
Abstract
Backgrounds
Mitochondria plays a critical role in the development and pathogenesis of nonalcoholic fatty liver disease (NAFLD). Neohesperidin (NHP) could lower blood glucose and prevent obesity in mice. However, the direct effect of NHP on hepatic steatosis has not been reported.
Methods
Mice were fed with either a chow diet or HFD with or without oral gavage of NHP for 12 weeks. A variety of biochemical and histological indicators were examined. In vitro cell culture model was utilized to demonstrate underlying molecular mechanism of the effect induced by NHP treatment.
Results
NHP increases mitochondrial biogenesis, improves hepatic steatosis and systematic insulin resistance in high fat diet (HFD) fed mice. NHP elevates hepatic mitochondrial biogenesis and fatty acid oxidation by increasing PGC-1α expression. Mechanistically, the activation of AMP-activated protein kinase (AMPK) is involved in NHP induced PGC-1α expression.
Conclusions
PGC-1α-mediated mitochondrial biogenesis plays a vital role in the mitigation of hepatic steatosis treated by NHP. Our result suggests that NHP is a good candidate to be dietary supplement for the auxiliary treatment of NAFLD.
Similar content being viewed by others


Introduction
Hypercaloric diet is one of the main factors leading to metabolic syndrome, mainly manifested as obesity, hyperlipidemia, insulin resistance and fatty liver, among others1. Nonalcoholic fatty liver disease (NAFLD), which is characterized by the accumulation of excess fat in the liver of people who drink little or no alcohol2, is increasingly recognized as the hepatic manifestation of metabolic syndrome. The progression of NAFLD leads to liver cirrhosis and liver cancer1,3. However, the treatment for NAFLD is still limited.
It has been known that mitochondria play a critical role in the development and pathogenesis of NAFLD4,5. Mitochondrial biogenesis is essential to augment mitochondrial capacity, which helps relieving lipid accumulation in liver6. Proliferator-activated receptor γ coactiva-tor-1α (PGC-1α) is a key regulator of energy homeostasis by transcriptional regulation of genes involved in fatty acid oxidation and mitochondrial biology5,7,8. Previous researches have shown a 40% decrease in hepatic PGC-1α expression in NAFLD patients, accompanied by mitochondrial dysfunction, lipid accumulation, and insulin resistance9,10. Furthermore, the treatment that stimulates mitochondrial function can delay the progression of obesity and diabetes5. Therefore, PGC-1α-mediated mitochondrial biogenesis is essential for the improvement of NAFLD.
Traditional medicine and complementary have gained more attention for long-term use in treating metabolic diseases like obesity and diabetes due to less side-effects compared with synthetic chemical drugs11,12. Clinical studies reveal that dietary intake of flavonoids can reduce the risk of NAFLD13,14. Our previous study has shown that Quzhou Fructus Aurantii, which is rich in flavonoids, could ameliorate fatty liver and insulin resistance in high-fat diet (HFD) fed mice15. However, its mechanism is still unclear. Neohesperidin is the main component of Quzhou Fructus Aurantii15. It is reported that neohesperidin could inhibit lipid accumulation in adipocytes, lowers blood glucose and lipid and prevent obesity in mice16,17. However, the direct effect of neohesperidin on hepatic steatosis has not been reported.
In current study, we found that NHP elevated PGC-1α expression and hepatic mitochondrial biogenesis in HFD fed mice. It also enhanced fatty acid oxidation, alleviated hepatic steatosis and insulin resistance. Mechanistically, NHP induced AMPK activation is involved in this process.
Materials and methods
The research reported here was approved by the Ethics Committee of Quzhou People’s Hospital. All methods were carried out in accordance with the relevant guidelines and regulations.
Animal experiments
The protocol of animal experiment was approved by the Ethics Committee of Animal Experiments of Quzhou people’s hospital, China. Animal experiment was conducted at the Experimental Animal Center of Zhejiang University of Traditional Chinese Medicine, China. Eight-week-old male C57BL/6 male were purchased from GemPharmatech Co., Ltd., Jiangsu, China (license number of animal production: SYXK 2015-0001). All animals were kept under standard conditions with having free access to distilled water and common pelleted food. After one week of acclimation, the mice were randomly distributed into 3 groups of 12 mice: chow group, which were fed a chow diet (Provided by the Experimental Animal Center of Zhejiang University of Traditional Chinese Medicine) and received intragastrically administered distilled water; HFD group, which were fed a high fat diet with 60 kcal% fat (Research diet D12492, Research Diet, NJ) and received intragastrically administered distilled water; HFD + NHP group, which were fed HFD and treated with 50 mg/kg per day of intragastrically administered neohesperidin (NHP, CAS# 13241-33-3, Shanghai Yuanye Biological Technology, China) for 12 weeks. Food intake, body weight and fasting blood glucose (FBG) were measured once a week.
Biochemical testing
Fasting blood glucose (FBG) levels were measured by blood glucose meter (Johnson & Johnson, USA). Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglyceride (TG), nonesterified fatty acid (NEFA), total cholesterol (TC) levels were detected by biochemical analyzer according to the manufacturer’s instruction (DiaSys Diagnostic Systems, Shanghai, China). Hepatic triglyceride (TG) and cholesterol (TC) contents were assessed using enzymatic reactions with commercial kits (Dongou Diagnostics Co., LTD, Zhejiang, China). Hepatic malondialdehyde (MDA), reactive oxygen species (ROS), superoxide dismutase (SOD), catalase (CAT), reduced glutathione (GSH) were tested by using commercially available kits according to the manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute, China).
Oral glucose tolerance test (OGTT) and insulin tolerance test (ITT)
At the 8th week of the experiment, 5 mice were taken from each group for the OGTT and ITT. For OGTT, the mice were fasted for 12 h and then oral d-glucose (2 g/kg). For ITT, the mice were fasted for 6 h and then injected i.p. with insulin (0.75 U/kg). Blood glucose levels were measured at 0, 30, 60, 90, and 120 min, which was measured by tail vein using a standard glucometer (Johnson & Johnson, USA).
Histopathological analysis
Liver tissues were fixed in 10% formalin and processed into paraffin sections. Then the sections were stained with hematoxylin and eosin (H&E) staining. NAFLD activity score (NAS) was scored in a blinded manner according to Kleiner et al.18 Macrovesicular steatosis: score 0: <5% (minimal), score 1: 5–33% (mild), score 2: 34–66% (moderate), score 3: >66% (severe); lobular inflammation: score 0: none, score 1: <2 foci/200× field, score 2: 2–4 foci/200× field, score 3: >4 foci/200× field; hepatocellular ballooning: score 0: none, score 1: few, score 2: many. These three values were then added to obtain the NAS score (range 0–8). Mean scores were evaluated through calculating five different 200× microscopic fields per mouse section by two independent trained observers.
Liver tissues were processed into frozen sections. The sections were stained with oil red O staining from each group according to the manufacturer’s instructions (Solarbio Life Science, China) and the intensity of Oil Red O was quantified with ImageJ software (U.S. National Institutes of Health, Bethesda, MD). The final count represented the mean of percentage of stained area from five randomly selected 400× microscopic fields per mouse section.
Immunohistochemical staining
The liver tissues sections were deparrafinized and antigen retrieval were also performed as previously described19. MaxVision HRP-Polymer antiRabbit IHC Kit (MXB Biotechnologies, Fuzhou, China) was used to develop signal. The antibody used in this part was antiMPO (1: 50, ab9535, Abcam). The number of MPO-positive cells was calculated in 5 randomly selected 200× microscopic fields per mouse section using ImageJ software (U.S. National Institutes of Health, Bethesda, MD).
Cell culture and treatment
Human hepatoma HepG2 cell line was obtained from the Shanghai Bank of Cell Lines (Shanghai, China) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS, BBI Life Sciences Corporation, China), 100 U/mL penicillin, and 100 U/mL streptomycin at 37 °C in a humidified atmosphere with 5% CO2. The source of the cell line was Identified by STR profiling and tested for mycoplasma contamination. HepG2 cells were intervened by 0.4 mM palmitate (PA) after starving in serum-free DMEM for 24 h to establish the hepatic steatosis model. The cells were treated with DMSO, 0.4 mM PA, 0.4 mM PA + 100 μM NHP, 0.4 mM PA + 100 μM NHP + 20 μM SR-18292 (CAS#2095432-55-4, Sigma-Aldrich, USA) or 0.4 mM PA + 100 μM NHP + 100 nM Compound C (CAS#866405-64-3, Merch Millipore, USA) for 16 h, respectively. Then the cells were lysed, RNA and protein were collected for further measurement.
Mitochondrial function assay
Succinate dehydrogenase activity was detected by using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay, as our previously described20. ATP content was measured with ATP Determination Kit (Molecular Probes) according to the manufacturer’s instructions (#A22066, Thermo Fisher Scientific, USA).
Mitochondrial staining
HepG2 cells were treated with DMSO, 0.4 mM PA, and 0.4 mM PA + 100 μM NHP for 16 h. Then, the cells were stained with 200 nM Mito-Tracker Red (#M7512, ThermoFisher, USA) for 60 min at 37 °C, according to the manufacturer’s instruction. The fluorescence was visualized by a SUNNY RX50 fluorescence microscope. Fluorescence intensity was detected by using Microplate Reader (BioTek, USA) at Ex 579/Em 599 nm.
Real-time PCR and mtDNA analysis
Total RNA from liver tissues or hepatic cells, was collected using Trizol Reagent (#DP424, Tiangen Biotech Co. Ltd., Beijing, China) according to the instruction of the manufacturer. The cDNA was prepared by Maxima Reverse Transcriptases (#EP0751, Thermo Fisher Scientific, USA). The mitochondria extraction and purification of liver tissue was using QIAamp DNA Mini Kit (#51304, Qiagen) according to the manufacturer’s protocol21. Real-time PCR was performed in triplicate using SGExcel FastSYBR Mixture (#B532955-0005, Sangon Biotech Co., Ltd., Shanghai, China) by Roche LightCyclerR 480 Quantitative PCR System (Indianapolis, USA). Normal relative expression analysis was normalized to the internal control Gapdh or β-actin. mtDNA analysis was quantified by qPCR using primers specific for the mitochondrial cytochrome c oxidase subunit 2 (COX2) gene and normalized to genomic DNA by amplification of the ribosomal protein s18 (Rps18) nuclear gene. Primers are listed in Table 1.
Western blot analysis
The protein extraction method was according to our previous study19,22. Equal amounts of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Then, the separated proteins were transferred to polyvinylidene fluoride (PVDF) membrane and blocked with 1% casein at room temperature for 1 h. Subsequently, the membrane incubated overnight at 4 °C with the primary antibodies against antiphospho-AMPKα (1: 1000, #2535, Cell Signaling Technology), antiAMPKα (1: 1000, #5831, Cell Signaling Technology), and antiβ-actin (1:3000, A1978, Millipore Sigma). After washing with TBST for four times, the membranes were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. Immunoreactive bands were visualized using Tanon 4200SF system (Tanon Biotechnology, Shanghai, China) and quantified densitometry using Image J software (U.S. National Institutes of Health, Bethesda, MD).
Statistical analysis
All of data are presented as mean ± SD values. Statistical significance was evaluated using Student’s unpaired two-tailed t-test and among more than two groups by analysis of one-way ANOVA with Bonferroni’s post hoc test. p values of <0.05 were considered statistically significant and p values of <0.01 were considered statistically highly significant. Analysis was performed using GraphPad Prism software, version 5.0 (GraphPad Software, La Jolla, CA).
Results
NHP improves liver function in HFD-fed mice
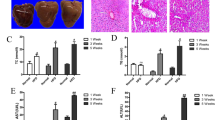
Long-term feeding of high-fat diet can cause hepatic lipid accumulation and impaired liver function. Compared to chow diet fed mice, the HFD mice displayed evidently more body weight gain and adipose tissue weight (Fig. S1). Administration of NHP reduced body weight gain significantly and adipose tissue weight to some extent, but had no effect on food intake in HFD-fed mice (Fig. S1). Meanwhile, NHP treatment decreased serum and liver ALT and AST levels in mice fed with HFD (Fig. 1a, b), suggesting improved liver function. In addition, administration of NHP obviously relieved hepatic steatosis in HFD mice indicated by H&E staining (Fig. 1c) and NAFLD activity score (NAS) evaluation (Fig. 1d).

C57BL/6 mice were fed either a chow or a high fat diet (HFD) for 12 weeks. Mice were treated with daily oral doses of NHP (50 mg/kg). Water was gavaged as control. a Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. b Hepatic alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels (n = 12). c The representative images of H&E staining in livers from each group. Scale bar = 300 μm. Black arrow represents macrovesicular steatosis; red arrow represents hepatocellular ballooning; blue arrow represents lobular inflammation. d The NAFLD activity score was shown. Data were expressed as the mean ± SD (n = 12). *p < 0.05, **p < 0.01, versus chow group; #p < 0.05, ##p < 0.01, versus HFD group.
NHP inhibits hepatic lipid accumulation in HFD-induced mice
To further characterize the effect of NHP on hepatic pathophysiology in HFD fed mice, we assessed serum and hepatic indicators for lipid metabolism. NHP treatment reduced TC, TG, and NEFA levels in serum (Fig. 2a, c). Moreover, hepatic TC and TG as well as liver weight were decreased upon NHP administration (Fig. 2d, f). The hepatic lipid accumulation examined by Oil Red O staining revealed that the percentage of stained area reduced to nearly the half by NHP in HFD mice (Fig. 2g).

a Serum total cholesterol (TC) level. b Serum triglyceride (TG) level. c Serum nonesterified fatty acid (NEFA) level. d Liver weight (g). e Hepatic total cholesterol (TC) level. f Hepatic triglyceride (TG) level. Data were expressed as the mean ± SD (n = 12). g The representative images of Oil Red O staining in livers from each group. Scale bar = 300 μm. The quantification of Oil Red O-stained areas was shown. Data were expressed as the mean ± SD (n = 12). *p < 0.05, **p < 0.01, versus chow group; #p < 0.05, ##p < 0.01, versus HFD group.
Insulin resistance is one of the most important features in HFD-induced mice15. NHP treatment obviously improved the levels of fasting blood glucose (FBG), fasting serum insulin (FINS) and HOMA-IR in HFD fed mice (Fig. S2A to C). The results from oral glucose-tolerance tests (OGTT) and insulin-tolerance tests (ITT) showed that NHP also markedly improved peripheral insulin resistance and glucose intolerance in HFD mice (Fig. S2D and E).
NHP relieves hepatic inflammation and oxidative stress in HFD-induced mice
Long-term excessive lipid accumulation in liver can cause inflammatory response and oxidative stress23,24. Our results showed that the number of MPO positive neutrophils were less in NHP treated group compared to their HFD counterparts (Fig. 3a). The mRNA expression of inflammatory factors such as Il-6, Il-1β, Tnf-α were also reduced by NHP treatment (Fig. 3b). NHP treatment significantly reduced hepatic MDA and ROS level, markers of oxidative stress (Fig. 3c, d). Additionally, the mRNA expression of antioxidant genes Cat, Sod1, Gpx1, and Ucp2 was increased by NHP treatment (Fig. 3e). At the same time, the levels of intracellular antioxidant enzymes (SOD, CAT) and ROS scavenger (GSH) were also increased by NHP in the liver of HFD induced mice (Fig. 3f).

a The representative images of myeloperoxidase (MPO) immunohistochemical staining in liver from each group. Scale bar = 300 μm. Black arrow points to MPO-positive cells. Quantification and statistical analysis of MPO-positive cells in liver from each group. Values were expressed as mean ± SD (n = 12). b The mRNA expressions of Il-6, Il-1β and Tnf-α were determined by RT-PCR. Values were expressed as mean ± SD (n = 5). c Hepatic malondialdehyde (MDA) level. d Hepatic reactive oxygen species (ROS) level. Data were expressed as the mean ± SD (n = 12). e The mRNA expressions of Cat, Sod1, Gpx1, and Ucp2 were determined by RT-PCR. Values were expressed as mean ± SD (n = 5). f Hepatic superoxide dismutase (SOD) activity, catalase (CAT) activity and reduced glutathione (GSH) level in each group. Data were expressed as the mean ± SD (n = 12). *p < 0.05, **p < 0.01, versus chow group; #p < 0.05, ##p < 0.01, versus HFD group.
NHP suppresses fatty acid synthesis and promotes fatty acid oxidation in liver
Under high-fat diets, increased lipid synthesis and reduced fatty acid oxidation promote hepatic steatosis5,25. We examined the expression of critical genes involved in fatty acid synthesis and fatty acid oxidation in liver. The result showed that NHP upregulated fatty acid oxidation gene expression, such as Pparα, Acaa2, Cpt-1, Pdk4, and Acox1 (Fig. 4a), while downregulated the expression of lipogenic genes including Srebf1, Fasn, Scd1, and Acc1 (Fig. S3) in the liver of HFD mice. Compared with fatty acid synthesis genes, NHP modulated fatty acid oxidation genes to a higher degree, which can be seen from the multiples of gene expression changes between HFD and HFD + NHP groups (Fig. 4a and S3).

a The mRNA expression of fatty acid oxidation genes was determined by real-time PCR. b Mitochondrial DNA content analyzed by quantitative PCR in liver from each group. c The ATP content was detected in liver. d Nrf-1 and Tfam mRNA analyzed by RT-PCR. All values were expressed as mean ± SD (n = 5). *p < 0.05, **p < 0.01, versus chow group; #p < 0.05, ##p < 0.01, versus HFD group.
NHP elevates mitochondrial biogenesis in liver of HFD mice
In order to further clarify the mechanism by which NHP regulates fatty acid oxidation, we examined mitochondrial function in liver of HFD fed mice. Treatment with NHP markedly increased mtDNA copy number (Fig. 4b) and ATP content (Fig. 4c) in the liver of HFD mice, compared to their control counterparts. Of note, the expression of genes associated with mitochondrial biogenesis (Nrf-1 and Tfam) was significantly up-regulated by NHP (Fig. 4d). These results suggested that the elevation fatty acid oxidation by NHP might be due to increased mitochondrial biogenesis in liver.
NHP improves mitochondrial capacity by promoting PGC-1α expression
Since PGC-1α is a crucial coactivator for fatty acid oxidation and mitochondrial biogenesis5,26, we next examined its expression in the liver of HFD mice. The treatment of NHP markedly increased the protein abundance of PGC-1α in the liver of HFD mice (Fig. 5a). The mRNA expression of Pgc-1α was also upregulated by NHP (Fig. 5b). Palmitic acid (PA) incorporation in culture medium was previously reported to disrupt mitochondrial function and increase lipid accumulation in HepG2 cells27,28. To determine whether NHP increases mitochondrial biogenesis via upregulation of PGC-1α, we used SR-18292, a selective PGC-1α inhibitor, to block the transcriptional activity of PGC-1α29. As shown in Fig. 6c, administration of NHP obviously increased the mRNA expression of PGC-1α target genes, which was inhibited by PA in HepG2 cells. The addition of SR-18292 counteracted NHP-induced increase of NRF-1 and TFAM (Fig. 5c). Mitochondrial mass is a crucial parameter for cellular oxidative capacity. NRF-1 and TFAM are critical regulators for mitochondrial fragmentation and mass30. We examined the alteration of mitochondrial mass using Mito-Tracker Red in PA induced HepG2 cells treated with NHP alone or plus SR-18292. NHP treatment prominently increased PA induced reduction of mitochondrial mass, but it was largely decreased by SR-18292 (Fig. 5d, e). In line with this observation, SR-18292 also counteracted the increase of ATP generation and succinate dehydrogenase activity treated by NHP in PA-induced HepG2 cells (Fig. 5f, g), which were considered as important indicators for mitochondrial capacity20. Moreover, the reduction of TG level induced by NHP in PA treated HepG2 cells was also diminished by PGC-1α inhibition (Fig. 5h). These data suggested that NHP elevates mitochondrial biogenesis by promoting PGC-1α expression.

a The protein level of PGC-1α in liver of HFD-induced mice was assessed by western blot analysis. The density of bands was normalized to β-actin. (b) The mRNA expression of Pgc-1α in liver of HFD-induced mice was determined by real-time PCR. Values were expressed as mean ± SD (n = 5). *p < 0.05, **p < 0.01, versus chow group; ##p < 0.01, versus HFD group. The HepG2 cells were treated with DMSO, 0.4 mM PA, 0.4 mM PA + 100 μM NHP or 0.4 mM PA + 100 μM NHP + 20 μM SR-18292 for 16 h, respectively, after starving in serum-free DMEM for 24 h. c The mRNA expressions of NRF-1 and TFAM were determined by RT-PCR. Values were expressed as mean ± SD (n = 3). d The HepG2 cells were stained with Mito-Tracker (red) and visualized by fluorescence microscopy. Scale bar = 300 μm. e The fluorescence intensity (mitochondrial mass) was determined by Microplate reader. f The ATP content was detected in the HepG2 cells. g The level of succinate dehydrogenase in cellular mitochondria was determined using MTT. h The content of triglycerides (TG) in the HepG2 was assessed using the commercial kits. Values were expressed as mean ± SD (n = 6). *p < 0.05, **p < 0.01.

a The protein level of phospho-AMPK and AMPK in livers of HFD-induced mice were assessed by western blot analysis. The density of bands was normalized to AMPK. b The HepG2 cells were treated with various concentrations of NHP (0, 5, 10, 25, 50, 100 μM) for 16 h. The amount of phospho-AMPK and AMPK was measured by western blotting. c The HepG2 cells were treated with DMSO, 0.4 mM palmitate (PA), 0.4 mM PA + 50 μM NHP or 0.4 mM PA + 100 μM NHP for 16 h respectively, after starving in serum-free DMEM for 24 h. The amount of phospho-AMPK and AMPK was measured by western blotting. d The HepG2 cells were treated with DMSO, 0.4 mM PA, 0.4 mM PA + 100 μM NHP or 0.4 mM PA + 100 μM NHP + 100 nM Compound C (CompC) for 16 h, respectively. The amount of phospho-AMPK and AMPK was measured by western blotting. e The mRNA expression of PGC-1α was determined by real-time PCR. Values were expressed as mean ± SD (n = 3). *p < 0.05, **p < 0.01.
NHP promotes PGC-1α expression by increasing AMPK activity
It has been shown that PGC-1α gene expression is induced by chemical activation of AMPK31. In this study, we showed that the phosphorylation of hepatic AMPK was consistently increased by NHP treatment in HFD-fed mice (Fig. 6a). Moreover, under regular culture condition, NHP stimulated AMPK phosphorylation in a dose-dependent manner in HepG2 cells (Fig. 6b). The addition of PA in culture medium downregulated AMPK phosphorylation while NHP treatment significantly reversed it (Fig. 6c). To examine whether AMPK is responsible for NHP induced PGC-1α, we re-inhibited increased AMPK activity by Compound C, a reversible AMPK inhibitor, after NHP treatment. We found that the mRNA of PGC-1α was obviously reduced after Compound C administration, compared to NHP treatment alone in PA-treated HepG2 cells (Fig. 6d, e). These results suggested that the increase of PGC-1α expression by NHP was via the simulation of AMPK activity.
Discussion
Here, we identify, for the first time, that NHP has the effect on hepatic mitochondrial biogenesis in HFD-induced mice. We showed that NHP ameliorate hepatic steatosis and systematic insulin resistance in HFD fed mice. NHP elevated hepatic mitochondrial biogenesis and fatty acid oxidation by increasing PGC-1α expression. The promotion of PGC-1α expression by NHP was achieved by the activation of AMPK. Overall, PGC-1α-mediated mitochondrial biogenesis plays a vital role in the mitigating effect of NHP on hepatic steatosis.
Fatty acid oxidation, fatty acids uptake, de novo synthesis, and the secretion of very low density lipoprotein (VLDL) are the major pathways influencing liver lipid content32,33. Although the clinical significance of mitochondrial β-oxidative impairment in the progression of NAFLD is inconclusive, studies have shown that fatty acid oxidation plays an important role in reducing liver lipid accumulation32. Mice deficient in MCAD and VLCAD, the acyl-CoA dehydrogenases involved in the beta dehydrogenation of the acyl-CoA ester derived from fatty acid in the process of β-oxidation, develop hepatic steatosis even fed by chow diet, emphasizing the role of these proteins and fatty acid β-oxidation in the regulation of hepatic TG content34. Targeting peroxisomes, alternative organelles of nonmitochondrial compartments of the cell for fatty acid oxidation, either by hepatocyte-specific depletion of peroxisomes or by deficiency in ACOX (which catalyzes the initial step in peroxisomal fatty acid oxidation) expression results in hepatic lipid accumulation, arguing the role of fatty acid oxidation in NAFLD35. In our study, we found that NHP-induced fatty acid oxidation was a major way by which NHP improved fatty liver. This observation further supports that fatty acid oxidation has a beneficial effect on improving lipid accumulation in liver.
PGC-1α mediated mitochondrial biogenesis is essential for the enhancement of mitochondrial capacity and fatty acid oxidation36. PGC-1α acts as a cardinal transcriptional regulator of mitochondrial biogenesis by activating nuclear respiratory factor-1 (NRF-1), which induce the transcription of mitochondrial transcription factor A (TFAM) expression thereby upregulating mtDNA replication and transcription26,37. Besides, PGC-1α promotes fatty acid β-oxidation by acting as a co-activator of PPARα, which in turn promotes the expression of genes related to mitochondrial fatty acid catabolism38. In the present study, we confirm that the increase of PGC-1α is required for mitochondrial biogenesis after NHP treatment.
Increased fatty acid oxidation not only reduces the lipid overload and lipotoxicity under HFD, but also produces ROS. Excessive fatty acid oxidation may overwhelm the capacity of the antioxidant defense system and induce oxidative stress and hepatic inflammation39. Intriguingly, NHP repressed oxidative stress and inflammatory response in liver induced by HFD, counteracting the adverse effect of enhanced fatty acid oxidation. In addition to the role on mitochondrial biogenesis, PGC-1α is also reported to be a regulator of antioxidant enzymes in response to oxidative stress40,41. PGC-1α reduces mitochondrial ROS production through upregulation of antioxidant genes expression such as Cat, Sod1, Gpx1, and Ucp241. In current study, we find that the expression of these antioxidant genes is elevated by NHP in vivo. Overproduction of ROS can lead to mitochondrial damage, including mutations in mitochondrial DNA, damage to the mitochondrial respiratory chain and mitochondrial membrane permeability42. We speculate the elevation of mitochondrial capacity is at least partially due to the relief of oxidative stress by NHP treatment, and the up-regulation of PGC-1α promoted antioxidant enzyme expression could be one of the underlying mechanisms.
The energy sensor AMPK is a master regulator in the control of energy metabolism in liver. At the posttranslational level, AMPK could activate PGC-1α by direct phosphorylation of its Ser538 residue31 or indirectly- by activating NAD+-dependent deacetylase SIRT143. At the transcriptional level, AMPK also promotes PGC-1α expression41,44. AMPK activator, 5-aminoimidazole-4-carboxamide-1-b-d-ribofuranoside (AICAR), induces PGC-1α transcription in rat muscles45. Additionally, metformin increases PGC-1α mRNA and protein expression via AMPK dependent manner in primary hepatocytes46. In this study, we find that NHP elevates the level of PGC-1α is also AMPK dependent.
Taken together, our results demonstrate that NHP alleviates hepatic steatosis and insulin resistance in HFD fed mice. NHP increases hapatic mitochondrial biogenesis by promoting PGC-1α expression. The activation of AMPK could be the underlying mechanism through which NHP regulates PGC-1α expression. This suggests that NHP has the potential to become a dietary supplement for the auxiliary treatment of NAFLD.
References
Cohen, J. C., Horton, J. D. & Hobbs, H. H. Human fatty liver disease: old questions and new insights. Science 332, 1519–1523 (2011).
Jiao, N. et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 67, 1881–1891 (2018).
Kim, Y. J. & Jung, U. J. Honokiol improves insulin resistance, hepatic steatosis, and inflammation in type 2 diabetic db/db mice. Int. J. Mol. Sci. 20, 2303 (2019).
Dey, A. & Swaminathan, K. Hyperglycemia-induced mitochondrial alterations in liver. Life Sci. 87, 197–214 (2010).
Price, N. L. et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15, 675–690 (2012).
Begriche, K., Massart, J., Robin, M. A., Bonnet, F. & Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58, 1497–1507 (2013).
Whitaker, R. M., Corum, D., Beeson, C. C. & Schnellmann, R. G. Mitochondrial biogenesis as a pharmacological target: a new approach to acute and chronic diseases. Annu. Rev. Pharm. Toxicol. 56, 229–249 (2016).
Scarpulla, R. C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278 (2011).
Besse-Patin, A. et al. Estrogen signals through peroxisome proliferator-activated receptor-gamma coactivator 1alpha to reduce oxidative damage associated with diet-induced fatty liver disease. Gastroenterology 152, 243–256 (2017).
Westerbacka, J. et al. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 56, 2759–2765 (2007).
Yeh, G. Y., Eisenberg, D. M., Kaptchuk, T. J. & Phillips, R. S. Systematic review of herbs and dietary supplements for glycemic control in diabetes. Diabetes Care 26, 1277–1294 (2003).
Xu, L., Zhao, W., Wang, D. & Ma, X. Chinese medicine in the battle against obesity and metabolic diseases. Front. Physiol. 9, 850 (2018).
Cicero, A. F. G., Colletti, A. & Bellentani, S. Nutraceutical approach to non-alcoholic fatty liver disease (NAFLD): the available clinical evidence. Nutrients 10, 1153 (2018).
Zhong, S. et al. The therapeutic effect of silymarin in the treatment of nonalcoholic fatty disease: a meta-analysis (PRISMA) of randomized control trials. Medicine 96, e9061 (2017).
Bai, Y. F. et al. The flavonoid-rich Quzhou Fructus Aurantii extract modulates gut microbiota and prevents obesity in high-fat diet-fed mice. Nutr. Diabetes 9, 30 (2019).
Shen, C. Y., Wan, L., Wang, T. X. & Jiang, J. G. Citrus aurantium L. var. amara Engl. inhibited lipid accumulation in 3T3-L1 cells and Caenorhabditis elegans and prevented obesity in high-fat diet-fed mice. Pharm. Res. 147, 104347 (2019).
Jia, S. et al. Hypoglycemic and hypolipidemic effects of neohesperidin derived from Citrus aurantium L. in diabetic KK-A(y) mice. Food Funct. 6, 878–886 (2015).
Kleiner, D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005).
Wang, S. W. et al. Cinobufacini ameliorates dextran sulfate sodium-induced colitis in mice through inhibiting M1 macrophage polarization. J. Pharm. Exp. Ther. 368, 391–400 (2019).
Wang, S. W. et al. Astilbin ameliorates cisplatin-induced nephrotoxicity through reducing oxidative stress and inflammation. Food Chem. Toxicol. 114, 227–236 (2018).
Rooney, J. P. et al. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 1241, 23–38 (2015).
Ou, Q. et al. Silybin alleviates hepatic steatosis and fibrosis in NASH mice by inhibiting oxidative stress and involvement with the Nf-kappaB pathway. Dig. Dis. Sci. 63, 3398–3408 (2018).
Toney, A. M. et al. Urolithin A, a gut metabolite, improves insulin sensitivity through augmentation of Mitochondrial function and biogenesis. Obesity 27, 612–620 (2019).
Wang, K. et al. Chronic administration of Angelica sinensis polysaccharide effectively improves fatty liver and glucose homeostasis in high-fat diet-fed mice. Sci. Rep. 6, 26229 (2016).
Li, Y. et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 (2011).
Wu, Z. et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 (1999).
Sun, Y. et al. Berberine attenuates hepatic steatosis and enhances energy expenditure in mice by inducing autophagy and fibroblast growth factor 21. Br. J. Pharm. 175, 374–387 (2018).
Galmes-Pascual, B. M. et al. 17beta-estradiol ameliorates lipotoxicity-induced hepatic mitochondrial oxidative stress and insulin resistance. Free Radic. Biol. Med 150, 148–160 (2020).
Sharabi, K. et al. Selective chemical inhibition of PGC-1alpha gluconeogenic activity ameliorates type 2 diabetes. Cell 169, 148–160 (2017).
Piantadosi, C. A. & Suliman, H. B. Redox regulation of mitochondrial biogenesis. Free Radic. Biol. Med. 53, 2043–2053 (2012).
Jager, S., Handschin, C., St-Pierre, J. & Spiegelman, B. M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl Acad. Sci. USA 104, 12017–12022 (2007).
Koo, S. H. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 19, 210–215 (2013).
Postic, C. & Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 118, 829–838 (2008).
Tolwani, R. J. et al. Medium-chain acyl-CoA dehydrogenase deficiency in gene-targeted mice. PLoS Genet. 1, e23 (2005).
Dirkx, R. et al. Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology 41, 868–878 (2005).
Lei, P. et al. Sulforaphane improves lipid metabolism by enhancing mitochondrial function and biogenesis in vivo and in vitro. Mol. Nutr. Food Res. 63, e1800795 (2019).
Chen, Y. et al. Synergistic effects of cilostazol and probucol on ER stress-induced hepatic steatosis via heme oxygenase-1-dependent activation of mitochondrial biogenesis. Oxid. Med. Cell Longev. 2016, 3949813 (2016).
Sergi, D. et al. Mitochondrial (Dys)function and insulin resistance: from pathophysiological molecular mechanisms to the impact of diet. Front. Physiol. 10, 532 (2019).
Chen, Z., Tian, R., She, Z., Cai, J. & Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 152, 116–141 (2020).
St-Pierre, J. et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408 (2006).
Rabinovitch, R. C. et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 21, 1–9 (2017).
Guo, C., Sun, L., Chen, X. & Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 8, 2003–2014 (2013).
Canto, C. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 (2009).
Fernandez-Marcos, P. J. & Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, 884S–890S (2011).
Suwa, M., Nakano, H. & Kumagai, S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J. Appl. Physiol. 95, 960–968 (2003).
Aatsinki, S. M. et al. Metformin induces PGC-1alpha expression and selectively affects hepatic PGC-1alpha functions. Br. J. Pharm. 171, 2351–2363 (2014).
Acknowledgements
We thank Prof. Jin-qi Fu for great advices to this work. This work was supported by National Natural Science Foundation of China [81903873], Medical and Health Technology Projects of Zhejiang Province, China [2020PY087, 2019PY089], the Chinese medicine science foundation of Zhejiang Province, China [2018ZB134], Quzhou technology projects, China [2019K34, 2018K20, 2016J016].
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Sw., Sheng, H., Bai, Yf. et al. Neohesperidin enhances PGC-1α-mediated mitochondrial biogenesis and alleviates hepatic steatosis in high fat diet fed mice. Nutr. Diabetes 10, 27 (2020). https://doi.org/10.1038/s41387-020-00130-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41387-020-00130-3
This article is cited by
-
Role of liver FGF21-KLB signaling in ketogenic diet-induced amelioration of hepatic steatosis
Nutrition & Diabetes (2024)
-
The potential of therapeutic strategies targeting mitochondrial biogenesis for the treatment of insulin resistance and type 2 diabetes mellitus
Archives of Pharmacal Research (2024)
-
Natural compounds proposed for the management of non-alcoholic fatty liver disease
Natural Products and Bioprospecting (2024)
-
Mini-encyclopedia of mitochondria-relevant nutraceuticals protecting health in primary and secondary care—clinically relevant 3PM innovation
EPMA Journal (2024)
-
UPLC-QTOF-MS with a chemical profiling approach for holistic quality evaluation between a material reference of Wen Dan decoction and its commercial preparations
Chinese Medicine (2023)
