C for 20 min. After being boiled, the proteins were resolved by SDS-PAGE, and subjected to autoradiograph.
Statistics analysis
Data are expressed as the mean ± standard deviation (SD) from at least three separate experiments. The difference among groups was determined by One-way ANOVA and post hock least significance difference test and the difference between groups was analyzed by Student’s t-test using GraphPad prism version7 (GraphPad Software, USA). P-values < 0.05 was considered statistically significant.
References
Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599–616.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Wang J, Li WT, Zheng YX, Zhao SS, Li N, Huang Y, et al. The association between helicobacter pylori infection and chronic hepatitis C: a meta-analysis and trial sequential analysis. Gastroenterol Res Pract. 2016;2016:8780695.
Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64:S84–101.
Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci. 2006;31:137–55.
Ding F, Xiao H, Wang M, Xie X, Hu F. The role of the ubiquitin-proteasome pathway in cancer development and treatment. Front Biosci (Landmark Ed). 2014;19:886–95.
Liu X, Guo H, Wang H, Markham R, Wei W, Yu XF. HIV-1 Vpr suppresses the cytomegalovirus promoter in a CRL4(DCAF1) E3 ligase independent manner. Biochem Biophys Res Commun. 2015;459:214–9.
Lin GY, Paterson RG, Richardson CD, Lamb RA. The V protein of the paramyxovirus SV5 interacts with damage-specific DNA binding protein. Virology. 1998;249:189–200.
Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, et al. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016;16:2846–54.
Decorsiere A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531:386–9.
Gao Y, Feng J, Yang G, Zhang S, Liu Y, Bu Y, et al. Hepatitis B virus X protein-elevated MSL2 modulates hepatitis B virus covalently closed circular DNA by inducing degradation of APOBEC3B to enhance hepatocarcinogenesis. Hepatology. 2017;66:1413–29.
Delogu S, Wang C, Cigliano A, Utpatel K, Sini M, Longerich T, et al. SKP2 cooperates with N-Ras or AKT to induce liver tumor development in mice. Oncotarget. 2015;6:2222–34.
Campagnolo M, Pesaresi A, Zelezetsky I, Geremia S, Randaccio L, Bisca A, et al. Structural studies on Pax-8 Prd domain/DNA complex. J Biomol Struct Dyn. 2007;24:429–41.
Poleev A, Fickenscher H, Mundlos S, Winterpacht A, Zabel B, Fidler A, et al. PAX8, a human paired box gene: isolation and expression in developing thyroid, kidney and Wilms' tumors. Development. 1992;116:611–23.
Wang L, Bo X, Zheng Q, Ge W, Liu Y, Li B. Paired box 8 suppresses tumor angiogenesis and metastasis in gastric cancer through repression of FOXM1 via induction of microRNA-612. J Exp Clin Cancer Res. 2018;37:159.
Ghannam-Shahbari D, Jacob E, Kakun RR, Wasserman T, Korsensky L, Sternfeld O, et al. PAX8 activates a p53-p21-dependent pro-proliferative effect in high grade serous ovarian carcinoma. Oncogene. 2018;37:2213–24.
Hung N, Chen YJ, Taha A, Olivecrona M, Boet R, Wiles A, et al. Increased paired box transcription factor 8 has a survival function in Glioma. BMC Cancer. 2014;14:159.
Ma S, Yang J, Song C, Ge Z, Zhou J, Zhang G, et al. Expression quantitative trait loci for PAX8 contributes to the prognosis of hepatocellular carcinoma. PLoS ONE. 2017;12:e0173700.
Chai HJ, Ren Q, Fan Q, Ye L, Du GY, Du HW, et al. PAX8 is a potential marker for the diagnosis of primary epithelial ovarian cancer. Oncol Lett. 2017;14:5871–5.
Suzuki A, Hirokawa M, Takada N, Higuchi M, Yamao N, Kuma S, et al. Diagnostic significance of PAX8 in thyroid squamous cell carcinoma. Endocr J. 2015;62:991–5.
de Cristofaro T, Mascia A, Pappalardo A, D'Andrea B, Nitsch L, Zannini M. Pax8 protein stability is controlled by sumoylation. J Mol Endocrinol. 2009;42:35–46.
Zannini M, Francis-Lang H, Plachov D, Di Lauro R. Pax-8, a paired domain-containing protein, binds to a sequence overlapping the recognition site of a homeodomain and activates transcription from two thyroid-specific promoters. Mol Cell Biol. 1992;12:4230–41.
Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–9.
Gong J, Zhou Y, Liu D, Huo J. F-box proteins involved in cancer-associated drug resistance. Oncol Lett. 2108;15:8891–900.
Wang Z, Gao D, Fukushima H, Inuzuka H, Liu P, Wan L, et al. Skp2: a novel potential therapeutic target for prostate cancer. Biochim Biophys Acta. 2012;1825:11–7.
Chen L, Wu T, Wei TQ, Wei X, Li SF, Wang KJ, et al. Skp2-mediated degradation of p27 regulates cell cycle progression in compressed human bladder smooth muscle cells. Kaohsiung J Med Sci. 2014;30:181–6.
Kalra N, Kumar V. The X protein of hepatitis B virus binds to the F box protein Skp2 and inhibits the ubiquitination and proteasomal degradation of c-Myc. FEBS Lett. 2006;580:431–6.
Lee S, Kim W, Ko C, Ryu WS. Hepatitis B virus X protein enhances Myc stability by inhibiting SCFSkp2 ubiquitin E3 ligase-mediated Myc ubiquitination and contributes to oncogenesis. Oncogene. 2016;35:1857–67.
Hou D, Che Z, Chen P, Zhang W, Chu Y, Yang D, et al. Suppression of AURKA alleviates p27 inhibition on Bax cleavage and induces more intensive apoptosis in gastric cancer. Cell Death Dis. 2018;9:781.
Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–8.
Lu L, Han H, Tian Y, Li W, Zhang J, Feng M, et al. Aurora kinase A mediates c-Myc's oncogenic effects in hepatocellular carcinoma. Mol Carcinog. 2015;54:1467–79.
Di Palma T, Filippone MG, Pierantoni GM, Fusco A, Soddu S, Zannini M. Pax8 has a critical role in epithelial cell survival and proliferation. Cell Death Dis. 2013;4:e729.
Li CG, Nyman JE, Braithwaite AW, Eccles MR. PAX8 promotes tumor cell growth by transcriptionally regulating E2F1 and stabilizing RB protein. Oncogene. 2011;30:4824–34.
Varshavsky A. The ubiquitin system, autophagy, and regulated protein degradation. Annu Rev Biochem. 2017;86:123–8.
Xu Q, Gu S, Liang J, Lin Z, Zheng S, Yan J. The biological function of Hepatitis B virus X protein in hepatocellular carcinoma. Oncol Res. 2019;27:509–14.
Li J, He J, Fu Y, Hu X, Sun LQ, Huang Y, et al. Hepatitis B virus X protein inhibits apoptosis by modulating endoplasmic reticulum stress response. Oncotarget. 2017;8:96027–34.
Von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell. 2003;11:1189–200.
Minor MM, Slagle BL. Hepatitis B virus HBx protein interactions with the ubiquitin proteasome system. Viruses. 2014;6:4683–702.
Jamal A, Swarnalatha M, Sultana S, Joshi P, Panda SK, Kumar V. The G1 phase E3 ubiquitin ligase TRUSS that gets deregulated in human cancers is a novel substrate of the S-phase E3 ubiquitin ligase Skp2. Cell Cycle. 2015;14:2688–700.
Mattiroli F, Sixma TK. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat Struct Mol Biol. 2014;21:308–16.
VerPlank JJS, Goldberg AL. Regulating protein breakdown through proteasome phosphorylation. Biochem J. 2017;474:3355–71.
Kivinummi K, Urbanucci A, Leinonen K, Tammela TLJ, Annala M, Isaacs WB, et al. The expression of AURKA is androgen regulated in castration-resistant prostate cancer. Sci Rep. 2017;7:17978.
Jacobsen A, Bosch LJW, Martens-de Kemp SR, Carvalho B, Sillars-Hardebol AH, Dobson RJ, et al. Aurora kinase A (AURKA) interaction with Wnt and Ras-MAPK signalling pathways in colorectal cancer. Sci Rep. 2018;8:7522.
Hoeck JD, Jandke A, Blake SM, Nye E, Spencer-Dene B, Brandner S, et al. Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nat Neurosci. 2010;13:1365–72.
Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78.
Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell. 2016;30:357–8.
Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62.
Liu Z, Wang J, Xing W, Peng Y, Quan J, Fan X. LPS binding to HMGB1 promotes angiogenic behavior of endothelial cells through inhibition of p120 and CD31 via ERK/P38/Src signaling. Eur J Cell Biol. 2017;96:695–704.
Chen RC, Wang J, Kuang XY, Peng F, Fu YM, Huang Y, et al. Integrated analysis of microRNA and mRNA expression profiles in HBx-expressing hepatic cells. World J Gastroenterol. 2017;23:1787–95.
Acknowledgements
I would like to express my special thanks to my boyfriend Dr Yan Liang for his positive encouragement and supportive suggestions during the whole research work. My grateful thanks are also extended to Prof. Lun-Quan Sun for his assistance in proofreading with on this project. I would like to express my appreciation and gratitude to Prof. Wei Guo in University of Pennsylvania and Prof. Dao-Lin Tang in Southwestern Medical Center for their kind advice in the revision.
Funding
This work was supported by grant from International Scientific and Technology Cooperation Program of China (2015DFA31490), grant from National Natural Sciences Foundation of China (No.81700561, No.81873574, No.81402623), the National Science and Technology Major Project of China (2017YFC0908104, 2018ZX10732202) and Independent Exploration and Innovation Project of Graduate Student of Central South University (No.2016zzts519).
Author information
Authors and Affiliations
Contributions
X-GF and X-WH conceived and designed the project and approved the final manuscript. JW conducted the experiment and wrote the paper. NL and ZB. Y.H. helped with edit the manuscript. SF, S-MY and Y-MF contributed with cell culture and plasmid construction. R-CC and P-CZ helped with mice experiment. R-RZ and YH helped with analyze the data.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Li, N., Huang, ZB. et al. HBx regulates transcription factor PAX8 stabilization to promote the progression of hepatocellular carcinoma. Oncogene 38, 6696–6710 (2019). https://doi.org/10.1038/s41388-019-0907-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-0907-2
This article is cited by
-
Integrated analysis of intratumoral biomarker and tumor-associated macrophage to improve the prognosis prediction in cancer patients
BMC Cancer (2023)
-
VCP interaction with HMGB1 promotes hepatocellular carcinoma progression by activating the PI3K/AKT/mTOR pathway
Journal of Translational Medicine (2022)
-
Aerobic glycolysis enhances HBx-initiated hepatocellular carcinogenesis via NF-κBp65/HK2 signalling
Journal of Experimental & Clinical Cancer Research (2022)
-
HBx promotes hepatocarcinogenesis by enhancing phosphorylation and blocking ubiquitinylation of UHRF2
Hepatology International (2021)
