
Abstract
Helminths are remarkably successful parasites that can invade various mammalian hosts and establish chronic infections that can go unnoticed for years despite causing severe tissue damage. To complete their life cycles, helminths migrate through multiple barrier sites that are densely populated by a complex array of hematopoietic and non-hematopoietic cells. While it is clear that type 2 cytokine responses elicited by immune cells promote worm clearance and tissue healing, the actions of non-hematopoietic cells are increasingly recognized as initiators, effectors and regulators of anti-helminth immunity. This review will highlight the collective actions of specialized epithelial cells, stromal niches, stem, muscle and neuroendocrine cells as well as peripheral neurons in the detection and elimination of helminths at mucosal sites. Studies dissecting the interactions between immune and non-hematopoietic cells will truly provide a better understanding of the mechanisms that ensure homeostasis in the context of helminth infections.
Similar content being viewed by others
Introduction
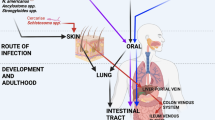
Helminths are parasitic worms that infect invertebrate intermediate hosts that serve as vectors necessary to complete their life cycles (e.g., Schistosoma mansoni cercariae emerge from aquatic snails to infect humans or other vertebrates) as well as diverse mammalian hosts in which they can establish persistent chronic infections. Most helminths migrate through several barrier tissues to complete their life cycles, including the skin, lungs, and gastrointestinal (GI) tract (Table 1). As they pass through these compartments, helminths secrete several excretory/secretory (E/S) products and cause tissue damage that has lasting consequences for homeostasis, including fibrotic lesions, or altered microbiota. It is well-established that colonization with helminths stimulates type 2 immune responses characterized by the secretion of IL-4, IL-5, lL-9, and IL-13 from group 2 innate lymphocyte cells (ILC2) and type 2T helper (Th2) lymphocytes.1 These cytokines induce the differentiation of M2 macrophages and the activation of eosinophils, basophils, neutrophils and mast cells that collectively clear worms and heal damaged tissue.1 However, we now understand that non-hematopoietic cells are also central components of anti-helminth immunity that mediate and regulate these immune responses.
Barrier tissues are composed of continuous layers of epithelial cells (ECs) that were initially considered to mainly initiate type 2 cytokine responses by releasing alarmins (IL-25, IL-33, and thymic stromal lymphopoietin; TSLP) after being damaged by tissue-dwelling helminths.2 This is supported by the fact that IL-25 and IL-33 are increased during infection, loss of IL-25 via Tuft cell absence and global genetic deletion of the TSLP receptor impairs type 2 cytokine responses and the ability to control parasite burdens.3,4,5,6,7 However, more recent evidence indicates that tissue damage is not the only way anti-helminth responses are initiated. The epithelium is also composed of specialized chemosensory cells, such as tuft cells, that uniquely contribute to parasite recognition. Underneath this dynamic epithelium, IL-33 enriched stromal cell (SC) niches are key initiators of type 2 inflammation in the skin, lungs, and fat.8,9 In addition to triggering helminth-induced inflammation, the coordinated actions of smooth muscle cells (SMCs) under the mucosa, mucus-producing goblet cells (GCs) and mast cell-induced regulation of chloride channels and breakdown of tight junctions between the epithelium increasing intestinal permeability10 are critical effectors of the ‘weep and sweep’ response required to dislodge and clear certain GI nematodes.11,12 Further, increased epithelial cell proliferation regulated by the intestinal stem cell (ISC) niche displace worms through an “epithelial escalator effect”.13,14 Paneth cells, which increase in number during helminth infection, secrete molecules that direct immune cell-mediated killing of helminths and contribute to the maintenance of the ISC niche.15 Finally, mucosal-associated neurons and enteroendocrine cells (EECs) are recognized to tightly regulate innate and adaptive immune cells to promote host-protective responses upon helminth infection.16 This review will highlight these rapidly growing notions and reaffirm the collective actions of specialized ECs, SCs, muscle layer and neurons as initiators, effectors and regulators required to combat helminth invasion or at least co-exist with minimal damage to the host.
Non-hematopoietic cells are initiators of anti-parasitic responses
Our current understanding of host-protective mechanisms against helminths mainly comes from animal models (Table 1) that either use the rodent helminth species from the same genus as a human helminth (e.g., Strongyloides ratti to model Strongyloides stercoralis) or use a promiscuous helminth that can undergo part or all of its life cycle in a range of mammalian hosts (e.g., Schistosoma mansoni). Some of these helminths naturally enter the body through the skin (N. brasiliensis (Nb), S. ratti (Sr), or S. mansoni (Sm)) while others are ingested by the host (H. polygyrus (Hp), T. muris (Tm), T. spiralis (Ts)). Despite these two entry points, all these species affect the mucosa in the GI tract. It is also important to note that some of these species can be completely expelled by a murine host with an intact immune system (e.g., Nb and Sr) and some are not and can maintain a low level, chronic infection, similar to the clinical state in humans (e.g., Hp or Sm). A long-standing question in the field is how the host’s body detects the initial presence of helminths (Fig. 1). The prevailing theory is that the host reacts to the damage of the epithelium more than direct detection of the helminth products, although some recent work has challenged the latter assumption. In contrast to bacteria, viruses, fungi and some protozoan parasites, we have not been able to identify a receptor that recognizes a helminth pathogen associated molecular pattern. Interestingly, some helminth E/S molecules share structural homology with mammalian danger associated molecular patterns (DAMPs). These include the S. mansoni-derived high mobility group box 1 (HMGB1) homolog and the secreted antioxidant enzyme peroxiredoxin, which can induce type 2 cytokine secretion and accumulation of M2 macrophages.17,18 However, the exact role of many E/S products remains unknown, including the identity of any host homologs or target receptors. While the role of alarmins (IL-25, IL-33, and TSLP) as early initiators of type 2 inflammation is clearly established, their secretion may not occur solely from dying cells in damaged epithelium. Indeed, recent work suggests that chemosensory TCs in the GI epithelium can quickly sense the presence of helminths and initiate type 2 inflammation.

1. Established evidence indicates that helminth-induced type 2 inflammation is initiated by alarmins (IL-25, IL-33, and TSLP) and DAMPs released by dying epithelial cells. 2. However, recent evidence suggests that tuft cells (TC), specialized chemosensory cells in mucosal barriers may potentially respond to helminth-derived signals or metabolites from a helminth-altered microbiome, which causes TC release of IL-25 and cysLT. 3. There is also evidence suggesting that stromal cell (SC) niches can trigger type 2 inflammation by an IL-33-dependent mechanism. However, it is unclear if IL-33 is released by SC, either while still alive (teal) or during cellular death (gray). 4. Cholinergic neurons (Neu) exposed to helminth-derived products in vitro can release the neuropeptide NMU. But, it is unclear if neighboring cells (e.g., TC) can signal to neurons during helminth infection.
Can tuft cells taste helminths?
TCs are a rare population of chemosensory ECs whose development from ISCs is mediated by the transcription factors Pou2f3 and GFI1b. They are characterized by the expression of surface marker doublecortin like kinase 1 (DCLK1) and several chemosensory receptors, including Transient Receptor Potential channel Melastatin 5 (TRPM5).5,6 TCs are one of the main producers of IL-254 and lipid inflammatory mediators like cysteinyl leukotriene (cysLT) and prostaglandin D2 (PGD2).19,20 Further, TCs are in close proximity to endocrine cells, sensory and acetylcholine-secreting neurons (termed “cholinergic”) in several mucosal tissues and TCs can synthesize acetylcholine suggesting that they can signal to these and other cell types that express acetylcholine receptors.21,22,23,24,25,26 Intestinal colonization by several helminth species (Nb, Hp, and Ts) induces the expansion of TCs early in infection,4,5,6 although Tm infection only induces a mild increase in TCs found in the cecum when a repeated low egg dose “trickle” infection is used.27 Critically, Pou2f3-deficient mice have impaired Nb clearance associated with reduced mucus and TC responses.28 Interestingly, while TC expansion induced by Nb and Hp infection requires IL-13 secretion from ILC2s which is also required for worm clearance,4,6 suggesting a feedforward TC-ILC2 interaction that is an active area of research; depletion of ILC2s does not affect worm burden following Tm trickle infection where TC expansion is not robustly induced.27
More recently, TCs have been further divided into two subsets based on single cell RNA sequencing, in both the trachea and the intestine.29,30 In the intestine, type 1 TCs express genes related to neuronal development, while type 2 TCs express immune-related genes and have higher Tslp and Dclk1 expression, but both subsets express Il25 equally.29 In the trachea, type 1 TC transcriptional profile relates to taste transduction, while type 2 TCs express genes related to leukotriene biosynthesis.30 Noteworthy, these two independent studies indicate that type 1 TCs express the transcription factor Pou2f3 and taste-related gene Gng13, while type 2 TCs express Alox5, one of the leukotriene biosynthesis genes as well as the Ptprc gene that codes for the leukocyte surface protein CD45.29,30 In the intestine, type 1 TCs are more abundant than TC progenitors or type 2 TCs at steady state, but upon Hp infection, TC progenitors spike at 3 dpi, and by 10 dpi type 2 TCs are the most abundant.29 It would be interesting to examine the relative contribution of each TC subtype in other helminth infection models including those that affect the respiratory tract.
TCs express several taste chemosensory G protein-coupled receptors (GPCRs) that sense sweet, bitter and umami compounds, which couple to the non-selective cation channel TRPM5 permeable to calcium.6,31 Similar to Pou2f3-deficient mice, genetic deletion of TRPM5 impaired Hp clearance, and IL-25 secretion by TCs and ILC2 expansion during Tm infection.6 Further, stimulation of TRPM5-expressing TCs with Ts-derived E/S products or larval extracts resulted in intracellular calcium influx and subsequent IL-25 release.32 Together these results suggest that TCs may “taste” helminth E/S or cuticle components, which causes calcium-dependent cytokine release via TRPM5. Whether TRPM5 or other GPCRs directly detect these helminth components is unclear. Genetic deletion of chemosensory GPCRs like Succinate receptor 1 (Sucnr1) or Taste receptor type 1 member 3 (Tas1r3) was unable to prevent helminth (Nb, Hp) -induced TC hyperplasia despite these receptors being necessary for TCs to react to metabolites like succinate or infection with protozoans from the Tritrichomonas family.33,34,35 Of note, TC stimulation with Ts-derived molecules was sufficient to induce the expression of several Taste type 2 (Tas2) receptors, suggesting that TCs may respond to diverse signals upon helminth infection.32 Nevertheless, whether chemosensory receptors that bind to helminth E/S products or body wall components actually exists remains highly controversial (Fig. 1).
In addition to IL-25, TCs also promote anti-helminth responses by their rapid and robust secretion of lipid mediators such as cysLT or PGD2.19,20 TCs are a significant source of cysLT during the acute phase of Hp and Nb intestinal colonization.19 Loss of TC-specific cysLT synthesis impaired the activation of ILC2s as early as 16 h post-Hp infection, associated with fewer TCs and GCs and increased worm burdens. Similarly, Nb infection induced the expression of PGD2 and enzymes required for its synthesis in TCs.20 Given the labile nature of these lipid mediators, it is possible that cysLTs and PGD2 represent a rapid mechanism for TCs to stimulate nearby ISCs or cholinergic neurons. Based on the data discussed so far, and the fact that Ach synergizes with IL-13 to drive sinonasal TC differentiation in culture,36 it may be possible that rapid crosstalk among helminth-sensing TCs, ILC2s, and cholinergic neurons synergistically drives intestinal TC hyperplasia. The close proximity of TCs with neurons could have significant beneficial effects to control worm burdens but may be detrimental during co-infection. For example, TCs and IL-4Rα signaling mediated increased mortality, viral dissemination, intestinal permeability and loss of enteric neurons during co-infection with Hp and West Nile virus,37 indicating that fine modulation of TC responses is essential to maintain homeostasis.
Collectively, these studies highlight TCs as helminth-sensors and key initiators of anti-helminth immunity in the intestine. A similar circuit might operate in the airways as Nb or Sr migrate through the respiratory tract where other TC-like populations, such as solitary chemosensory cells, reside. This is suggested by the fact that exposure to the allergen Alternaria alternata causes TCs secretion of cysLTs and IL-25, which synergize to promote ILC2 activation and eosinophilia.38
IL-33 rich stromal cell niches are strategically positioned in barrier sites
Like IL-25, mice deficient in IL-33 have delayed worm expulsion following infection with Nb, Hp, and Sr,39,40,41,42 but not with Ts or Sm,43,44 suggesting that anti-helminth mechanisms can vary between infection with different helminth species. IL-33 is a nuclear protein highly expressed in the ECs of the skin, lungs, and GI tract.45 Further, IL-33 expression has been identified in other non-hematopoietic cells such as the endothelium and neurons in the central nervous system, as well as hematopoietic cells including myeloid cells.40,46,47,48 Hung et al. demonstrated that epithelial-derived IL-33 was required to induce worm clearance whereas deletion of IL-33 from myeloid cells accelerated clearance of Nb adults compared to wildtype counterparts, indicating the cellular sources of IL-33 can have distinct roles for anti-helminth immunity.40 As with other alarmins, IL-33 is not only released from broken nuclei of necrotic cells, but also by intact cells, via cleavage of its nuclear localization signal and receptor-binding domains by several intracellular and secreted proteases, and potentially through perforin-2 pore formation.49 During helminth infections, IL-33 rapidly activates myeloid and lymphoid cells that express the ST2 receptor such as mast cells, basophils, macrophages, ILC2s, T regulatory cells and Th2 cells.45 This IL-33-ST2 signaling induces cytokine release and secretion of tissue remodeling factors such as amphiregulin (Areg) that promote worm expulsion and wound healing.
In addition to these well-defined mechanisms, recent work indicates that IL-33 is highly expressed in SC niches within the skin, lungs and fat. The first study to demonstrate that IL-33 is expressed by fat associate lymphoid cluster (FALC) SCs50 used a rodent helminth Litomosoides sigmodontis (Ls) that upon subcutaneous injection travels through the blood stream and colonizes the pleural cavity around the lungs and heart to mature into adulthood and reproduce. Ls infection caused IL-33 secretion from FALC stroma, which stimulated IL-5 secretion from ILC2s that in turn activates B cells and induces IgM secretion, critical for parasite clearance.50 Boothby, et al. identified a subpopulation of IL-33-enriched fibroblasts in the subcutaneous fascia of dorsal skin that supports the development of Th2 cells, which the authors termed TH2-interacting fascial fibroblasts (TIFFs).8 Interestingly, TIFFs had an enriched expression of IL-4Rα and treatment with IL-4 and IL-13 stimulated TIFF accumulation suggesting a mutualistic crosstalk between TIFFs and Th2 cells. While TIFFs were absent in ear or tail mouse skin, fibroblasts with the TIFF transcriptional signature were present in healthy human skin and elevated in lesioned skin from a patient suffering with eosinophilic fasciitis. The potential contribution of TIFFs to anti-helminth immunity was suggested by experiments showing that subcutaneous infection with Nb resulted in the accumulation of TIFFs and Th2 cells,8 however, further genetic loss- and gain-of-function strategies are required to dissect the contributions of TIFFs to immunity against skin-penetrating helminths.
The location of TIFFs and their enrichment for IL-33 expression resembles that of other stroma located in adventitial cuffs surrounding body cavities, including adventitial stromal cells (ASCs).9 ASCs are in close proximity to ILC2s in the lungs and other non-barrier tissues during homeostasis.9 Critically, ASCs were sufficient to support the growth of ILC2 and Th2 cells in vitro by their secretion of TSLP and IL-33.9 In turn, lymphocyte-derived type 2 cytokines promoted IL-33 expression in ASCs,9 similar to the Th2 – TIFF crosstalk in the skin under homeostatic conditions. Infection with Nb resulted in accumulation of both ILC2s and Th2 cells around the ASC niche in the lungs. Remarkably, depletion of ASCs resulted in reduced expansion of ILC2s and Th2 cells following Nb infection, although the impact on worm clearance or lung repair was not reported.9 However, deletion of IL-33 in the ASC compartment only prevented Th2 cell expansion, but did not affect ILC2 responses during Nb infection, suggesting IL-33-independent mechanisms whereby ASCs support ILC2 expansion.
Similarly, a subpopulation of SCs in visceral white adipose tissue (WAT) is highly enriched for IL-33, which supports type 2 cytokine release from ILC2s and Treg survival.51,52 Notably, WAT SCs rapidly release IL-33 following Nb infection, inducing WAT-resident ILC2s to secrete type 2 cytokines and recruit eosinophils. Collectively, these studies unveil a dynamic role of similar yet tissue-specific SC niches that highly express IL-33, which maintains the growth and survival of ILC2s and Th2 cells. In turn, ILC2s and Th2 cells provide positive feedback signals to promote SC survival. However, the spatiotemporal contributions of these tissue-specific SC populations to immunity against helminths and the tissue remodeling that follows upon worm clearance remains unknown. There is potential for non-hematopoietic cells, particularly SCs supporting the lymph node, to also act as antigen-presenting cells.53,54,55 This has not been fully explored in the context of helminth infection, but could be a possibility in species such as Nb, Sr, or Sm that travel through many different tissues and the circulation. Similarly, cells in the ISC niche have been proposed to present antigen and shape T cell responses in the local microenvironment which may affect the differentiation of specialized epithelial cell subsets such as TCs.56
Despite advances in understanding how epithelial and SCs contribute to anti-helminth responses in the respiratory and GI tract, it remains unclear whether similar mechanisms operate in the skin during invasion by helminths. This may be due to the common use of subcutaneous injection of helminth larvae to initiate infection, which bypasses the natural entry process. The skin is comprised of several layers of keratinocytes at different developmental stages in the epidermis, which forms an impermeable barrier, protecting the dermis and subcutaneous tissue. Keratinocytes are highly enriched for IL-33 and TSLP expression, which can initiate allergic inflammation and itch.57,58 Human hosts are generally unaware of infection by skin-penetrating helminths until the parasites have established themselves in the viscera and produce respiratory or GI symptoms. Further, it is unclear if repeated exposure to skin-invading helminths results in cutaneous tissue remodeling or increased alarmin expression by keratinocytes. Previous exposure to helminths can change the response of local immune cells to trap and prevent helminth migration.59,60 Nb larvae are trapped in the skin of previously exposed mice by the action of basophils and M2 macrophages,60 while Sr larvae are effectively prevented from leaving the infection site by eosinophils and neutrophils during secondary infection.59 How these innate immune responses are established is unclear, as is the potential contribution of keratinocytes, SCs, and skin-innervating neurons to this process. It is also uncertain if helminth species have evolved strategies to bypass established host-protective mechanisms. It is entirely plausible that exposure to helminths could have long term effects on the architecture of the skin that can impact the development of helminth-induced inflammation.
Non-hematopoietic cells mediate worm clearance and tissue repair
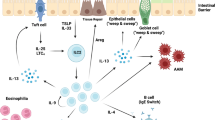
Type 2 cytokines mainly provoke three major host-protective mechanisms: parasite killing, parasite expulsion, and tissue remodeling (Fig. 2). These effects are actively enabled by the coordinated actions of specialized cells in the epithelium that secrete mucus (GCs), anti-microbial peptides (PCs), hormones and neurotransmitters (e.g., EECs and neurons). Under basal conditions, TCs and GCs are sparse, but expand following helminth infection by differentiation from basal cells in the ISC niche. In addition, increased EC proliferation and sloughing through what has been described as an “epithelial escalator effect” has been proposed to facilitate worm clearance and tissue remodeling during some helminth infections. Interestingly, some of the inflammatory mediators induced by helminth infection act on the ISC niche and drive the specification of distinct EC lineages. Moreover, underlying SMCs can mechanically contract and induce expulsion of helminths from the mucosal epithelium. Tissue remodeling growth factors can also act on non-hematopoietic cells to control hemorrhage, deposition of extracellular matrix proteins and reepithelization. This section will highlight the importance of the coordinated events that contribute to host-protective immunity.

A Epithelial (EC), goblet (GC), and Paneth cells (PC) secrete factors that directly affect helminths. B Worm expulsion from the GI tract is accomplished by several coordinated mechanisms: mucus secretion by GC, smooth muscle cell (SMC) contraction controlled by cholinergic neurons (Neu) and serotonin (5-HT) from enterochromaffin cells (ECCs), among other cell types, and growth of the villi to push worms off the “epithelial escalator”. The intestinal stem cell niche (ISC) at the base of the villi controls cellular expansion and differentiation of specialized secretory cell types (e.g., tuft cells, TC, and PC) needed for the epithelial escalator and mucus secretion effector functions. C GCs contribute to tissue repair by secreting TFFs that can promote EC proliferation. ECs and macrophages (Mac) synergistically promote collagen deposition by myofibroblasts to repair damaged tissues.
Goblet cells and mucus
Perhaps the most conserved consequence of GI helminth infection is an increased number of GCs and secretion of mucus (part of the “weep”) in epithelium of the lungs and GI tract, which are type 2 cytokine-dependent. Mucus is a carbohydrate-rich layer composed of membrane-bound glycocalyx, which lines the apical surface of ECs and consists of membrane-bound mucins, and an overlying mucus gel, consisting of secreted mucins.11 Mucins are high molecular-weight O-linked glycoproteins and the main constituents of mucus.11 Of the 20 mucin proteins, Muc2 is the main component of intestinal mucus that has been extensively studied in the context of helminth infection. Increased intestinal expression of Muc2 is observed following infection with Tm, Ts, Hp and Nb.61,62,63,64 However, only in the context of Tm infection is Muc2 required for worm clearance during acute infection,63 indicating that other mucins can compensate and ensure timely expulsion. Muc5ac, which is not expressed in the GI tract during homeostasis, becomes upregulated and is necessary to clear Nb, Tm, and Ts infections.27,62 Muc5ac is upregulated in the colon when mice become resistant to Tm “trickle” infection, which consists of repeated exposures to low-larval numbers.27,62 Consistent with a protective role of Muc5ac, serine proteases secreted by Tm degraded Muc2, but not Muc5ac,65 indicating that some mucins may have distinct anti-helminthic properties.
Altered glycosylation patterns of mucins also affects their anti-helminth properties. A switch from sulphomucins to syalomucins establishes chronic Tm infection.66 Mucin sulphation induced by IL-13 conferred resistance to mucin degradation by Tm E/S products, demonstrating that type 2 inflammation can modify mucus properties to confer helminth resistance.66 Muc5ac is elevated in the lungs after Nb larvae have exited the tissue,67 suggesting that mucin expression can be sustained by residual inflammatory mediators and/or parasite components left behind. Whether this elevated mucin response also actively participates in the wound healing or trained immunity following initial parasite clearance is unknown. Interestingly, strictly enteric intestinal nematodes (i.e., Ts and Hp) can promote airway GC responses and increase the lung trapping of larvae upon a subsequent Nb infection.68 The distal activation of airway GCs was potentially mediated by migratory ILC2s that recirculated from the intestines to the lungs.68 However, the chemotactic signals that directed ILC2s into the lungs remain to be determined.
The protective role of mucus is also associated with factors other than mucins. For example, resistin-like molecule β (RELM-β) is also secreted by GCs during helminth infection and is required for clearance of Nb or Hp.69 Moreover, trefoil factors (TFF1, TFF2, and TFF3) are GC-derived tissue-reparative mediators that promote anti-helminth immunity. Genetic deletion of either TFF2 or TFF3 delays the clearance of Nb from the intestine,70,71 although the two TFFs appear to accomplish this by distinct pathways. TFF2 deficiency was associated with reduced secretion of type 2 cytokines and RELM-β expression by GCs, which may be because TFF2 promotes IL-33 release from ECs.71 In contrast, delayed worm expulsion in TFF3 deficient mice is not associated with reduced type 2 cytokine production. Instead TFF3 binds to LINGO2 and releases LINGO2 suppression of EGFR signaling to maintain intestinal integrity and promote anti-helminth immunity.70 Taken together these exciting findings illustrate how signals from GCs are critical for helminth expulsion, immune regulation, and wound repair. However, there is still more to uncover regarding the regulation of GCs and mucus production. Canonically, IL-13 and IL-4Rα signaling induces increased mucus secretion in multiple models of helminth infection.72 However, IL-22 deficient mice had delayed Tm expulsion and blunted GC responses despite having an intact type 2 cytokine response.73 Future studies should also address whether different signals promote GC hyperplasia while others increase the rate of mucus secretion by GCs and if other non-hematopoietic cells adjacent to GCs regulate this process.
Although it was first hypothesized that mucus exerts its anti-helminth properties simply by preventing attachment of helminths to the epithelium, it appears that mucus may also become directly toxic to some helminth species. Host intestinal mucus was found in the intestine of Nb adults and was associated with damage to the worm gut cells.74 Moreover, mucus from previously exposed sheep had paralyzing activity on sheep-infecting helminths in vitro,75 suggesting that specific components of the mucus may induce direct damage to helminths. Particularly, RELM-β binds to structures located on the integument of nematodes and inhibits their chemotaxis.76 Critically, RELM-β pre-treatment was sufficient to impede the ability of Hp adult worms to sense and ingest host tissues which directly diminished their survival and fecundity,69 demonstrating the potential of mucus-associated proteins to directly interfere with worm survival.
Paneth cells, do they secrete anti-helminthic peptides?
Mucus also contains anti-microbial peptides (AMPs), such as phospholipase A2 (PLA2) secreted by PCs, which were not thought to affect helminths until recently. Like TCs and GCs, PCs are specialized ECs that differentiate from ISCs and may actually regulate proliferation within the ISC niche.15 PC hyperplasia was initially observed following Nb, Hp, Ts and Sm infection at times post-infection that correlate with induction of type 2 cytokine responses.77 Related to their support of the ISC niche, Ts-induced PC expansion was associated with increased proliferative crypt ECs.78 Interestingly, no alterations in the number of PCs were reported by single cell analysis of intestinal ECs from Hp infected mice, despite expansion of TC and GC numbers.29 However, the lack of genetic strategies that specifically target PCs has hindered scientists from determining if they have non-redundant contributions to anti-helminth immunity.
A recent study by Entwistle, et al. has identified epithelial-derived PLA2g1b, an enzyme that regulates lipid metabolism by hydrolysis of phospholipids, as an endogenous anthelmintic.79 PLA2g1b and other lipid-metabolism genes were enriched in duodenal tissue from mice resistant to secondary Hp infection. Moreover, PLA2g1b-deficient mice failed to acquire resistance to secondary Hp infection despite developing intact type 2 and mucus responses. Interestingly, helminth-induction of Pla2g1b expression required intact innate and adaptive lymphocyte responses but type 2 cytokine treatment in vitro downregulated Pla2g1b in intestinal organoids, reflecting a complex transcriptional regulation of this pathway. These findings suggest that some aspect of the helminth’s presence or other cell types not present in vitro are required for upregulation of Pla2g1b, while type 2 cytokines could negatively regulate it to check lipid metabolism that may be detrimental to host healing. Remarkably, treatment of Hp iL3 with PLA2g1B reduced their capacity to reach adulthood, which correlated with reduced phospholipid abundance and membrane integrity, suggesting that PLA2g1b causes direct damage to helminths. These exciting studies begin to answer the long-standing question of whether helminths are directly damaged and killed by the host before being expelled and if non-hematopoietic cells contribute to this process. Helminths are outstandingly known to secrete several E/S products, thus it will not be surprising that they have evolved different pathways to avoid the actions of such mediators. This is perhaps why this question has been so difficult to answer. Potentially related to this modulation, Hp-induced type 2 inflammation induces the expression of the AMP small-proline rich protein 2A (SPRR2A), which exhibit gram-positive bactericidal properties and is required to protect against bacterial invasion of intestinal tissues during Hp infection.80 This suggests that helminths may modulate the secretion of AMPs to shape the composition of the microbiota perhaps to make the intestinal microenvironment more suitable for their survival.
Modulation of the intestinal stem cell niche during helminth infection
The cells of epithelium, both non-specialized and specialized (e.g., TCs, GCs, PCs and others) are constantly renewed by their differentiation from ISCs located in the base of intestinal crypts. ISCs are characterized by the expression of Leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5). During helminth infection, ISC activity is modulated by type 2 cytokines and helminth-derived factors. The type 2 cytokines IL-4 and IL-13 induce differentiation of GCs and TCs from ISCs.4,5,6,20,56,81 Further, constitutive activation of STAT6, which acts downstream of IL-4/IL-13 signaling increases the numbers of specialized secretory cells and reduces adult worm burden following Nb or Hp infection.82 Conversely, IL-10 or stimulation with Tregs, which are also generated during helminth infections, increased the proportion of ISCs and their expression of a ‘stem-like’ gene signature.56 In addition to cytokines, MHC-II-expressing ISCs interact with T cells to promote TC differentiation, which is required to control Hp worm burden, at the expense of depleting the ISC pool.56 Tregs are required to maintain the renewal of Lgr5+ ISCs, so perhaps their increase during helminth infection helps to re-establish the ISC pool in the face of TC and GC hyperplasia.56 ILC2-derived IL-13 also promotes the renewal of ISCs, which required the expression of the circular RNA from the pan-gene transcript 3 (circPan3) to stabilize and increase protein expression of IL-13Rα1 in ISCs at baseline,83 suggesting different non-redundant roles of innate and adaptive lymphocytes located at the vicinity of the ISC niche. These findings demonstrate the profound influence of immune signals on the fate of differentiated IECs and the maintenance of the ISC pool.
In addition to cytokine regulation, recent studies revealed that other inflammatory mediators such as PGD2 and helminth-derived E/S products also regulate ISC differentiation. Type 2 cytokine stimulation induces the expression of the PGD2 receptor CRTH2 in ISCs, GC and TCs.20 PGD2 signaling via CRTH2 in ISCs was necessary to limit differentiation of ISCs into GCs following type 2 cytokine stimulation. Type 2 cytokines induce PGD2 secretion from TCs, which can neighbor ISCs in the epithelium. This group also found that CRTH2 expression in non-hematopoietic cells was required to limit GC and TC responses during Nb infection,20 revealing a novel mechanism of for how PGD2 signals can regulate the magnitude of helminth-induced type 2 cytokine inflammation. Similar to the regulatory role of PGD2, exposure of intestinal organoids to Hp E/S products or iL3 suppressed the differentiation of TCs and GCs.84 Consistent with these findings in vitro, mice pre-infected with Hp presented with reduced TC and GC responses induced by a subsequent Nb infection or succinate treatment. Importantly, exposure of small intestinal organoids to Hp E/S molecules resulted in an altered, immature spheroid morphology that correlated with downregulation of the master regulator of secretory-cell differentiation Atohl and upregulation of Hes1 that preserves stem cell status.84 Related to these studies, crypts with granulomas from Hp disruption have a fetal-like transcriptional program characterized by reduced expression of ISC-signature genes including Lgr5 and olfactomedin 4 (Olfm4) and increased expression of interferon (IFN)-induced genes including Ly6a that encodes stem cell antigen-1 (Sca-1).14 Noteworthy, the elevated expression of Sca-1 in intestinal crypts was abrogated in Hp-infected IFNγ-deficient mice, suggesting a Type 1 cytokine-dependent mechanism. Due to their fetal-like reversion, Hp-induced Sca-1-expressing crypts failed to form intestinal organoids and instead gave rise to spheroids lacking expression of differentiated epithelium markers.14 Collectively, these studies may reflect an evolutionary strategy for helminths to counteract Type 2 immune signals and re-shape the ISC microanatomic niche to prevent hyperplasia of specialized anti-helminthic ECs.
Epithelial cells contribute to worm clearance and wound healing
Infection with Tm causes intestinal epithelial cell (IEC) proliferation to move an embedded larva up and off the epithelium of the colon before it reaches adulthood, an effect known as the ‘epithelial escalator’.13,20 This would be accomplished by increased proliferation in the ISC niche and differentiation into IECs to grow the epithelial layer. However, this can occur at the expense of TC and GC hyperplasia, which are critical for helminth clearance. Despite the fact that IL-13 and constitutive activity of STAT6 promote helminth-induced IEC proliferation in vivo,13,82 in vitro treatment of intestinal organoids with IL-4 and IL-13 does not increase cell proliferation.20,56 This suggests additional mechanisms contribute to this process in vivo. One of these mechanisms may be CRTH2 signaling; since ablation of CRTH2 impaired the ‘epithelial escalator’ while increasing the number of GCs.20 These studies reflect a delicate balance between IEC proliferation and induction of GC hyperplasia, both of which are required for proper worm clearance. For example, lysine-specific histone demethylase 1 A (LSD1), which seems to be required for PC differentiation,85 limits IEC proliferation at baseline,85 but renders mice more susceptible to Tm infection because of impaired mucus secretion.86 Likely, this may be due to a reduced potential for GC differentiation. In apparent contrast to these findings, ablation of the lysine methyltransferase SETD7, which regulates Hippo and Wnt pathways in IECs, increased IEC turnover and accelerated clearance of Tm infection with no apparent alterations of type 2 cytokine or mucus responses,87 highlighting the complex signaling pathways the regulate IEC proliferation and differentiation.
As mentioned above, type 2 cytokine responses are important to promote wound healing even after helminths have been cleared from mucosal sites, but the mediators between IL-4/IL-13 signaling and the healing outcome are still being uncovered. This has been most studied in helminth models where larvae cause acute lung damage and hemorrhage en route to the GI tract, especially Nb infection (see Table 1, skin-penetrating helminths: Sr/Ss, Nb/Ad or Na, Sm). Relm-α expression in ECs is necessary to control lung damage and hemorrhage following Nb infection, and while it requires IL-13 for effective healing,88 it can also be induced by chitinase-3 like 3 protein, also known as eosinophil chemotactic factor-lymphocyte (Ym1), that is mostly secreted by myeloid cells.89 IL-13 is also required for epithelial cell secretion of surfactant protein D (SP-D),88 a molecule that can bind L4 Nb larvae in the lungs, and promote killing by M2 macrophages.90 SP-D also promotes Relm-α expression, suggesting that it can also promote wound healing. Relm-α is required for lysyl hydroxylase 2b-dependent assembly of collagen fibrils to promote rapid wound healing in the lung.91 Further, the DAMP ATP induces Areg expression in macrophages, which then acts on pericytes to evoke their activation of TGFβ and subsequent differentiation into collagen-producing myofibroblasts that promote revascularization in the lung after Nb infection.92 Altogether, these studies demonstrate the active involvement of the epithelium in the repair of the lungs affected by migratory larvae.
As we have mentioned, the nature of healing and remodeling in the skin following penetration by helminths is largely understudied, especially with respect to non-hematopoeitic cells. Sm cercariae penetration does cause keratinocyte proliferation, and a subset of keratinocyte progenitors to expand and accumulate in hair follicles.93 In addition, keratinocytes exhibit increased expression of stress-associated genes such as Il1a, Il1b, Il33, and Tslp within hours of exposure to Sm cercariae, however, whether exposure to Sm or other skin-penetrating helminths alters epidermal architecture or transcriptional profile to prevent future entry or heal damage requires further investigation.
Smooth muscle cells contribute to multiple anti-helminth mechanisms
Hypercontraction of SMCs underlying intestinal tissue promotes the ‘sweep’ or physical removal of worms from these sites. Muscle cells express the IL-25 receptor and contract when treated with IL-25, which drives Nb clearance.94 Contraction can also be induced by IL-4 and IL-13, which induce STAT6 intracellular signaling in Nb and Hp infections.12 IL-4Rα signaling in SMCs is required to produce adequate contractile expulsion of Sm eggs, or mortality results from GI blockage.95 Helminth-induced (Hp or Nb) SMC contraction requires neurotransmitters and neuropeptides such as acetylcholine and substance P (SP),12 suggesting the involvement of cholinergic and non-cholinergic enteric neurons. It has been postulated that acetylcholinesterase secreted by helminths (Sr, Nb) may serve to degrade and limit host cholinergic signaling and anti-helminth action in the GI tract.96,97,98,99 In addition, helminth (Nb, Hp) infection causes IL-13-dependent upregulation of the serotonin receptor 5-HT2A in SMCs, which increases serotonin-evoked contraction.100 Surprisingly, IL-17 to IL-17ra signaling in SMCs promoted contraction and Ts expulsion in a type 2 cytokine-independent manner,101 revealing that multiple cytokine pathways can stimulate SMC hypercontractility.
In addition to their contraction, IL-4Rα signaling in SMCs has also been shown to promote type 2 cytokine secretion and GC differentiation in the lungs and SI,102,103 suggesting a more complex contribution of SMCs than originally thought. The altered GC differentiation may be associated with a novel regulatory pathway by which SMC support the regeneration of ISC niche following tissue damage by regulating extracellular matrix components using muscle-derived matrix metalloproteinases.104 In summary, these studies highlight additional pleiotropic functions that require further investigation.
Helminth clearance is regulated by neuroendocrine cells, neurons, and their associated products
Sensory neurons detect damage in the skin or mucosal tissues and may release a variety of neuropeptides and neurotransmitters105 that can communicate with immune cells.106 This release by sensory, autonomic, and enteric neurons can contribute to wound healing by activating mast cells, recruiting immune mediators, fibroblasts, and promoting angiogenesis in large scale wounds.107,108,109 Loss of appropriate innervation of the skin, as in diabetic neuropathy, can impair wound healing and prolong inflammation.108 Hoeffel et al. have recently shown that UV skin damage evokes secretion of the neuropeptide TAFA4 from sensory neurons expressing the Gαi-interacting protein (GINIP), which promotes tissue regeneration and reduces skin inflammation by promoting IL-10 secretion in dermal macrophages, as well as their expression of tissue-reparative transcripts.110 While these exciting studies demonstrate that sensory neurons can contribute to the response and healing of damage in the skin, the contribution of these pathways to parasite detection, tissue healing and/or immune modulation during skin helminth infections remains unclear. There is evidence that neuropeptides and neurotransmitters promote clearance of helminths, and repair in the lung and GI tract. While neurons are thought of as the major source of neurotransmitters or neuropeptides, other specialized neuroendocrine or EECs, and immune cells found in mucosal tissues can produce them as well. Often, their cognate receptors can be found on all these cell types, which may be in close proximity to each other in the tissue, setting up regulatory feedback loops that could enhance or dampen downstream inflammatory or contractile responses. This section will discuss the current literature supporting a role for neuroendocrine cells, neurons, and neuron-associated molecules in regulating anti-helminth responses in the lungs and GI tract, and their potential involvement at the site of skin penetration (Fig. 3).

a Most work in the GI tract has examined multiple cellular sources of neurotransmitters serotonin (5-HT) and acetylcholine (Ach), including ILC2s, tuft cells, and ECCs. IL-33-evokes 5-HT release in ILC2 and ECCs. Serotonergic neurons (purple) activate cholinergic neurons (orange), which regulates the ISC. Ach can act on and be released by ILC2s, which also receive NMU signals from peptidergic neurons (red). Both inputs evoke the release of IL-13 from ILC2s. It is possible that some helminths may secrete AChEs to deplete Ach gradients in tissues. b Non-peptidergic neurons of the Mrgpr class (E, F) are reduced in density around encysted Sm eggs, but their role is unclear. c In the airway, ILC2s are positively regulated by NMU, while NMB and CGRP tend to inhibit their proliferation and type 2 cytokine secretion (although CGRP does not inhibit IL-5 secretion). Basophils induce the upregulation of the NMBR on ILC2s. The exact cellular source of CGRP and NMB are not clear. d Very little is known regarding multicellular crosstalk during helminth skin penetration, or if this is modulated by helminth E/S products.
EECs, satiety, and GI-motility neuropeptides: CCK, Leptin, gastrin, and SST
The heterogeneous EEC population plays an important role in signaling nutrient content and satiety signals to the brain, as well as regulating gut motility, insulin, and local pH.111 The former is controlled by neuropeptides like cholecystokinin (CCK), and the latter by somatostatin (SST) and gastrin, among others.111 Not surprisingly, helminth infections can alter these cells and the secretion of certain neuropeptides, although the effects are not generalizable across all helminths.111 Nb infection reduces serum CCK levels in rats,112 while Ts is associated with hyperplasia of CCK + cells and hypersecretion of CCK that corresponds with hypophagia and weight loss in mice.113,114 Fat tissue secretes the satiety hormone leptin, thus weight loss during Ts infection likely corresponds to reduced fat mass and measured reduction in serum leptin levels, while exogenous leptin delays expulsion of Ts.114 Similarly, mice on a protein deficient diet have increased serum leptin likely due to increased consumption and fat accumulation, which delays clearance of Hb infection.115 Ts is also associated with increased gastrin levels in serum,116 which could correspond to enhanced acidity within the GI tract.
In addition to affecting the feeding behavior of the host, and driving helminth expulsion via changes in gut motility or pH, these neuropeptides can regulate immune responses.111 Of specific relevance to helminth infections, CCK promotes the development of Th2 or Tregs in vitro117 and Th2 cells promote CCK + cell hyperplasia and CCK-associated hypophagia in Ts infection,114 indicating a positive feedback loop between EECs and Th2 cells. Conversely, leptin promotes type 1 T cell responses111 and may reduce secretion of type 2 cytokines.114,116 CCK also drives acetylcholine production from B cells to recruit neutrophils.118 Although neutrophils are important mediators of helminth killing, it is unclear if this mechanism may contribute to neutrophil recruitment in the context of helminth infection.
Sm is associated with decreased SST + EECs in the intestine, and upregulation of the SST receptor SSTR2A on cholinergic neurons, corresponding to a reduced inhibitory effect of SST on neurogenic contractions.119 Further, in humans with Sm infection, low serum SST levels were associated with greater pathology, including liver fibrosis, while higher SST levels were found in infected, but asymptomatic individuals.120 Indeed, the SST analog octreotide suppressed experimentally induced liver fibrosis likely via inhibition of the Wnt/β-catenin pathway in hepatic stellate cells.121 SST is inhibitory to T cell proliferation,122 dendritic cell activation123 and stabilizes mast cells during Ts infection,124 preventing their degranulation, although the direct role of SST on immune function in Sm infection has not been fully explored. Although closely associated with the digestive system, these neuropeptides can potentially affect immune regulation and tissue repair in other parts of the body affected by helminth infection, such as the airway mucosa. Indeed, there are pulmonary neuroendocrine cells known to regulate goblet cell mucous secretions in the context of type 2 allergic inflammation,125,126 but their role during helminth infection is unexplored.
Canonical neurotransmitters: serotonin and acetylcholine
IL-33-TRPA1 evoked serotonin secretion and multicellular crosstalk
Smooth muscle contraction is controlled by neurotransmission, under both normal conditions and during helminth expulsion. IL-33-dependent serotonin production during helminth infection can emanate from multiple cell types.127,128 EECs contain a subtype called enterochromaffin cells (ECCs)129 that are thought to produce 90% of peripheral serotonin,130 but factors that regulate their production of serotonin are unclear. During Tm infection, IL-33 binding to its receptor ST2 expressed on ECCs induces phospholipase C γ (PLCγ) to release intracellular calcium stores in a My88-independent manner, which in turn causes activation of the transient receptor potential channel ankyrin 1 (TRPA1),127 a non-selective cation channel found on sensory neurons, epithelial, and immune cells in barrier tissues that responds to noxious chemical mediators and painfully cold stimuli.131 TRPA1 activation allows the intracellular influx of calcium required to release serotonin from ECCs that enhances firing of enteric neurons, increases peristalsis, and enhances Tm clearance.127 The authors posit that this IL-33-mediated activation of TRPA1 could also occur in enteric neurons.127 TRPA1 activation may have additional positive actions independent of serotonin release during helminth infection as suggested by a study showing that treating Hp infected mice with cinnamaldehyde, a TRPA1 agonist, alleviated weight loss and increased antioxidant responses in ECs in the context of Hp infection.132 ILC2s also express tryptophan hydroxylase 1 (Tph1), the rate limiting enzyme for serotonin biosynthesis in an IL-33 dependent manner during Nb infection.128 Deleting Tph1 from ILC2s abrogated their ability to adopt an inflammatory phenotype and migrate to the MLN, due to downregulation of activating cell receptors such as ICOS.128 The potential for multiple cell types, including migratory immune cells to secrete and potentially respond to serotonin gradients suggests that local gradients may provide a signal to orchestrate activation and migration to sites of infection.
Acetylcholine modulates “Weep” and type 2 inflammation
In addition to its anti-helminth peristaltic function, acetylcholine can control the permeability of the epithelial barrier (contributing to the ‘weep and sweep’) and modulate local inflammatory processes to enhance helminth clearance and tissue healing. Expression of the muscarinic acetylcholine receptor 3 (m3ACR) on cultured ECs, in the absence of muscle, is associated with improved barrier function and knock out of the channel delays clearance of Nb.133 Similar to the role of Thp1 in ILC2s,128 ILC2s also upregulate the enzyme required to make acetylcholine, choline acetyltransferase (ChAT), in response to Nb or exogenous treatment with alarmins (IL-25, IL-33, TSLP).134 ILC2s are found in close proximity to cholinergic neurons in the gut and also express multiple mACRs and several alpha and beta nicotinic ACR subunits,134 suggesting that both ILC2-derived and neuronal acetylcholine initiate a positive feedback loop in ILC2s to maintain type 2 responses. Acetylcholine induces ILC2 secretion of IL-13 and reduces worm burden during Nb infection by enhancing type 2 cytokine and cellular cascades.134 Conversely, removing ChAT from IL-7R + ILC2s results in impaired helminth-induced inflammation which impaired worm clearance.134 The latter suggests that acetylcholine from ILC2s is critical for enhanced peristalsis to expel worms from the gut. It is worth noting that this work focused mostly on ILC2s from the intestinal lamina propria, but also found upregulation of ChAT in lung ILC2s of infected mice. In the context of allergy, secretion of type 2 cytokines and ILC2 expansion was abrogated by acetylcholine and agonists of the α7nACR found on ILC2s.135 This suggests that inhibitory feedback could be more prominent in the lung, although such a mechanism may be unique to allergens and perhaps not engaged by helminths. Understanding regional tissue regulation of acetylcholine signaling and its effects on type 2 inflammation will be important in advancing our understanding of host-protective immunity.
Neuronal contribution to growth and specialization of the GI epithelial mucosa
Cholinergic signaling also has ties to stem cell function,136 which could have implications for tissue repair in the context of GI nematodes. Neuronal serotonin, not mucosal serotonin, activates cholinergic neurons innervating the GI mucosa to control cell proliferation.137 This was under steady-state conditions, but it may be informative to determine if this dynamic is altered in the context of helminth infections when other cell types may actively secrete serotonin in proximity to cholinergic neurons bearing serotonin receptors. TCs are an additional non-neuronal cell type with ChAT expression, capable of secreting acetylcholine but whether this mechanism regulates ISC fate in the context of anti-helminth immunity is unclear.26 Pharmacological antagonism of muscarinic receptors reduces the number of ISCs, potentially by acting on enteric ECs that express m3ACR.138 Prox1-positive endocrine cells respond to this by inducing TC expansion but with an enteroendocrine gene signature. These findings could have important implications considering that genetic deficiency of m3ACR impairs GC expansion during Nb infection,133 requiring a more granular dissection of how serotonergic and cholinergic signaling regulate homeostasis of the GI epithelial barrier. It is possible that during helminth infection the combined effects of neuron- and non-neuron-derived neurotransmitters and other neuropeptides contribute to enhanced epithelial proliferation and/or specialization into secretory cells that promote wound healing and helminth clearance but this has yet to be investigated in greater detail. It has been postulated that acetylcholinesterase secreted by helminths (Sr, Nb) may serve to degrade and limit host cholinergic signaling and anti-helminth actions in the GI tract,96,97,98,99 suggesting that helminths countermeasure mammalian neurotransmission.
Canonical neuropeptides: NMU, NMB, and CGRP
Neuromedin U from cholinergic neurons acts on ILC2s to promote type 2 inflammation
As is the case for neurotransmitters, neuropeptides can also come from and act on both neuronal and non-neuronal cells to modulate neural activity and inflammatory processes.139 There are many neuropeptides that can be released by sensory afferents associated with the somatosensory dorsal root ganglia (DRG) or nodose ganglia (NG) to influence neighboring cells, but only some have been specifically investigated in the context of helminth infection. So far, neuromedin U (NMU), neuromedin B (NMB), and calcitonin gene-related peptide (CGRP) are suggested to both promote (NMU: Nb, Tm, Hp)140,141 and limit (NMB, CGRP: Nb)142,143 type-2 inflammation associated with helminth infection. Other neuropeptides such as SP and neuropeptide Y contribute to allergen associated type 2 inflammation in skin and lung respectively,144,145 but whether they play a role during any stage of helminth infection is currently unknown.
Recently, two groups independently demonstrated that cholinergic, Ret+ enteric neurons are proximal to ILC2s, and that their release of NMU enhances proliferation and cytokine release by ILC2s through NMU Receptor 1 (NMUR1).140,141 In the context of Nb infection or E/S product exposure, NMU expression is increased in target organs and NMUR1 expression is exclusively increased in ILC2s in the lungs and small intestines.140,141 NMU upregulation also occurred in Tm and Hp infection.141 Nb-derived E/S products evoked NMU release from enteric neuronal organoids in a Myd88-dependent manner, suggesting that enteric neurons can directly react to the presence of helminths. This NMU promotion of ILC2 function is important for controlling Nb infection, at least at early stages;140,141 late-stage expulsion of worms was intact,140 perhaps due to intact acetylcholine signaling processes mentioned previously. This work mainly focused on NMU released by cholinergic neurons in the lung and GI tract, which would be part of NG and enteric nervous system. Nevertheless, NMU and NMUR1 are also expressed in DRG sensory neurons146 and in the trigeminal system,147 which innervate the skin, musculature, vasculature, and connective tissues throughout the body. NMU signaling via NMUR1 can reduce the activation threshold of nociceptive neurons and increased pain sensitivity in experimental animals,146,148 but whether these signals operate during helminth skin penetration is unknown.
NMB and CGRP limit type 2 inflammation and helminth clearance
While NMU promotes type 2 inflammation and anti-helminth actions in the lung and GI tract, NMB and CGRP appear to limit this inflammation. In the context of Nb infection, basophils contribute to limit type 2 inflammation in the lung, preventing excess damage and dysfunction.142 The presence of basophils causes upregulation of the NMB receptor on ILC2 cells.142 Exogenous NMB inhibits type 2 immune responses, likely by downregulation of purinergic 2x receptor 7 (P2rx7), which is also associated with increased worm burden.142 The cellular source of NMB is not clear, nor has evidence of endogenous NMB release during helminth infection been demonstrated. Of note, these effects are seen primarily in the lung, so whether NMB signaling acts similarly in the GI tract to limit inflammatory tissue damage remains to be determined. NMB in the CNS controls neural circuits related to sighing, particularly in response to hypoxia, so it stands to reason that there may be a peripheral neuronal source in the airway mucosa. NMB is found in TRPV1/CGRP + trigeminal neurons149 and NMB contributes to histaminergic itch,150 supporting evidence that skin somatosensory neurons may release NMB. Thus, there is a possibility NMB could play some role in regulating immune responses during helminth skin penetration, but requires specific examination.
Like NMB, CGRP also seems to be largely inhibitory toward type 2 inflammation, by direct opposition of NMU’s actions on ILC2s during Nb infection.143 As with serotonin and acetylcholine, ILC2s can express CGRP and all the components of its receptor, indicating potential for self or neuronally regulated feedback.143 CGRP causes cAMP-dependent abrogation of ILC2 proliferation and IL-13 secretion, while enhancing IL-5 expression.143 This indicates that CGRP can regulate the relative levels of IL-5 vs other type 2 cytokines (IL4, 13) expressed by ILC2s, which may have implications for other diseases where specific type 2 cytokines play a critical role.143 How the balance between the actions of NMU vs NMB or CGRP is controlled during infection would be an important area of study to optimize healing and anti-helminth therapies. CGRP and SP also signal a number of functions critical for wound healing107,108 that may be important for host survival during helminth infection. CGRP is also found in skin sensory neurons, and its release can influence inflammatory responses to fungal and bacterial skin infections,151,152 but the role of CGRP in the percutaneous stage of helminth infection is currently unknown. Interestingly, CGRP is dispensable for papain-associated allergic responses, mediated by SP-evoked dendritic cell migration to the draining lymph node.145 Because allergy and helminth immunity both engage type 2 immune pathways, this might indicate that CGRP may not be a significant contributor, positively or negatively, but further study on this matter is required.
Is there a role for non-peptidergic neurons in helminth infection?
There are sensory afferents traditionally classified as “non-peptidergic” found in the skin,153 NG, and the enteric nervous system,154 which are only now being explored in terms of their ability to modulate local immune responses. A large subset of these “non-peptidergic” afferents express members of the Mas-related G protein coupled receptor (Mrgpr) family, which transmit itch sensation, contribute to both skin mechanical pain and visceral pain, and modulation of gut motility.153,155 Itch is the only documented sensation associated with helminth or hookworm skin penetration indicated by human subjects.156,157,158 Thus far, only MrgprE and MrgprF expression in enteric neurons has been examined during helminth infection.159 In schistosomiasis, MrpgrE and MrgprF+ cholinergic secretomotor neurons in the GI submucosa are reduced, especially in proximity to encysted Sm eggs.159 What this means functionally remains to be seen, but other members (MrgprA3, C11, D) have been implicated in enhancing visceral hypersensitivity (MrgprA3, C11, D)160,161 and slowing gastric motility (MrgprD).162 Notably MrgprD+ neurons in the skin stabilize mast cells via glutamate release, which alleviates dermatitis.163 Further work may uncover a novel role for Mrgpr+ neurons in helminth infection.
Conclusion
Helminths colonize multiple barrier tissues where hematopoietic and non-hematopoietic cells are in constant communication. Although sometimes underappreciated, these non-hematopoietic cells are sentinels that promote the recognition and elimination of helminth parasites and initiate tissue repair to reestablish proper organ function. These active processes define non-hematopoietic cells as ‘non-traditional’ immune cells that contribute to host-protective responses against helminths. It is clear that TCs and ISC niches potentially sense helminth infections and initiate type 2 cytokine responses. These signals also operate in multiple effector cells that execute a variety of anti-helminth mechanisms including toxic mucus secretion, cellular outgrowth to push larvae from the epithelium, peristaltic movements to propel them out of the body, hemorrhage control, and wound healing. These processes are also influenced by regulators, including EECs and peripheral neurons that act as rheostats to fine tune the magnitude and timing of anti-helminth inflammation as well as the ISC niche in barrier tissues. It is therefore fundamental that we consider the participation of these barrier-resident cells in sensing and initiating anti-helminth responses and controlling inflammation during infection. Most of our understanding regarding anti-helminth immunity is based on studies in the GI tract and to some degree in the lower airways. While advances have been made in terms of understanding how the ISC niche is regulated during helminth infection, we still do not know if these mechanisms also apply to the stem cell niche in the airway mucosa or other sites. Many helminth species enter the host by penetrating the skin, invading lymphatic vessels, skeletal muscles and even the brain yet our understanding of how anti-helminth immunity operates at these sites is remarkably limited. Such knowledge remains critical for developing effective therapies to clear helminth infections and heal tissue damage in humans and animals. Further, these models may provide insight to allergic inflammation, given that they engage common mechanisms. While animal studies have advanced our understanding of helminth infection and host responses and led to vaccine candidates,164 mice and humans do have some differences immune and sensory neuron function,165,166 which will warrant future studies in culture systems derived from human tissues. As such, a deep appreciation of the communication pathways between ‘traditional’ and ‘non-traditional’ immune cells will provide critical insight(s) that could be translated into novel prevention and treatment strategies for helminth infections. For example, a better understanding of skin penetration mechanisms, could indicate preventative therapeutic strategies for vulnerable populations in endemic areas or occupations with soil-transmitted helminth exposure (e.g., Ss), which do not currently exist.
References
Inclan-Rico, J. M. & Siracusa, M. C. First responders: innate immunity to helminths. Trends Parasitol. 34, 861–880 (2018).
Coakley, G. & Harris, N. L. The Intestinal Epithelium at the Forefront of Host & Helminth Interactions. Trends Parasitol. 36, 761–772 (2020).
Humphreys, N. E., Xu, D., Hepworth, M. R., Liew, F. Y. & Grencis, R. K. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J. Immunol. (Baltim., Md: 1950) 180, 2443–2449 (2008).
von Moltke, J., Ji, M., Liang, H. E. & Locksley, R. M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225 (2016).
Gerbe, F. et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226 (2016).
Howitt, M. R. et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333 (2016).
Taylor, B. C. et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J. Exp. Med. 206, 655–667 (2009).
Boothby, I. C. et al. Early-life inflammation primes a T helper 2 cell–fibroblast niche in skin. Nature 599, 667–672 (2021).
Dahlgren, M. W. et al. Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches. Immunity 50, 707–722.e706 (2019).
McDermott, J. R. et al. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc. Natl Acad. Sci. USA 100, 7761–7766 (2003).
Sharpe, C., Thornton, D. J. & Grencis, R. K. A sticky end for gastrointestinal helminths; the role of the mucus barrier. Parasite Immunol. 40, e12517 (2018).
Zhao, A. et al. Dependence of IL-4, IL-13, and Nematode-Induced Alterations in Murine Small Intestinal Smooth Muscle Contractility on Stat6 and Enteric Nerves. J. Immunol. 171, 948–954 (2003).
Cliffe, L. J. et al. Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion. Science 308, 1463–1465 (2005).
Nusse, Y. M. et al. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 559, 109–113 (2018).
Sato, T. et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418 (2011).
Veiga-Fernandes, H. & Artis, D. Neuronal–immune system cross-talk in homeostasis. Science 359, 1465 (2018).
Donnelly, S. et al. Helminth 2-Cys peroxiredoxin drives Th2 responses through a mechanism involving alternatively activated macrophages. FASEB J. 22, 4022–4032 (2008).
Gnanasekar, M., Velusamy, R., He, Y. X. & Ramaswamy, K. Cloning and characterization of a high mobility group box 1 (HMGB1) homologue protein from Schistosoma mansoni. Mol. Biochem. Parasitol. 145, 137–146 (2006).
McGinty, J. W. et al. Tuft-Cell-Derived Leukotrienes Drive Rapid Anti-helminth Immunity in the Small Intestine but Are Dispensable for Anti-protist Immunity. Immunity 52, 528–541.e527 (2020).
Oyesola, O. O. et al. PGD2 and CRTH2 counteract Type 2 cytokine–elicited intestinal epithelial responses during helminth infection. J. Exp. Med. 218,e20202178 (2021).
Inaba, A. et al. Interleukin-4 Promotes Tuft Cell Differentiation and Acetylcholine Production in Intestinal Organoids of Non-Human Primate. Int. J. Mol. Sci. 22, 7921 (2021).
Cheng, X., Voss, U. & Ekblad, E. Tuft cells: Distribution and connections with nerves and endocrine cells in mouse intestine. Exp. Cell Res. 369, 105–111 (2018).
Krasteva, G. et al. Cholinergic chemosensory cells in the trachea regulate breathing. Proc. Natl Acad. Sci. USA 108, 9478–9483 (2011).
Panneck, A. R. et al. Cholinergic epithelial cell with chemosensory traits in murine thymic medulla. Cell Tissue Res. 358, 737–748 (2014).
Saunders, C. J., Christensen, M., Finger, T. E. & Tizzano, M. Cholinergic neurotransmission links solitary chemosensory cells to nasal inflammation. Proc. Natl Acad. Sci. USA 111, 6075–6080 (2014).
Schütz, B. et al. Chemical coding and chemosensory properties of cholinergic brush cells in the mouse gastrointestinal and biliary tract. Front. Physiol. 6, 87–87 (2015).
Glover, M., Colombo, S. A. P., Thornton, D. J. & Grencis, R. K. Trickle infection and immunity to Trichuris muris. PLOS Pathog. 15, e1007926 (2019).
Gerbe, F. et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 (2016).
Haber, A. L. et al. A single-cell survey of the small intestinal epithelium. Nature 551, 333–339 (2017).
Montoro, D. T. et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018).
Zhang, Y. et al. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell 112, 293–301 (2003).
Luo, X.-C. et al. Infection by the parasitic helminthTrichinella spiralis activates a Tas2r-mediated signaling pathway in intestinal tuft cells. Proc. Natl Acad. Sci. 116, 5564–5569 (2019).
Nadjsombati, M. S. et al. Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 49, 33–41.e37 (2018).
Schneider, C. et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell 174, 271–284.e214 (2018).
Howitt, M. R. et al. The Taste Receptor TAS1R3 Regulates Small Intestinal Tuft Cell Homeostasis. Immunohorizons 4, 23–32 (2020).
Deng, J. et al. Solitary chemosensory cells are innervated by trigeminal nerve endings and autoregulated by cholinergic receptors. Int. Forum Allergy Rhinol. 11, 877–884 (2021).
Desai, P. et al. Enteric helminth coinfection enhances host susceptibility to neurotropic flaviviruses via a tuft cell-IL-4 receptor signaling axis. Cell 184, 1214–1231.e1216 (2021).
Ualiyeva, S. et al. Tuft cell-produced cysteinyl leukotrienes and IL-25 synergistically initiate lung type 2 inflammation. Sci. Immunol. 6, eabj0474 (2021).
Hung, L.-Y. et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc. Natl Acad. Sci. 110, 282–287 (2013).
Hung, L.-Y. et al. Cellular context of IL-33 expression dictates impact on anti-helminth immunity. Sci. Immunol. 5, eabc6259 (2020).
Meiners, J. et al. IL-33 facilitates rapid expulsion of the parasitic nematode Strongyloides ratti from the intestine via ILC2- and IL-9-driven mast cell activation. PLOS Pathog. 16, e1009121 (2020).
Osbourn, M. et al. HpARI Protein Secreted by a Helminth Parasite Suppresses Interleukin-33. Immunity 47, 739–751.e735 (2017).
Mukendi, J. P. K., Nakamura, R., Uematsu, S. & Hamano, S. Interleukin (IL)-33 is dispensable for Schistosoma mansoni worm maturation and the maintenance of egg-induced pathology in intestines of infected mice. Parasites Vectors 14, 70 (2021).
Scalfone, L. K. et al. Participation of MyD88 and Interleukin-33 as Innate Drivers of Th2 Immunity to Trichinella spiralis. Infect. Immun. 81, 1354–1363 (2013).
McSorley, H. J. & Smyth, D. J. IL-33: A central cytokine in helminth infections. Semin. Immunol. 53, 101532 (2021).
Vainchtein, I. D. et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Sci. (N.Y., NY) 359, 1269–1273 (2018).
Nguyen, P. T. et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 182, 388–403.e315 (2020).
Küchler, A. M. et al. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am. J. Pathol. 173, 1229–1242 (2008).
Hung, L. Y. et al. Cellular context of IL-33 expression dictates impact on anti-helminth immunity. Sci. Immunol. 5, eabc6259 (2020).
Jackson-Jones, L. H. et al. Fat-associated lymphoid clusters control local IgM secretion during pleural infection and lung inflammation. Nat. Commun. 7, 12651 (2016).
Spallanzani, R. G. et al. Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci. Immunol. 4, eaaw3658 (2019).
Mahlakõiv, T. et al. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci. Immunol. 4, eaax0416 (2019).
Hirosue, S. & Dubrot, J. Modes of Antigen Presentation by Lymph Node Stromal Cells and Their Immunological Implications. Front. Immunol. 6, 446–446 (2015).
Koyama, M. et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat. Med. 18, 135–142 (2012).
Torti, N., Walton, S. M., Brocker, T., Rülicke, T. & Oxenius, A. Non-hematopoietic cells in lymph nodes drive memory CD8 T cell inflation during murine cytomegalovirus infection. PLoS Pathog. 7, e1002313–e1002313 (2011).
Biton, M. et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 175, 1307–1320.e1322 (2018).
Trier, A. M. et al. IL-33 signaling in sensory neurons promotes dry skin itch. J. Allergy Clin. Immunol. 149,1473–1480.e6. (2021).
Wilson, S. R. et al. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 155, 285–295 (2013).
Ehrens, A. et al. Eosinophils and Neutrophils Eliminate Migrating Strongyloides ratti Larvae at the Site of Infection in the Context of Extracellular DNA Trap Formation. Front. Immunol. 12, 715766 (2021).
Obata-Ninomiya, K. et al. The skin is an important bulwark of acquired immunity against intestinal helminths. J. Exp. Med. 210, 2583–2595 (2013).
Hashimoto, K. et al. Depleted intestinal goblet cells and severe pathological changes in SCID mice infected with Heligmosomoides polygyrus. Parasite Immunol. 31, 457–465 (2009).
Hasnain, S. Z. et al. Muc5ac: a critical component mediating the rejection of enteric nematodes. J. Exp. Med. 208, 893–900 (2011).
Hasnain, S. Z. et al. Mucin gene deficiency in mice impairs host resistance to an enteric parasitic infection. Gastroenterology 138, 1763–1771 (2010).
Inagaki-Ohara, K., Sakamoto, Y., Dohi, T. & Smith, A. L. γδ T cells play a protective role during infection with Nippostrongylus brasiliensis by promoting goblet cell function in the small intestine. Immunology 134, 448–458 (2011).
Hasnain, S. Z., McGuckin, M. A., Grencis, R. K. & Thornton, D. J. Serine Protease(s) Secreted by the Nematode Trichuris muris Degrade the Mucus Barrier. PLOS Neglected Tropical Dis. 6, e1856 (2012).
Hasnain, S. Z. et al. Immune-driven alterations in mucin sulphation is an important mediator of Trichuris muris helminth expulsion. PLOS Pathog. 13, e1006218 (2017).
Inclan-Rico, J. M. et al. Basophils prime group 2 innate lymphoid cells for neuropeptide-mediated inhibition. Nat. Immunol. 21, 1181–1193 (2020).
Campbell, L. et al. ILC2s mediate systemic innate protection by priming mucus production at distal mucosal sites. J. Exp. Med. 216, 2714–2723 (2019).
Herbert, D. B. R. et al. Intestinal epithelial cell secretion of RELM-β protects against gastrointestinal worm infection. J. Exp. Med. 206, 2947–2957 (2009).
Belle, N. M. et al. TFF3 interacts with LINGO2 to regulate EGFR activation for protection against colitis and gastrointestinal helminths. Nat. Commun. 10, 4408–4408 (2019).
Wills-Karp, M. et al. Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J. Exp. Med. 209, 607–622 (2012).
Oeser, K., Schwartz, C. & Voehringer, D. Conditional IL-4/IL-13-deficient mice reveal a critical role of innate immune cells for protective immunity against gastrointestinal helminths. Mucosal Immunol. 8, 672–682 (2015).
Turner, J.-E., Stockinger, B. & Helmby, H. IL-22 Mediates Goblet Cell Hyperplasia and Worm Expulsion in Intestinal Helminth Infection. PLOS Pathog. 9, e1003698 (2013).
Miller, H. R. Gastrointestinal mucus, a medium for survival and for elimination of parasitic nematodes and protozoa. Parasitology 94(Suppl), S77–100 (1987).
Douch, P. G. C., Harrison, G. B. L., Buchanan, L. L. & Greer, K. S. In vitro bioassay of sheep gastrointestinal mucus for nematode paralysing activity mediated by substances with some properties characteristic of SRS-A. Int. J. Parasitol. 13, 207–212 (1983).
Artis, D. et al. RELMβ/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl Acad. Sci. USA 101, 13596–13600 (2004).
Kamal, M., Dehlawi, M. S., Brunet, L. R. & Wakelin, D. Paneth and intermediate cell hyperplasia induced in mice by helminth infections. Parasitology 125, 275–281 (2002).
Walsh, R., Seth, R., Behnke, J., Potten, C. S. & Mahida, Y. R. Epithelial stem cell-related alterations in Trichinella spiralis-infected small intestine. Cell Prolif. 42, 394–403 (2009).
Entwistle, L. J. et al. Epithelial-Cell-Derived Phospholipase A2 Group 1B Is an Endogenous Anthelmintic. Cell Host Microbe 22, 484–493.e485 (2017).
Hu, Z. et al. Small proline-rich protein 2A is a gut bactericidal protein deployed during helminth infection. Science 374, eabe6723 (2021).
McKenzie, G. J., Bancroft, A., Grencis, R. K. & McKenzie, A. N. J. A distinct role for interleukin-13 in Th2-cell-mediated immune responses. Curr. Biol. 8, 339–342 (1998).
Schubart, C. et al. Selective expression of constitutively activated STAT6 in intestinal epithelial cells promotes differentiation of secretory cells and protection against helminths. Mucosal Immunol. 12, 413–424 (2019).
Zhu, P. et al. IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nat. Immunol. 20, 183–194 (2019).
Drurey, C. et al. Intestinal epithelial tuft cell induction is negated by a murine helminth and its secreted products. J. Exp. Med. 219, e20211140 (2021).
Zwiggelaar, R. T. et al. LSD1 represses a neonatal/reparative gene program in adult intestinal epithelium. Sci. Adv. 6, eabc0367 (2020).
Parmar, N. et al. Intestinal-epithelial LSD1 controls goblet cell maturation and effector responses required for gut immunity to bacterial and helminth infection. PLOS Pathog. 17, e1009476 (2021).
Oudhoff, M. J. et al. Intestinal Epithelial Cell-Intrinsic Deletion of Setd7 Identifies Role for Developmental Pathways in Immunity to Helminth Infection. PLOS Pathog. 12, e1005876 (2016).
Chenery, A. L. et al. IL-13 deficiency exacerbates lung damage and impairs epithelial-derived type 2 molecules during nematode infection. Life Sci. Alliance 4, e202001000 (2021).
Sutherland, T. E. et al. Ym1 induces RELMα and rescues IL-4Rα deficiency in lung repair during nematode infection. PLOS Pathog. 14, e1007423 (2018).
Thawer, S. et al. Surfactant Protein-D Is Essential for Immunity to Helminth Infection. PLOS Pathog. 12, e1005461 (2016).
Knipper, J. A. et al. Interleukin-4 Receptor alpha Signaling in Myeloid Cells Controls Collagen Fibril Assembly in Skin Repair. Immunity 43, 803–816 (2015).
Minutti, C. M. et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 50, 645–654.e646 (2019).
Bourke, C. D. et al. Epidermal keratinocytes initiate wound healing and pro-inflammatory immune responses following percutaneous schistosome infection. Int. J. Parasitol. 45, 215–224 (2015).
Zhao, A. et al. Critical Role of IL-25 in Nematode Infection-Induced Alterations in Intestinal Function. J. Immunol. 185, 6921–6929 (2010).
Marillier, R. G. et al. IL-4Rα-responsive smooth muscle cells increase intestinal hypercontractility and contribute to resistance during acute Schistosomiasis. Am. J. Physiol.-Gastrointest. Liver Physiol. 298, G943–G951 (2010).
Hunt, V. L. et al. The genomic basis of parasitism in the Strongyloides clade of nematodes. Nat. Genet. 48, 299–307 (2016).
Hunt, V. L., Tsai, I. J., Selkirk, M. E. & Viney, M. The genome of Strongyloides spp. gives insights into protein families with a putative role in nematode parasitism. Parasitology 144, 343–358 (2017).
Sanderson, B. E. Acetylcholinesterase activity in Nippostrongylus brasiliensis (nematoda). Comp. Biochem. Physiol. 29, 1207–1213 (1969).
Sanderson, B. E. & Ogilvie, B. M. A study of acetylcholinesterase throughout the life cycle of Nippostrongylus brasiliensis. Parasitology 62, 367–373 (1971).
Zhao, A. et al. Contribution of 5-HT2A Receptor in Nematode Infection-Induced Murine Intestinal Smooth Muscle Hypercontractility. Gastroenterology 131, 568–578 (2006).
Steel, N. et al. TGFβ-activation by dendritic cells drives Th17 induction and intestinal contractility and augments the expulsion of the parasite Trichinella spiralis in mice. PLOS Pathog. 15, e1007657 (2019).
Horsnell, W. G. C. et al. IL-4Rα-responsive smooth muscle cells contribute to initiation of TH2 immunity and pulmonary pathology in Nippostrongylus brasiliensis infections. Mucosal Immunol. 4, 83–92 (2011).
Horsnell, W. G. C. et al. Delayed goblet cell hyperplasia, acetylcholine receptor expression, and worm expulsion in SMC-specific IL-4Ralpha-deficient mice. PLoS Pathog. 3, e1–e1 (2007).
Martín-Alonso, M. et al. Smooth muscle-specific MMP17 (MT4-MMP) regulates the intestinal stem cell niche and regeneration after damage. Nat. Commun. 12, 6741 (2021).
Basbaum, A. I., Bautista, D. M., Scherrer, G. & Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 139, 267–284 (2009).
Gao, Y., Mei, C., Chen, P. & Chen, X. The contribution of neuro-immune crosstalk to pain in the peripheral nervous system and the spinal cord. Int. Immunopharmacol. 107, 108700 (2022).
Cañedo-Dorantes, L. & Cañedo-Ayala, M. Skin acute wound healing: a comprehensive review. Int. J. Inflamm. 2019, 3706315 (2019).
Nowak, N. C., Menichella, D. M., Miller, R. & Paller, A. S. Cutaneous innervation in impaired diabetic wound healing. Transl. Res. 236, 87–108 (2021).
Ten Hove, A. S., Seppen, J. & de Jonge, W. J. Neuronal innervation of the intestinal crypt. Am. J. Physiol. Gastrointest. Liver Physiol. 320, G193–g205 (2021).
Hoeffel, G. et al. Sensory neuron-derived TAFA4 promotes macrophage tissue repair functions. Nature 594, 94–99 (2021).
Worthington, J. J., Reimann, F. & Gribble, F. M. Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity. Mucosal Immunol. 11, 3–20 (2018).
Ovington, K. S., Bacarese-Hamilton, A. J. & Bloom, S. R. Nippostrongylus brasiliensis: changes in plasma levels of gastrointestinal hormones in the infected rat. Exp. Parasitol. 60, 276–284 (1985).
McDermott, J. R. et al. Immune control of food intake: enteroendocrine cells are regulated by CD4+ T lymphocytes during small intestinal inflammation. Gut 55, 492–497 (2006).
Worthington, J. J., Samuelson, L. C., Grencis, R. K. & McLaughlin, J. T. Adaptive immunity alters distinct host feeding pathways during nematode induced inflammation, a novel mechanism in parasite expulsion. PLoS Pathog. 9, e1003122 (2013).
Tu, T., Koski, K. G. & Scott, M. E. Mechanisms underlying reduced expulsion of a murine nematode infection during protein deficiency. Parasitology 135, 81–93 (2008).
Castro, G. A., Copeland, E. M., Dudrick, S. J. & Johnson, L. R. Serum and antral gastrin levels in rats infected with intestinal parasites. Am. J. Trop. Med. Hyg. 25, 848–853 (1976).
Zhang, J. G. et al. Cholecystokinin octapeptide regulates the differentiation and effector cytokine production of CD4(+) T cells in vitro. Int. Immunopharmacol. 20, 307–315 (2014).
Reardon, C. et al. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc. Natl Acad. Sci. USA 110, 1410–1415 (2013).
De Jonge, F. et al. Effects of Schistosoma mansoni infection on somatostatin and somatostatin receptor 2A expression in mouse ileum. Neurogastroenterol. Motil. 15, 149–159 (2003).
Chatterjee, S. et al. Circulating levels of the neuropeptide hormone somatostatin may determine hepatic fibrosis in Schistosoma mansoni infections. Acta Trop. 90, 191–203 (2004).
Zhang, C. et al. Octreotide attenuates hepatic fibrosis and hepatic stellate cells proliferation and activation by inhibiting Wnt/β-catenin signaling pathway, c-Myc and cyclin D1. Int. Immunopharmacol. 63, 183–190 (2018).
Tang, S. C., Braunsteiner, H. & Wiedermann, C. J. Regulation of human T lymphoblast growth by sensory neuropeptides: augmentation of cholecystokinin-induced inhibition of Molt-4 proliferation by somatostatin and vasoactive intestinal peptide in vitro. Immunol. Lett. 34, 237–242 (1992).
Kao, J. Y. et al. Somatostatin inhibits dendritic cell responsiveness to Helicobacter pylori. Regul. Pept. 134, 23–29 (2006).
Saavedra, Y. & Vergara, P. Somatostatin inhibits intestinal mucosal mast cell degranulation in normal conditions and during mast cell hyperplasia. Regul. Pept. 111, 67–75 (2003).
Barrios, J. et al. Pulmonary Neuroendocrine Cells Secrete γ-Aminobutyric Acid to Induce Goblet Cell Hyperplasia in Primate Models. Am. J. Respir. Cell Mol. Biol. 60, 687–694 (2019).
Lu, Y. et al. Eosinophil extracellular traps drive asthma progression through neuro-immune signals. Nat. Cell Biol. 23, 1060–1072 (2021).
Chen, Z. et al. Interleukin-33 Promotes Serotonin Release from Enterochromaffin Cells for Intestinal Homeostasis. Immunity 54, 151–163.e156 (2021).
Flamar, A.-L. et al. Interleukin-33 Induces the Enzyme Tryptophan Hydroxylase 1 to Promote Inflammatory Group 2 Innate Lymphoid Cell-Mediated. Immun. Immun. 52, 606–619.e606 (2020).
Yu, Y., Yang, W., Li, Y. & Cong, Y. Enteroendocrine Cells: Sensing Gut Microbiota and Regulating Inflammatory Bowel Diseases. Inflamm. bowel Dis. 26, 11–20 (2020).
Gershon, M. D. & Tack, J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology 132, 397–414 (2007).
Maglie, R. et al. The Role of TRPA1 in Skin Physiology and Pathology. Int. J. Mol. Sci. 22, 3065 (2021).
Zhu, L. et al. The phytonutrient cinnamaldehyde limits intestinal inflammation and enteric parasite infection. J. Nutr. Biochem. 100, 108887 (2021).
McLean, L. P. et al. Type 3 muscarinic receptors contribute to intestinal mucosal homeostasis and clearance of Nippostrongylus brasiliensis through induction of TH2 cytokines. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G130–141 (2016).
Chu, C. et al. The ChAT-acetylcholine pathway promotes group 2 innate lymphoid cell responses and anti-helminth immunity. Sci. Immunol. 6, eabe3218 (2021).
Galle-Treger, L. et al. Nicotinic acetylcholine receptor agonist attenuates ILC2-dependent airway hyperreactivity. Nat. Commun. 7, 13202 (2016).
Takahashi, T. Multiple Roles for Cholinergic Signaling from the Perspective of Stem Cell Function. Int. J. Mol. Sci. 22, 666 (2021).
Gross, E. R. et al. Neuronal serotonin regulates growth of the intestinal mucosa in mice. Gastroenterology 143, 408–417.e402 (2012).
Middelhoff, M. et al. Prox1-positive cells monitor and sustain the murine intestinal epithelial cholinergic niche. Nat. Commun. 11, 111 (2020).
Jean, E. E., Good, O., Rico, J. M. I., Rossi, H. L. & Herbert, D. R. Neuroimmune regulatory networks of the airway mucosa in allergic inflammatory disease. J. Leukoc. Biol. 111, 209–221 (2022).
Cardoso, V. et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281 (2017).
Klose, C. S. N. et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286 (2017).
Inclan-Rico, J. M. et al. Basophils prime group 2 innate lymphoid cells for neuropeptide-mediated inhibition. Nat. Immunol. 21, 1181–1193 (2020).
Nagashima, H. et al. Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity 51, 682–695.e686 (2019).
Oda, N. et al. Requirement for neuropeptide Y in the development of type 2 responses and allergen-induced airway hyperresponsiveness and inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 316, L407–l417 (2019).
Perner, C. et al. Substance P Release by Sensory Neurons Triggers Dendritic Cell Migration and Initiates the Type-2 Immune Response to Allergens. Immunity 53, 1063–1077.e1067 (2020).
Yu, X. H. et al. Pro-nociceptive effects of neuromedin U in rat. Neuroscience 120, 467–474 (2003).
Honzawa, M., Sudoh, T., Minamino, N., Tohyama, M. & Matsuo, H. Topographic localization of neuromedin u-like structures in the rat brain: An immunohistochemical study. Neuroscience 23, 1103–1122 (1987).
Wang, F. et al. Neuromedin U inhibits T-type Ca2+ channel currents and decreases membrane excitability in small dorsal root ganglia neurons in mice. Cell Calcium 49, 12–22 (2011).
Mishra, S. K., Holzman, S. & Hoon, M. A. A nociceptive signaling role for neuromedin B. J. Neurosci. 32, 8686–8695 (2012).
Meng, Q. T. et al. BNP facilitates NMB-encoded histaminergic itch via NPRC-NMBR crosstalk. Elife 10, e71689 (2021).
Cohen, J. A. et al. Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 178, 919–932.e914 (2019).
Kashem Sakeen, W. Riedl Maureen S, Yao C, Honda Christopher N, Vulchanova L, Kaplan Daniel H. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous. Immun. Immun. 43, 515–526 (2015).
Xing, Y. et al. Visualizing the Itch-Sensing Skin Arbors. J. Investig. Dermatol. 141, 1308–1316 (2021).
Inclan-Rico, J. M., Kim, B. S. & Abdus-Saboor, I. Beyond somatosensation: Mrgprs in mucosal tissues. Neurosci. Lett. 748, 135689 (2021).
Steele, H. R. & Han, L. The signaling pathway and polymorphisms of Mrgprs. Neurosci. Lett. 744, 135562 (2021).
Arthur, R. P. & Shelley, W. B. Larva currens; a distinctive variant of cutaneous larva migrans due to Strongyloides stercoralis. AMA Arch. Derm. 78, 186–190 (1958).
Daveson, A. J. et al. Effect of hookworm infection on wheat challenge in celiac disease-a randomised double-blinded placebo controlled trial. PLoS ONE 6, e17366–e17366 (2011).
Feary, J. R. et al. Experimental hookworm infection: a randomized placebo-controlled trial in asthma. Clin. Exp. Allergy 40, 299–306 (2010).
Avula, L. R. et al. The effect of inflammation on the expression and distribution of the MAS-related gene receptors MrgE and MrgF in the murine ileum. Histochem Cell Biol. 136, 569–585 (2011).
Castro, J. et al. Activation of pruritogenic TGR5, MrgprA3, and MrgprC11 on colon-innervating afferents induces visceral hypersensitivity. JCI Insight 4,e131712 (2019).
Bautzova, T. et al. 5-oxoETE triggers nociception in constipation-predominant irritable bowel syndrome through MAS-related G protein-coupled receptor D. Sci. Signal 11, eaal2171 (2018).
Xu, M. et al. Mas-related G protein-coupled receptor D is involved in modulation of murine gastrointestinal motility. Exp. Physiol. 106, 2502–2516 (2021).
Zhang, S. et al. Nonpeptidergic neurons suppress mast cells via glutamate to maintain skin homeostasis. Cell 184, 2151–2166.e2116 (2021).
Montaño, K. J., Cuéllar, C. & Sotillo, J. Rodent Models for the Study of Soil-Transmitted Helminths: a proteomics approach. Front Cell Infect. Microbiol. 11, 639573–639573 (2021).
Mestas, J. & Hughes C. C. Of mice and not men: differences between mouse and human immunology. J. Immunol. (Baltimore, Md: 1950) 172: 2731–2738 (2004).
Wangzhou, A. et al. Pharmacological target-focused transcriptomic analysis of native vs cultured human and mouse dorsal root ganglia. Pain 161, 1497–1517 (2020).
Acknowledgements
This work was supported by the National Institutes of Health (grant nos. T32 AI007532-24, RO1 AI164715-01, U01 AI163062-01, RO1 AI123173-05 to D.R.H). J.M.I.R. is supported by the Life Sciences Research Foundation. We used Biorender to draft the figures in this review paper.
Author information
Authors and Affiliations
Contributions
J.M.I. and H.L.R. drafted the paper and figures, all authors were involved in revision and approved the final version of this review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Inclan-Rico, J.M., Rossi, H.L. & Herbert, D.R. “Every cell is an immune cell; contributions of non-hematopoietic cells to anti-helminth immunity”. Mucosal Immunol 15, 1199–1211 (2022). https://doi.org/10.1038/s41385-022-00518-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00518-7
This article is cited by
-
Lessons from helminths: what worms have taught us about mucosal immunology
Mucosal Immunology (2022)
