
Abstract
Background/objectives
Bone-derived fibroblast growth factor 23 (FGF23) is a hormone that suppresses renal phosphate reabsorption and calcitriol (i.e., 1,25(OH)2D3) formation together with its co-receptor Klotho. FGF23- or Klotho-deficient mice suffer from rapid aging with multiple age-associated diseases, at least in part due to massive calcification. FGF23 is considered as a disease biomarker since elevated plasma levels are observed early in patients with acute and chronic disorders including renal, cardiovascular, inflammatory, and metabolic diseases. An energy-dense diet, which induces sequelae of the metabolic syndrome in humans and mice at least in part by enhancing pro-inflammatory TNFα formation, has recently been demonstrated to stimulate FGF23 production.
Methods
We investigated the relevance of TNFα for high-fat diet (HFD)-induced FGF23 formation in wild-type (tnf+/+) and TNFα-deficient (tnf−/−) mice.
Results
Within 3 weeks, HFD feeding resulted in a strong increase in the serum FGF23 level in tnf+/+ mice. Moreover, it caused low-grade inflammation as evident from a surge in hepatic Tnfα transcript levels. TNFα stimulated Fgf23 transcription in UMR106 osteoblast-like cells. Serum FGF23 was significantly lower in tnf−/− mice compared to tnf+/+ mice following HFD. Serum phosphate and calcitriol were not significantly affected by genotype or diet.
Conclusions
We show that HFD feeding is a powerful stimulator of murine FGF23 production through TNFα formation.
Similar content being viewed by others


Introduction
The hormone fibroblast growth factor 23 (FGF23) is mainly produced by osteoblasts and osteocytes in the bone1. Its renal effects include inhibition of phosphate reabsorption and calcitriol formation1, 2. Calcitriol is the biologically active form of vitamin D. The renal effects of FGF23 are mediated by a receptor which requires the protein α-Klotho (referred to as Klotho in the following) as an obligatory co-receptor1.
Klotho was originally discovered in 1997 as an anti-aging protein3,4,5. Klotho-deficient mice have an extremely short life span of a few weeks only and exhibit many disorders associated with aging in humans3. FGF23-deficient mice have a similar phenotype6. Both mouse strains suffer from drastically elevated plasma levels of phosphate and calcitriol due to the primary renal effect of FGF23 and Klotho. Importantly, the premature aging of Klotho- or FGF23-deficient mice is also a direct or indirect consequence of the hyperphosphatemia of the mice since maintaining them on a low phosphate or low vitamin D diet normalizes their life span7.
A high plasma FGF23 level has been found in patients with various acute and chronic disorders including renal (acute kidney injury, chronic kidney disease), cardiovascular (coronary heart disease, myocardial infarction, atrial fibrillation), inflammatory, and metabolic diseases8. The role of FGF23 in chronic kidney disease is established best: Plasma FGF23 is elevated before a marked decrease of glomerular filtration rate (GFR), and it exhibits a strong positive correlation with mortality, hypertrophy of the left ventricle, and disease progression9. Therefore, it is presently being considered as a valuable disease biomarker. However, it is yet incompletely understood whether and to which extent FGF23 contributes to pathophysiological processes rather than merely indicating them. At least, FGF23 has been shown to induce hypertrophy of the left ventricle independently of Klotho10.
Recently, inflammation has been shown to be a major trigger of FGF23 formation11,12,13,14. In line with this, pro-inflammatory cytokines including TNFα induce FGF23 production15.
Metabolic syndrome is characterized by hypertension, glucose intolerance, dyslipidemia, as well as obesity, and affects millions of patients world-wide and represents a significant health burden particularly in industrialized countries16. Although the complex pathophysiological processes have not yet been uncovered completely, it is clear that an imbalance between caloric needs and intake is the predominant factor. In mice, a diet rich in fats (high-fat diet (HFD)) induces metabolic syndrome17,18,19. Low-grade inflammation associated with metabolic syndrome is relevant especially for the development of glucose intolerance20. In this respect, pro-inflammatory cytokines derived from adipose tissue or the liver are a major source of inflammation in metabolic syndrome. Among those cytokines, TNFα has been found to play a predominant role21. Interestingly, an energy-dense diet has recently been demonstrated to upregulate the production of FGF23 in rats22.
Here, we sought to define the role of metabolic syndrome-associated TNFα production in HFD-induced FGF23 formation.
Materials and methods
Animals and treatments
All animal experiments were conducted according to the German law for the welfare of animals and were approved by the authorities of the state of Saxony-Anhalt. Experiments were performed in TNFα-deficient (tnf−/−) mice (from The Jackson Laboratory, Sulzfeld, Germany; Stock No: 005540; the generation and genotyping is available on the website of The Jackson laboratory) and in age- and sex-matched wild-type mice (tnf+/+) fed a control diet (Ssniff, Soest, Germany; standard diet for maintenance V1534).
At the age of 8–10 weeks, the mice were fed a HFD containing 70% kcal from fat (Altromin, Lage, Germany; C1090-70) for 3 weeks, and the body weight was recorded weekly. The animals had free access to food and tap water. Serum was taken before and on the last day of the treatment. The exact number of mice and the number of replications is provided in the figure legends. For all animal experiments, no randomization was used, no blinding was done, and no statistical test was applied to estimate the sample size.
Serum parameters
To obtain blood specimens, the animals were lightly anesthetized with ether, and blood was drawn into heparinized capillaries by puncturing the retro-orbital plexus. Since the entire procedure takes less than a minute, it is unlikely to have a significant impact on our study. Serum concentrations of intact FGF23 and calcitriol were determined by ELISA kits (Immutopics, San Clemente, CA, USA; IDS, Frankfurt am Main, Germany). Inorganic phosphate was measured by a photometric method (Biocon® Diagnostik, Vöhl/Marienhagen, Germany).
Tissue collection and quantification of liver and adipose tissue Tnfα mRNA expression
For the determination of Tnfα mRNA abundance, total RNA was extracted from the liver and gonadal adipose tissue using the peqGold Trifast™ reagent (Peqlab, Erlangen, Germany) according to the manufacturer’s protocol. The RNA integrity was assessed by agarose gel electrophoresis and the RNA purity by measurement of the optical density at 260 and 280 nm. Single-strand cDNA was synthesized from 1.2 μg of total RNA at 42 °C for 60 min by use of the RevertAidTM M-MuLV Reverse Transcriptase (MBI Fermentas, St. Leon-Rot, Germany) and oligo dT18 primers (Eurofins MWG Operon, Ebersberg, Germany). The mRNA expression level was determined by real-time polymerase chain reaction (RT-PCR) with the Rotor-Gene 6000 system (Corbett Research, Mortlake, Australia) using 2 µl cDNA templates, SYBR® Green I (Sigma-Aldrich, München, Germany), 1.25 U Taq DNA polymerase (Promega, Mannheim, Germany), 500 µM dNTP (Ares Bioscience, Köln, Germany), and 13.3 pmol of a primer pair specific for Tnfα (NM_013693.2; forward 5′-AGT CCG GGC AGG TCT ACT TT-3′, reverse 5′-GGT CAC TGT CCC AGC ATC TT-3′). The Tnfα expression was normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (Gapdh, forward 5′-AAC GAC CCC TTC ATT GAC-3′, reverse 5′-TCC ACG ACA TAC TCA GCA C-3′) (in liver) or 18S (forward 5′-GGG AGC CTG AGA AAC GGC-3′, reverse 5′-GGG TCG GGA GTG GGT AAT TT-3′) (in adipose tissue) using the ΔΔCt method.
Cell culture
Cell culture was performed as previously described23. Briefly, UMR106 rat osteosarcoma cells (ATCC, Manassas, VA, USA) were cultured in DMEM high glucose medium (Gibco, Grand Island, NY, USA) supplemented with 10% FCS (Gibco) and 100 U/ml penicillin/100 µg/ml streptomycin (Gibco) under standard culture conditions. After 24 h, the cells were treated with or without TNFα (Sigma-Aldrich) for different periods.
Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from the cells using Trifast reagent (Peqlab) according to the manufacturer’s instructions. Messenger RNA was transcribed with GoScript™ Reverse Transcription System (Promega) using 1.2 μg of total RNA and random primers. For qRT-PCR analysis, the final volume of the qRT-PCR reaction mixture was 20 µl and contained: 2 µl cDNA, 0.5–1 µM of a primer pair specific for rat Fgf23 (forward 5′-TAGAGCCTATTCAGACACTTC-3′, reverse 5′-CATCAGGGCACTGTAGATAG-3′) or the housekeeping gene TATA box-binding protein (Tbp, forward 5′-ACTCCTGCCACACCAGCC-3′, reverse 5′-GGTCAAGTTTACAGCCAAGATTCA-3′), 10 µl GoTaq® qPCR Master Mix (Promega), and sterile water up to 20 µl. PCR conditions were 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 57 °C for 30 s, and 72 °C for 30 s. Quantitative RT-PCR was performed on a Rotor-Gene Q (QIAGEN, Hilden, Germany).
Statistics
Data are provided as means ± SEM, n represents the number of independent experiments or number of mice per group, respectively. All data were tested for significance using the tests indicated in the figure legends. For serum FGF23, normal distribution was assumed. The data meet the assumptions of the respective tests. Variance was similar between the groups apart from the data in Fig. 2B and Fig. 4E. Therefore, Welch’s correction was applied in these cases. Only results with p < 0.05 were considered statistically significant.
Results
At the age of 8–10 weeks, we started to feed wild-type mice (tnf+/+) a HFD ad libitum for 3 weeks. Similar to what has recently been demonstrated in rats22, the HFD caused a strong increase (by almost four times) in the serum intact FGF23 level (Fig. 1).

Arithmetic means ± SEM of the serum intact FGF23 concentration (n = 3 mice, no replication) in tnf+/+ mice before (control) and after 3 weeks of feeding a high-fat diet (HFD); **p < 0.01 (paired, two-tailed t-test)
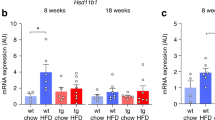
HFD feeding and subsequent adipose tissue accumulation are associated with subclinical inflammation and the generation of the key pro-inflammatory cytokine TNFα. Hence, we found that feeding HFD indeed resulted in a significant increase in liver Tnfα mRNA expression levels (Fig. 2A) in tnf+/+ mice pointing to HFD-associated low-grade inflammation. Moreover, also adipose tissue Tnfα mRNA expression levels (Fig. 2B) tended to be higher in tnf+/+ mice on HFD, a difference, almost reaching statistical significance (p = 0.106).

Arithmetic means ± SEM (n = 7 mice per group, one replication) of relative hepatic (A; unpaired, two-tailed t-test) and adipose tissue (AT). (B; unpaired, two-tailed t-test with Welch’s correction) Tnfα mRNA abundance (relative to Gapdh or 18S mRNA) in tnf+/+ mice on control diet and on high-fat diet (HFD) for 3 weeks. *p < 0.05
Next, we carried out cell culture experiments with UMR106 osteoblast-like cells to test whether TNFα is capable of stimulating FGF23 production as has been shown for IDG-SW3 cells15. According to Fig. 3a, a 24 h incubation with TNFα resulted in a dose-dependent upregulation of Fgf23 mRNA transcript levels in UMR106 cells with significance at 5 and 10 ng/ml TNFα. The time dependence for the effect of 5 ng/ml TNFα is illustrated in Fig. 3B.

Arithmetic means ± SEM of relative Fgf23 mRNA abundance (relative to Tbp mRNA) in UMR106 cells incubated for 24 h without (white bar) or with (black bars) TNFα at the indicated concentration (A; n = 5) or incubated with TNFα (5 ng/ml) for the indicated periods (B; n = 6); *p < 0.05 and **p < 0.01 (one-way ANOVA followed by Dunnett’s multiple comparisons test)
Our last series of experiments explored whether the HFD-induced FGF23 production is dependent on TNFα formation. To this end, we compared tnf+/+ mice with tnf−/− mice. On control diet, the serum intact FGF23 concentration was not significantly different between tnf+/+ mice and tnf−/− mice (Fig. 4A). However, after 3 weeks of feeding the HFD, the serum intact FGF23 level was significantly different between the genotypes being nearly 50% lower in tnf−/− mice compared to tnf+/+ mice (Fig. 4A). Serum calcitriol was not significantly different between tnf−/− and tnf+/+ mice on either control or HFD, but was significantly lower in a group of HFD-fed mice compared to mice on control diet (Fig. 4B). Similarly, the serum phosphate concentration was not significantly affected by neither genotype nor diet (Fig. 4C). On control diet, no significant difference between the body weight of tnf+/+ mice and tnf−/− mice could be observed (Fig. 4D). However, the HFD resulted in significantly stronger weight gain in tnf+/+ mice than in tnf−/− mice (Fig. 4D, E).

Arithmetic means ± SEM of the serum intact FGF23 concentration (A; n = 3 tnf+/+ mice and 7 tnf−/− mice, one replication) and serum phosphate concentration (C; n = 11 tnf+/+ mice, and 8 tnf−/− mice, two replications) in tnf+/+ mice (white bars) and tnf−/− mice (black bars) before (control) and after 3 weeks of feeding a high-fat diet (HFD). Arithmetic means ± SEM of the serum calcitriol concentration (B; n = 8 mice per group) in a group of tnf+/+ mice (white bars) and tnf−/− mice (black bars) on control diet or HFD fed for 3 weeks. Arithmetic means ± SEM (n = 13 tnf+/+ mice and 12 tnf−/− mice, two replications) of the total body weight (D) in dependence of the duration of HFD and relative weight gain after 3 weeks of HFD (E); *p < 0.05, **p < 0.01, and ***p < 0.001 indicate significant difference between the genotypes; ###p < 0.001 indicates significant difference between control and HFD. (A–C: one-way ANOVA followed by Tukey’s multiple comparisons test; D: Holm-Sidak method; E: unpaired, two-tailed t-test with Welch’s correction)
Discussion
According to our study, the stimulatory effect of a HFD on FGF23 formation was significantly blunted in gene-targeted mice devoid of pro-inflammatory TNFα (tnf−/−). This result suggests that a HFD stimulates FGF23 production in large part by inducing low-grade inflammation.
It is well established that energy-dense diets including a HFD favor the development of metabolic syndrome characterized by insulin resistance, dyslipidemia, obesity, and hypertension24, 25. This pathophysiological condition is associated with systemic low-grade inflammation26. In particular, a pivotal role for the pro-inflammatory cytokine TNFα in the development of obesity-induced insulin resistance has been demonstrated21.
Inflammation has emerged as a powerful factor driving FGF23 production11. Our study demonstrates that TNFα upregulated Fgf23 transcript levels in UMR106 osteosarcoma-like cells. Importantly, TNFα is effective through transcription factor NF-κB15 and in line with this, NF-κB has also been demonstrated to enhance FGF23 synthesis14.
Elevated serum FGF23 concentrations are observed in acute and chronic renal, metabolic, and cardiovascular diseases8. Most of these clinical conditions are associated with inflammation. Therefore, similar to HFD feeding, these disorders may at least in part be effective in stimulating FGF23 production by enhancing the production of pro-inflammatory cytokines.
On control diet, the serum concentration of intact FGF23 was not significantly different between tnf+/+ mice and tnf−/− mice although a tendency toward lower FGF23 in tnf−/− mice was apparent. A pro-inflammatory milieu in HFD-treated animals, however, resulted in strong TNFα-dependent FGF23 generation.
Since an increase in serum FGF23 has been observed very early in some chronic disorders including chronic kidney disease, FGF23 has been suggested as a biomarker27. According to our results, an increase in serum FGF23 by almost four times was observed after 3 weeks of HFD feeding, a relatively short period as evident from a moderate increase in total body weight by only some 10% in tnf+/+ mice. Therefore, lower FGF23 may also indicate a better metabolic profile of an individual.
A major effect of FGF23 is the inhibition of renal calcitriol formation thereby lowering the serum calcitriol concentration1. Elevated FGF23 formation in HFD-fed mice could therefore be expected to decrease the serum calcitriol concentration. We did not, however, observe a significant difference in the serum calcitriol between the genotypes, although a tendency toward higher values in tnf−/− mice was obvious.
Taken together, our study demonstrates that a HFD stimulates FGF23 production at least in part by inducing TNFα formation.
References
Kuro-O, M. & Moe, O. W. FGF23-alphaKlotho as a paradigm for a kidney-bone network. Bone. https://doi.org/10.1016/j.bone.2016.11.013 (2016).
Spichtig, D. et al. Renal expression of FGF23 and peripheral resistance to elevated FGF23 in rodent models of polycystic kidney disease. Kidney Int. 85, 1340–1350 (2014).
Kuro-o, M. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 (1997).
Maltese, G. et al. The anti-ageing hormone klotho induces Nrf2-mediated antioxidant defences in human aortic smooth muscle cells. J. Cell. Mol. Med. https://doi.org/10.1111/jcmm.12996 (2016).
Donate-Correa, J. et al. Influence of Klotho gene polymorphisms on vascular gene expression and its relationship to cardiovascular disease. J. Cell. Mol. Med. 20, 128–133 (2016).
Shimada, T. et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest. 113, 561–568 (2004).
Kuro-O, M. A potential link between phosphate and aging--lessons from Klotho-deficient mice. Mech. Ageing Dev. 131, 270–275 (2010).
Schnedl, C., Fahrleitner-Pammer, A., Pietschmann, P. & Amrein, K. FGF23 in acute and chronic illness. Dis. Markers 2015, 358086 (2015).
Wahl, P. & Wolf, M. FGF23 in chronic kidney disease. Adv. Exp. Med. Biol. 728, 107–125 (2012).
Faul, C. et al. FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 121, 4393–4408 (2011).
Sharaf El Din, U. A. A., Salem, M. M. & Abdulazim, D. O. FGF23 and inflammation. World J. Nephrol. 6, 57–58 (2017).
Francis, C. & David, V. Inflammation regulates fibroblast growth factor 23 production. Curr. Opin. Nephrol. Hypertens. 25, 325–332 (2016).
David, V. et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 89, 135–146 (2016).
Zhang, B. et al. NFkappaB-sensitive Orai1 expression in the regulation of FGF23 release. J. Mol. Med. (Berl) 94, 557–566 (2016).
Ito, N. et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol. Cell. Endocrinol. 399, 208–218 (2015).
Grundy, S. M. et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 112, 2735–2752 (2005).
Chen, H. et al. PI3K-resistant GSK3 controls adiponectin formation and protects from metabolic syndrome. Proc. Natl Acad. Sci. USA 113, 5754–5759 (2016).
Tsai, V. W. et al. Treatment with the TGF-b superfamily cytokine MIC-1/GDF15 reduces the adiposity and corrects the metabolic dysfunction of mice with diet-induced obesity. Int. J. Obes. 42, 561–571 (2017).
Choi, J. et al. Role of the histone deacetylase inhibitor valproic acid in high-fat diet-induced hypertension via inhibition of HDAC1/angiotensin II axis. Int. J. Obes. (Lond). 41, 1702–1709 (2017).
Tamakoshi, K. et al. The metabolic syndrome is associated with elevated circulating C-reactive protein in healthy reference range, a systemic low-grade inflammatory state. Int. J. Obes. Relat. Metab. Disord. 27, 443–449 (2003).
Uysal, K. T., Wiesbrock, S. M., Marino, M. W. & Hotamisligil, G. S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389, 610–614 (1997).
Raya, A. I. et al. Energy-dense diets increase FGF23, lead to phosphorus retention and promote vascular calcifications in rats. Sci. Rep. 6, 36881 (2016).
Fajol, A. et al. Enhanced FGF23 production in mice expressing PI3K-insensitive GSK3 is normalized by beta-blocker treatment. FASEB J. 30, 994–1001 (2016).
Pitsavos, C., Panagiotakos, D., Weinem, M. & Stefanadis, C. Diet, exercise and the metabolic syndrome. Rev. Diabet. Stud. 3, 118–126 (2006).
Gregoire, F. M. et al. Diet-induced obesity and hepatic gene expression alterations in C57BL/6J and ICAM-1-deficient mice. Am. J. Physiol. Endocrinol. Metab. 282, E703–E713 (2002).
Dandona, P. Inflammation. The link between insulin resistance, obesity and diabetes. Trends Immunol. 25, 4–7 (2004).
Fliser, D. et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J. Am. Soc. Nephrol. 18, 2600–2608 (2007).
Acknowledgements
The authors acknowledge the technical assistance of Heike Giese, Suzanne Roß, and Franziska Reipsch. The study was supported by the Deutsche Forschungsgemeinschaft.
Authors’ contribution
P.G., A.F., F.H., and M.F. performed the experiments. G.I.S. and J.V. provided the essential tools. P.G. and M.F. analyzed the data and wrote the paper. F.L. and M.F. designed the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Glosse, P., Fajol, A., Hirche, F. et al. A high-fat diet stimulates fibroblast growth factor 23 formation in mice through TNFα upregulation. Nutr & Diabetes 8, 36 (2018). https://doi.org/10.1038/s41387-018-0037-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41387-018-0037-x
This article is cited by
-
FGF-23 protects cell function and viability in murine pancreatic islets challenged by glucolipotoxicity
Pflügers Archiv - European Journal of Physiology (2023)
-
The regulation of FGF23 under physiological and pathophysiological conditions
Pflügers Archiv - European Journal of Physiology (2022)
-
Elevation of serum fibroblast growth factor 23 level in a pediatric patient with lupus nephritis
CEN Case Reports (2022)
-
Metabolic syndrome; Definition, Pathogenesis, Elements, and the Effects of medicinal plants on it’s elements
Journal of Diabetes & Metabolic Disorders (2022)
-
Regulation of FGF23: Beyond Bone
Current Osteoporosis Reports (2021)
