
Abstract
Airway macrophages (AMs) play key roles in the maintenance of lung immune tolerance. Tissue tailored, highly specialised and strategically positioned, AMs are critical sentinels of lung homoeostasis. In the last decade, there has been a revolution in our understanding of how metabolism underlies key macrophage functions. While these initial observations were made during steady state or using in vitro polarised macrophages, recent studies have indicated that during many chronic lung diseases (CLDs), AMs adapt their metabolic profile to fit their local niche. By generating reactive oxygen species (ROS) for pathogen defence, utilising aerobic glycolysis to rapidly generate cytokines, and employing mitochondrial respiration to fuel inflammatory responses, AMs utilise metabolic reprogramming for host defence, although these changes may also support chronic pathology. This review focuses on how metabolic alterations underlie AM phenotype and function during CLDs. Particular emphasis is given to how our new understanding of AM metabolic plasticity may be exploited to develop AM-focused therapies.
Similar content being viewed by others

Introduction
The respiratory mucosa is a unique site, as our airways are continually exposed to particulates, viruses, bacteria, and fungi, which challenge the pulmonary immune system1. To maintain pulmonary homoeostasis and ensure efficient gas exchange, a complex regulatory system is in place, of which airway macrophages (AMs) are a core component. AMs are the most numerous immune cell type present in healthy lungs, are strategically positioned at the interface of airways and environment2, and critical sentinels of barrier immunity. AMs form the first line of defence against inhaled particles, pathogens and antigens3. Although inherently suppressive, AMs exhibit significant functional and phenotypical specialisation, allowing efficient responses to environmental signals and rapid alterations in phenotype. Increasing evidence suggests that metabolic alterations provide an additional layer of functional plasticity to AM populations. Activation of macrophages in vitro with a range of inflammatory stimuli, induce profound metabolic adaptations, such as the switch from oxidative phosphorylation (OXPHOS) to glycolysis in oxygen-sufficient conditions, similar to the “Warburg effect” seen in some cancers4. It is now clear that how macrophages utilise energy dictates immune responses, and that manipulating cellular metabolism can alter the inflammatory response5. However in vivo, the unique oxygen rich environment of the airways coupled with specific local nutrient availabilities, shapes AM phenotype and function. Indeed, many recent studies have indicated that in chronic lung diseases (CLDs), such as asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis (IPF), and during infection such as with Mycobacterium Tuberculosis (Mtb), there are significant alterations in AM metabolic processes and that targeting these pathways could represent an exciting therapeutic approach6,7. This review focuses on how metabolic adaptations underlie AM phenotype and function during CLDs. Particular emphasis is given to how our new understanding of AM metabolic plasticity may be exploited to develop AM-focused therapies.
Airway macrophages: guardians of the lung environment
To maintain gas exchange, it is critical that AMs sustain a naturally hyporesponsive state whilst also reserving the ability to rapidly mount effective inflammatory responses. This balance is achieved through complex AM-airway epithelial cell (AEC) interactions via cell surface-expressed receptors and secreted products. AMs express transforming growth factor beta receptor (TGF-βR), interleukin (IL)-10 receptor (IL-10R), CD200 receptor (CD200R) and signal regulatory protein-α, key components mediating AM:AEC crosstalk and in turn, regulating AM activation8. For example, AM-AEC contact decreases AM phagocytosis and cytokine production in a TGF-β-dependent manner9. Conversely, loss of the integrin αvβ6 such as through loss of contact of AMs with AEC upon toll-like receptor (TLR) activation leads to initiation of the AM pro-inflammatory phenotype and inflammatory response10.
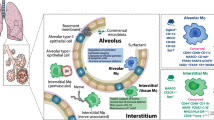
AMs are characterised by a distinct cellular phenotype. Human AMs highly express the lectin-binding transmembrane glycoprotein CD68, the adhesion molecule CD169, the integrin CD11c and mannose receptor CD20611 (Fig. 1). In mice, expression of the CD68+CD206+CD11chiCD11blo cell surface phenotype is conserved at steady state12,13,14, while murine AMs also express the Mer tyrosine kinase (MerTK), sialic acid dependent adhesion molecule SiglecF, hormone receptor F4/8, glycoprotein CD64 and the CD200 receptor15 (Fig. 1). Recent work in mice has indicated that many tissue resident macrophages, including those in the airways, are foetally derived and self-maintain locally with minimal contribution from circulating monocytes, during steady state conditions16,17,18,19,20. During murine prenatal development, foetal liver or yolk sac macrophages are the major contributing pool to AM populations21 and AM colonization of the lung occurs in sequential waves in the first week of life22. Furthermore, post birth and during maturation, circulating monocytes do not significantly contribute to lung macrophage populations at homoeostasis18. During pulmonary inflammatory responses however, monocytes are recruited to the lung23,24 and subsequently develop into AM-like cells18,25. Thus, post-injury murine airways contain at least two ontologically distinct AM populations, tissue resident AMs (TR-AM) which are prenatally derived and monocyte-derived AMs (Mo-AMs). Several groups have studied samples from lung transplant patients to investigate the origins of AMs in the human lung26,27,28,29,30. Utilizing bronchoalveolar lavage (BAL) from sex-mismatched lung transplant patients our group recently demonstrated that the majority of AMs in human lung post-transplant are derived from peripheral classical monocytes31. Thus, the unique airway niche combined with distinct ontological origins, age and environmental exposures results in remarkable AM plasticity and adaptability32,33,34. While the recruitment of monocytes to the lungs to replenish the TR-AM pool is relatively well understood in mice, the origins of AMs in the human lung during healthy aging or CLDs requires further investigation; in particular clear markers which distinguish Mo-AMs and TR-AM are not well established.

Murine AMs express the lectin-binding transmembrane glycoprotein CD68, the Mer tyrosine kinase (MERTK), the integrin alpha X chain protein CD11c, the type I membrane glycoprotein CD200 receptor, the mannose receptor CD206, the EGF-like module-containing mucin-like hormone receptor-like 1 (F4/80), the sialic acid binding lectin Siglec-F and the Fc receptor CD64. Human AMs express CD68, the adhesion molecule CD169, CD11c, CD206 as well as MHC class II receptor HLA-DR.
Airway macrophage metabolic phenotype at homoeostasis
Beyond delineation of macrophage populations based on anatomical location or ontological origin, macrophage populations are also classified according to their activation status. Analogous to the Th1/Th2 paradigm, in vitro cultured human monocyte derived macrophages (MDMs) or murine bone marrow derived macrophages (BMDMs) are categorised as M1 or M2 macrophages, respectively. Seminal studies have demonstrated that pro-wound healing M2 (IL-4 stimulated) macrophages in vitro rely on fatty acid oxidation (FAO), an intact tricarboxylic acid cycle (TCA) cycle, high rates of OXPHOS and increased expression of arginase 1 (Arg1), which catalyses the production of ornithine from arginine as precursor for collagen to facilitate wound healing5,35,36,37. Conversely, pro-inflammatory M1-macrophages rely on glycolysis and breaks in the TCA cycle lead to accumulation of metabolites, many of which have signalling functions such as citrate, succinate, fumarate and α-ketoglutarate35,38,39. However, although useful in defining the range of potential macrophage responses, in vitro derived cells do not recapitulate the core aspects of AM phenotypes which are shaped by the local niche11. As AMs are highly adapted to the unique environment of the airway lumen, it is perhaps unsurprising that the metabolic state of AMs is also distinct. Glucose concentrations in the alveolar lumen are less than 10% of blood glucose concentrations and AMs exhibit extremely low levels of glycolysis20; in stark contrast to BMDMs, AMs do not undergo glycolytic reprogramming in response to LPS40. Consequently, AMs readily engage OXPHOS and highly express the peroxisome proliferator-activated receptor gamma (PPARγ)41, which regulates lipid accumulation and promotes FAO to sustain OXPHOS.
AMs also play a major role in the catabolism of pulmonary surfactant, a monolayer composed mainly of phosphocholine-based lipids, phospholipids and cholesterol which lines the alveoli, lowers surface tension and prevents alveolar collapse during expiration42. Mice lacking GM-CSF and thus the AM compartment, develop pulmonary alveolar proteinosis (PAP), an inflammatory lung syndrome caused by the defective clearance of surfactant43,44,45. In humans, mutations in genes encoding for GM-CSF receptors, result in hereditary PAP as a result of progressive alveolar surfactant accumulation46,47,48,49. AM phenotype and behaviour are influenced by surfactant exposure, which has major implications for AM-mediated immune responses in pulmonary tissue. There are four principle surfactant proteins (SP-A-D) and SP-A and SP-D have been shown to directly influence AM functions such as cell migration, phagocytosis and activation phenotypes42. Both SP-A and SP-D bind carbohydrates, lipids, and nucleic acids and initiate phagocytosis of inhaled pathogens and apoptotic cells50. Furthermore, SP-A blocks the binding of TLR ligands to TLR2, TLR4 and TLR co-receptors and furthermore inhibits complement activation51,52.
Whilst the alveoli are covered with a monolayer of surfactant, a thin layer of mucus produced by goblet cells and ciliated epithelium protects the airways. Mucus serves as a barrier and facilitates clearance of microbes and pollutants. A major component of mucus are mucin glycoproteins, which may be categorized as polymerizing, nonpolymerizing and cell-surface associated. Of the cell-surface associated mucins, MUC1 is expressed in AMs and contributes to the resolution of inflammation by decreasing phagocytic potential and pro-inflammatory cytokine production53. The polymerizing mucins include MUC5AC and MUC5B; in particular, MUC5B deficiency has been linked to particle accumulation in the lung, mucus obstruction and impaired macrophage phagocytosis54. Pro-inflammatory macrophages induce MUC5B expression to aid mucociliary clearance55. Furthermore, a single nucleotide polymorphism in the MUC5B promotor has been strongly associated with the risk of developing IPF, highlighting the importance of mucins for the pulmonary environment56.
In addition to low glucose and a lipid rich environment, the airways also have a unique distribution of amino acids and central carbon metabolites. Surowiec et al. showed that whilst several glucogenic and ketogenic amino acids were present in the bronchial wash, only alanine is present in BAL57 (Fig. 2). In addition, the central airways contained key glycolytic and OXPHOS metabolites such as fructose, glucose-6-phosphate, fumarate and malate as well as oxidised gluthathione (GSSG, indicating oxidative stress, Fig. 2); interestingly, these could not be detected in the periphery, suggesting either minimal secretion, high utilisation or as a result of anatomical location (i.e. close proximity to nutrient rich pulmonary capillaries)57. Recently the lung microbiome has gained attention as a factor which modifies the pulmonary environment and directs immune responses by producing short chain fatty acids (SCFA). Whilst the airways and alveoli are colonised mainly by proteobacteria, bacteroidetes and firmicutes58,59, the nasal mucosa additionally hosts actinobacteria60,61 (Fig. 2). Proteobacteria, bacteroidetes and firmicutes produce large amounts of SCFA, including acetate, propionate and butyrate, which influence barrier function by regulating epithelial tight junctions62 and anti-inflammatory immune responses63. Recent advances in understanding the pulmonary microbiome during homoeostasis and CLDs are described in detail elsewhere58,59,64. Thus, at homoeostasis AMs are exposed to a unique environment, with minimal glucose availability and a distinct distribution of nutrients and SCFA, which depend on anatomical location (Fig. 2). However, despite the profound influence that local substrate availabilities may exert on macrophage development, activation and function, this is an understudied area. New knowledge, which further defines especially human AM substrate dependencies at homoeostasis is required in order to fully understand how local metabolic perturbations during CLDs may contribute to pathology. This is particularly relevant as already slight changes in nutrient availability during inflammation, such as succinate or citrate, can alter macrophage phenotypes through stabilization of Hif1α, post-translational modification of proteins and production of NO and ROS65, thereby contributing to a pathological development.

The respiratory mucosa stretches from the nasal cavity to the alveoli and its pseudostratified epithelium in the upper respiratory tract consists of mucus producing goblet cells, ciliated cells and progenitor basal cells on top of a basement membrane, covered with a layer of mucus. The alveoli are lined with type I alveolar epithelial cells (AEC) interspersed with surfactant producing type II AEC. Distinct metabolites of the key metabolic pathways have been detected in nasal secretions, bronchial washes and bronchoalveolar lavage (BAL) at the different respiratory mucosal sites. These key metabolic pathways are schematically represented here. Varying reactive oxygen species (ROS) and commensal bacteria have been detected in the nasal cavity, conducting airway and parenchyma, contributing to the unique environment in each compartment. The principal microbial phyla colonising the airway and alveoli are proteobacteria, bacteroidetes and firmicutes, while the nasal mucosa additionally hosts actinobacteria.
AM metabolism during CLDs
Chronic lung diseases affect a significant proportion of the world’s population, killing more than 100,000 people in the UK alone, each year66. Persistent inflammation, impaired repair processes and pulmonary remodelling are cardinal features of CLDs67,68,69. There are multiple overlaps in environmental exposures driving CLDs, such as smoking, pollution and environmental exposures; viral infection can also exacerbate symptoms in each disease2,70,71. Interestingly, AM metabolic adaptation may play a central role in dictating pathology during CLDs and present novel therapeutic opportunities (Fig. 3).

Several metabolic pathways are rewired during chronic lung disease. While this response exists to clear invading pathogens and launch an inflammatory response, long-term activation of these pathways has negative implications. The glycolysis pathway supports inflammatory responses of AM, while iron and metabolites produced in the TCA cycle can function as bacterial substrates and contribute to pathogen survival. While fatty acid synthesis and oxidation is useful as a way of storing energy and alternative energy source during times of macrophage activation, fatty acid synthesis can also contribute to mucus production. Leukotrienes contribute to the AM pro-inflammatory phenotype but also cause bronchial constriction and contribute to airway remodelling in asthmatics by causing smooth muscle thickening. The amino acid arginine is a proliferator for collagen via ornithine and proline and can thereby contribute to extracellular matrix deposition.
Asthma
Asthma is a heterogeneous disease of the airways characterized by airway remodelling, mucus production, airway hyperresponsiveness (AHR), and inflammation72. Although most patients have good control with standard medication, a proportion experience life-threatening, severe disease73. AMs are central to mediating type-2 inflammation against allergens and parasitic worms2. In vitro, macrophages respond to type-2 cytokines such as IL-4 that drive an ‘alternative’ M2 activation phenotype, linked to wound repair and type-2 pathology74,75. Manipulation of AM phenotype via genetic deletion of the transcription factor interferon regulatory factor 5 (Irf5), a master regulator of macrophage activation74, promoted pulmonary remodelling, AHR and mucus secretion in mice, in an IL-13 dependent manner76. Indeed, a recent study has shown that both CD206+ AM (activated by IL-4 and IL-13) and pro-inflammatory Irf5+ AM are increased in asthmatic patients77, highlighting the plasticity of macrophages and heterogeneity of human asthma. Roles of AMs during antigen induced airway inflammation include phagocytosis of apoptotic cells and eosinophils as well as triggering anti-inflammatory pathways to regulate airway hyper responsiveness, mucus secretion and matrix deposition76. In severe asthma however, this protective function is impaired, resulting in the loss of phagocytic ability and anti-inflammatory programme78, which can contribute to airway remodelling79. Thus, AMs are uniquely involved in responses to allergen and type-2 inflammation, and aberrant AM-phenotypes can directly influence respiratory pathology.
Numerous lines of evidence suggest that metabolic stress leading to the production of reactive oxygen species (ROS) plays a role in asthma. Increased ROS have been detected in AMs of asthmatic patients80, and contributes to lung injury81 and pro-inflammatory tumour necrosis factor-alpha (TNF-α) and IL-1β secretion by macrophages82. Furthermore, heme-oxygenase-1 (HO-1), which mediates ROS production in response to chemical and physical agents, is increased in AMs in asthmatics83. In addition to these pro-inflammatory characteristics, AMs show key features of a more anti-inflammatory phenotype in studies using ovalbumin to model allergic asthma. Using this model, Al-Khami et al. show that expression of carnitine palmitoyltransferase (CPT) is increased in AMs, shuttling fatty acids into the mitochondria, as well as increased gene expression of FAO related genes84.
Another functional pathway that is altered in asthmatic AMs and links to the underlying metabolic phenotype is the eicosanoid pathway, which is induced by Th2 cytokines IL-4 and IL-13. Eicosanoids, including prostaglandins and leukotrienes, are produced from the poly-unsaturated fatty acid arachidonic acid, which is released during asthma85. Increased production of the eicosanoid 5-HETE and leukotrienes B4 (LTB4) and E4 (LTE4) has been detected in AMs from asthmatic patients stimulated ex vivo86. This contributed to bronchial constriction and pro-inflammatory phenotype and failure to generate the anti-inflammatory eicosanoid 15-HETE and prostaglandin E2 (PGE2), which is associated with reduced AM phagocytosis78. LTE4 has been shown to cause AHR in subjects with aspirin-induced asthma87 and can be produced in AMs by γ-glutamyl transpeptidase85,88. IL-13 furthermore induces Arg1, which may further contribute to the asthmatic phenotype via metabolism of collagen precursors ornithine and proline to collagen and airway remodelling89,90.
Several of these observed alterations have been targeted therapeutically, attempting to rewire AM phenotype. These include the eicosanoid pathway, ROS, glycolysis and FAO. Administration of the anti-inflammatory eicosanoid 15-HETE inhibited leukotriene synthesis and reduced AHR in asthmatics91. Ex vivo, the corticosteroid dexamethasone decreased levels of thromboxane B2 and LTB4 in macrophages and asthmatic AMs92, while prednisone decreased LTB4 production in AMs93. Treatment with the antioxidant AD4 improved AHR and airway inflammation by decreasing ROS in the OVA-sensitised mouse model of allergic airway disease (AAD)94. Inhibiting glycolysis with 2-DG, Zhao et al. show altered AMs phenotype ameliorated AAD, while Al-Khami et al. reported improvements in AHR after treatment with FAO inhibitor etomoxir84,95.
Together, these studies indicate that there is significant disruption of AM metabolism during asthma and AAD, notably via dysfunction in eicosanoid, glycolysis and fatty acid pathways. In order to evaluate candidate therapies, it is crucial that studies utilise relevant preclinical models and ex vivo patient samples to understand disease. Models which more closely recapitulate the complex immune response to allergens, are more likely to reveal viable targets for intervention; in particular the ovalbumin model of AAD, which requires an adjuvant and a sensitization phase96, is a poor murine model of asthma. Furthermore, our new understanding of asthma heterogeneity has allowed the development of biologics which target “type-2 high” asthma97; delineation of how metabolic changes underlie distinct asthma phenotypes could lead to new treatments for other phenotypes, such as neutrophilic and paucigranulocytic asthma.
COPD
COPD is the 5th leading cause of death in high income countries98, affecting over 200 million people worldwide99. COPD is a heterogeneous disease, characterised by destruction of the parenchyma and emphysema, narrowing of the airways, remodelling and chronic inflammation driven by chronic exposure to cigarette smoke and particular matter100. AM numbers are increased in COPD BAL101 and contribute to COPD pathology through numerous pathways. During COPD, AMs are found in areas of lung destruction and produce pro-inflammatory cytokines102, chemokines100 and matrix metalloproteases (MMPs) with elastolytic properties103,104. At the same time, tissue inhibitor of metalloproteases (TIMP)-1 is decreased in AMs in COPD105 and furthermore, decreased phagocytic capacity and impaired bacterial killing have been described in COPD-AMs106,107,108.
AMs of COPD patients experience a high level of oxidant burden induced through cigarette smoke and subsequent increased ROS production is a key feature109. Compared to controls, COPD-AMs secrete increased levels of mitochondrial ROS (mtROS)110, superoxide and hydrogen peroxide81,108 whilst glutamyl cysteine ligase for GSH synthesis is downregulated111. Cigarette smoking also alters iron homoeostasis112 and AMs in COPD show increased sequestering of iron113, which can furthermore contribute to ROS production. Bewley et al. showed recently that increased generation of mtROS in COPD AMs results in impaired bacterial clearance108. This study also reported a decrease in the mitochondrial membrane potential114, which has recently been linked to AM exposure to particulate matter110. This may explain the impaired phagocytic capacity of AMs in COPD as decreased mitochondrial membrane potential results in energy failure in the cell, proton leakage and increased mtROS115. Another study by O’Beirne et al. further investigated the metabolic profile of AMs from healthy smokers, non-smokers and COPD patients. While all groups had similar baseline glycolysis rates, there was a decrease in coupling efficiency, maximal respiration and spare respiratory capacity in COPD-AMs, while proton leak was significantly increased116. In addition, expression of genes related to glutathione metabolism, mitochondrial transport, pyruvate metabolism, TCA cycle and electron transport chain were altered in smokers and COPD patients, compared to non-smoking healthy controls116.
Other metabolic alterations in COPD AMs include increased expression of inducible nitric oxide synthase (iNOS) contributing to increased levels of nitric oxide (NO)117 and increased levels of the adenosine receptor A2BR118, suggesting increased adenosine metabolism, which might be linked to the increased levels of Hif1α in COPD AMs119. While excessive ROS production through oxidant burden and iron accumulation has been identified as an important regulator of AM phenotype in COPD, it has only recently been linked to mitochondrial dysfunction and metabolic reprogramming. It would be interesting to follow up on these transcriptomic and metabolic alterations to understand their underlying disease driving role and to identify ways to rewire AM metabolism.
As corticosteroids have been found to be particularly ineffective in COPD, more specific pathways involved in AM function and metabolism have been investigated recently, such as the ROS pathway and iron accumulation. A study by Harvey et al. showed that treatment with sulforaphane in COPD AMs ex vivo improved bacterial clearance by activating the antioxidant and anti-inflammatory NRF2 pathway120, while Cloonan et al. found that treatment with an iron chelator or a low iron diet protected mice from cigarette smoke induced COPD121. Furthermore, procysteine, a precursor of GSH, increased AM efferocytosis in a mouse model of COPD122.
Overall, COPD is marked by distinct iron sequestration, ROS, NO production and energetic dysfunction in AMs; further delineation of how mitochondrial phenotype links to inflammatory processes and pathology in COPD will allow the identification of molecular targets for modulating mitochondria during the disease.
Cystic fibrosis
Cystic fibrosis (CF) is caused by mutation of the CF transmembrane conductance regulator (CFTR), a chloride channel, which regulates fluid homoeostasis in mucosal surfaces. In the lung, CFTR mutation and subsequent loss of function results in a reduced aqueous film covering the epithelium and mucus thickening, leading to impaired mucociliary clearance and frequent bacterial infection123. CF is furthermore characterised by hyper-inflammation of the lungs, airway obstruction, structural damage and progressive reduction of lung function124. During recurring airway inflammation, large numbers of neutrophils, macrophages and T lymphocytes infiltrate the lungs and secrete pro-inflammatory cytokines, while anti-inflammatory IL-10 is reduced125,126. Although AM numbers are increased in CF patients127,128, pathogen clearance is attenuated, leading to colonisation of the airways and chronic inflammation106,128. Meyer et al. report a more pro-inflammatory phenotype of AMs in a murine model of CF, even in the absence of infection129 and MDMs differentiated from CF patients show an increased inflammatory profile130, while others have shown that monocytes from CF patients had an impairment in activation upon IL-13 stimulation131. CF-AM phenotype can be heterogenous, depending on infection status and local environment. While AMs from P. Aeruginosa infected CF patients showed increased expression of mannose receptor CD206 and augmented arginase activity132, in CF sputum AMs a decrease in expression of CD206 and scavenger receptor MARCO was detected133. Furthermore, AMs are involved in the structural damage in CF airways by secreting serine- and metalloproteases, which subsequently degrade connective tissue components134. The lower volume of airway surface liquid in CF airways activates AMs to increase their release of MMP12, resulting in the cleavage of elastin and degradation of the airway and parenchyma135.
In CF airways, GSH is depleted136,137, while levels of iron, transferrin, haem and haemoglobin are increased138, resulting in high oxidative stress and ROS production. ROS in turn can induce TGF-β1139, which has recently been shown to be increased in CF-BAL and AMs and inhibits CFTR biogenesis and cellular trafficking to the surface of epithelial cells134, while also contributing to airway remodelling by recruitment and differentiation of myofibroblasts140. However, during infection with bacteria from the Burkholderia family, both MDMs and AMs from CF patients showed reduced superoxide production as well as decreased phosphorylation of NADPH oxidase (NOX) components p47phox and p40phox, suggesting an inherent deficit in CF-AMs generating oxidative bursts for pathogen defence141.
P. Aeruginosa is one of the most common pathogens to cause recurrent pulmonary infection in CF patients and exploits the host to maintain infection by inducing production of the TCA cycle metabolite itaconate in AMs. Itaconate exerts antimicrobial properties via inhibition of bacterial isocitrate lyase in the glyoxylate shunt142 and to evade this mechanism P. Aeruginosa has developed a way to use itaconate as an energy source143. Similarly succinate, which is secreted in high levels during CF and especially during bacterial infection144, can be utilised by P. Aeruginosa and S. Aureus as a substrate to generate oxidative stress.
Changes in lipid metabolism are a hallmark of CF and increased FAO, lipid turnover in cell membranes and eicosanoid production in AMs have been reported. Furthermore, sterol regulatory-element binding protein (SREBP), a regulator of lipid homoeostasis, has been linked to CFTR loss of function124. This results in altered plasma and tissue fatty acid profiles, and while levels of the omega-3 fatty acid docosahexaenoic acid (DHA) were unchanged in AMs upon loss of CFTR145, ex vivo treatment with DHA decreased TNF-α levels146. Furthermore, in CF AMs the anti-inflammatory lipoxin A4 (LXA4), which is synthesised from the fatty acid arachidonic acid, is reduced and the LXA4/LTB4 ratio in CF BAL is decreased147, while the fatty acid metabolite resolvin D1 (RvD1) has been suggested as a biomarker148.
Increased energy demand by AMs in CF, either by manipulation through bacterial pathogens or to fight sustained infection, results in increased utilization of all available metabolic pathways. Recently, Lara-Reyna et al. reported increased glycolysis, mitochondrial function, and production of TNF-α in CF macrophages is due to an alteration in the serine/threonine-protein kinase/endoribonuclease IRE1α pathway and this supports exacerbated inflammation149. While this study used PBMCs and monocyte-derived M1 macrophages from CF patients, it would be important to detect such a mechanism in AMs and to target this pathway specifically.
Several of the above described pathways have been identified as potential drug targets in CF, however yet there are no treatments targeting AMs. Delivery of GSH to the lower respiratory tract improves the antioxidant barrier of CF epithelium150, while treatment with cysteamine and restoration of MicroRNA 17 (MiR17) and MiR20 expression improves disease by restoring autophagy151,152. Several studies administered omega-3 fatty acids (DHA/EPA) to CF patients153,154,155,156, although only one trial reported improved FEV1 after 8 months treatment with DHA157. Treatment with DHA in a murine model of CF decreased liver inflammation but did not improve lung morphology158.
In conclusion, AM metabolic phenotype during CF is marked by increased energy expenditure to support exacerbated inflammation and is readily exploited by bacterial pathogens, leaving AMs deficient of oxidative burst capability during infection. It will be important to clarify the role of fatty acids in CF and furthermore, to target metabolic changes in AMs such as increased glycolysis, OXPHOS and FAO specifically to rewire AM phenotype and prevent exploitation through bacterial pathogens.
Idiopathic pulmonary fibrosis (IPF)
IPF is a chronic interstitial lung disease characterised by excessive extracellular matrix deposition in the lung parenchyma and has a particularly poor prognosis159. Repetitive alveolar injury in genetically susceptible individuals causes activation of mesenchymal cells, recruitment of fibroblasts and differentiation into myofibroblasts to replace damaged alveolar epithelial cells and provide a matrix for wound healing and tissue repair160. During IPF, the wound healing process is dysregulated, leading to fibrotic plaque formation and excessive build-up of extracellular matrix, resulting in impaired gas exchange. AMs have been identified as key contributors to the dysregulated wound healing process, by secreting large amounts of ROS and TGF-β161. Furthermore, AMs can shape the extracellular matrix by secreting factors contributing to the matrix (proline, collagen) and breaking down the matrix (plasmin, MMPs)162,163,164.
Several changes to the central carbon metabolism pathways have been identified recently in AMs of IPF patients, including dysmorphic mitochondria165. In murine models of pulmonary fibrosis, increased glucose consumption, glycolysis and enhanced expression of key glycolytic mediators was detected166, while in IPF AMs, expression of the pulmonary glucose transporter GLUT1 was increased167, which enabled augmented glucose uptake166. The increased glucose uptake via GLUT1 can furthermore sustain NADPH production in the pentose phosphate pathway and TCA cycle168 and is therefore a key substrate for ROS production via NOX169. Activation of macrophages results in the accumulation of endogenous metabolites capable of adopting immunomodulatory roles such as succinate170 and itaconate171,172,173. Recently, our laboratory identified itaconate as an endogenous anti-fibrotic in the human and murine lung. In patients with IPF, there were reduced levels of airway itaconate, and decreased expression of ACOD1 (which controls the synthesis of itaconate) in AMs compared to healthy controls. Acod1 deficiency in mice leads to more severe disease pathology and exogenous itaconate limits fibroblast activity174. These data indicate that AM metabolites may play a key role in the pathogenesis of lung fibrosis and may be exploited for the development of anti-fibrotic therapies.
ROS production is a key feature of AMs in IPF175 and can occur during OXPHOS, by the membrane bound NOX or by reaction of hydrogen peroxide with intracellular iron176. NOX, and subsequent superoxide production, is activated by binding GTP-bound Rac1177, which is secreted from AMs in IPF178 and can also activate the mTOR signalling hub179. Superoxide produced by NOX can further react with NO to form peroxynitrite (OONO-), another type of ROS. At the expense of NADPH, NO is produced in the mitochondria by iNOS, which is upregulated in pro-inflammatory macrophages180 and in IPF-AMs leading to increased levels of the cytotoxic OONO- in IPF AMs181. In the bleomycin mouse model of pulmonary fibrosis, increased levels of superoxide, NO and OONO- were measured in AMs182. MtROS is furthermore linked to expression of PPAR-γ coactivator 1-alpha (PGC-1α), which induces metabolic reprogramming to FAO and is regulated by the mitochondrial calcium uniporter (MCU), which is increased in IPF AMs183. MCU has furthermore been shown to regulate expression of the fatty acid transporter CPT-1, which is increased in AMs from IPF patients and bleomycin exposed mice183. While human IPF-AMs have increased levels of MCU, mitochondrial calcium and expression of PGC-1a, bleomycin exposed mice utilise increased FAO166, which is reduced in mice expressing dominant-negative MCU183. Furthermore, these mice were protected from bleomycin induced pulmonary fibrosis. These findings highlight calcium transport and FAO as pathways to target in IPF AMs; however, a better understanding of the linking mechanism will be necessary.
IPF AMs have also been shown to be iron laden184, which further induces oxidative stress and ROS production185. Using RNA-sequencing, Lee et al. show furthermore, that macrophage activation is increased in iron laden AMs in IPF, suggesting that iron accumulation plays a role in macrophage activation185. The proportion of AMs expressing transferrin receptor (CD71), importing transferrin bound iron into the cell, are decreased in IPF AMs, leading to an extracellular accumulation of transferrin. Furthermore, numbers of CD71-negative macrophages are an independent predictor of survival in IPF186. Iron metabolism is therefore likely a key pathway in IPF-AMs and targeting it would be a viable option to decrease ROS, oxidative stress and macrophage activation.
Recently, therapies targeting metabolic processes in IPF are of considerable interest. While antioxidant therapy in IPF was promising in vivo, the double-blind placebo controlled PANTHER trial, administering either N-acetylcysteine or placebo to IPF patients for 60 weeks did not show a change in lung function parameters187. Another arm of this study investigated the combined potential of corticosteroid prednisone, immunosuppressant azathioprine and N-acetylcysteine but was stopped prematurely due to increased mortality and adverse effects without evidence of benefit188. Another randomized, double-blind clinical trial assessed the safety and tolerability of N-acetylcysteine in patients already receiving pirfenidone anti-fibrotic therapy. While this trial showed that N-acetylcysteine in combination with pirfenidone was safe, no change in FVC, 6-minute walk test or occurrence of adverse effects was detected189. Another promising therapeutic avenue was the use of metformin, a potent metabolic remodelling drug often prescribed for type II diabetes. While on a global level metformin lowers the amount of blood sugar in diabetic patients, on a cellular level metformin activates AMP-activated protein kinase (AMPK) leading to inhibition of TGF-β induced NOX activity190. Sato et al., have shown that metformin inhibited TGF-β induced NOX activity via AMPK leading to inhibition of myo-fibroblast differentiation in vitro and reduced bleomycin induced collagen deposition in vivo191. Consistent with this, Rangarajan et al. showed that metformin treatment reversed bleomycin induced pulmonary fibrosis via AMPK activation, while in IPF patients AMPK phosphorylation was decreased192. A posthoc analysis study of the effect of metformin in IPF patients however showed no change in clinically outcomes193, once again showing the difficulty of translating in vitro and in vivo findings into the clinic. Another study investigating the NOX-NRF2 imbalance as a therapeutic target showed that in vivo knockdown of NOX4 and NOX1/4 inhibition restored the capacity of fibrosis resolution in aged mice194. Furthermore, treatment with nitrated fatty acids, reversed pulmonary fibrosis in a mouse model by promoting collagen uptake by AMs and dedifferentiating myofibroblasts195.
While these treatment approaches targeted metabolic changes during pulmonary fibrosis, none was specific to AMs. Targeting macrophage specific metabolic reprogramming, which sustains ROS and TGF-β production and contributes to dysregulated wound healing in IPF would therefore be a promising approach.
Respiratory tract infections
During respiratory tract infections, activation of pattern recognition receptors expressed by AM can elicit a variety of pro-inflammatory host responses2. For example, severe Coronavirus disease-19 (COVID-19) associated pneumonia patients may exhibit features of systemic hyper-inflammation also known as macrophage activation syndrome or “cytokine storm” which is associated with sustained elevation of macrophage/monocyte-derived pro-inflammatory cytokines (e.g., IL-6, IL-8, TNF-α, IL-1β) leading to acute respiratory distress syndrome (ARDS)196,197,198. Using single cell approaches a recent study demonstrated that highly inflammatory, monocyte recruited AMs, rather than quiescent pulmonary resident AMs, predominate in the BAL in COVID-19 patients with severe pathology, implicating these cells in COVID-19-associated ARDS199. Rather than direct infection of AMs, AM:AEC cross-talk has been identified as a major mechanism for control of many respiratory viral infections200 and AEC have been shown to be a key source of pro-inflammatory cytokines, modulating AM phenotype198,201. For example, Rhinovirus (RV), the causative agent of the common cold, primarily infects the upper airways, however prior infection with RV attenuates subsequent AM antibacterial responses202. Although AMs are susceptible to influenza A viral infection (IAV), replication within AMs has been shown to be minimal with the exception of several highly virulent strains203,204,205. Here, we will focus on Mycobacterium tuberculosis (Mtb) infection, as AMs are the primary infected cell type and metabolic changes in response to Mtb infection are well studied.
Tuberculosis
Tuberculosis (TB) is a contagious, chronic disease and one-third of the world’s population is infected with Mtb, the causative agent of TB, resulting in ~2 million deaths per year (2009 World Health Organization Report)206. During infection, Mtb colonises AMs intracellularly and disables innate intracellular defence mechanisms such as the phagolysosome and inflammasome and accesses macrophage intracellular nutrients207. AM host defence mechanisms against Mtb include production of ROS and reactive nitrogen species (RNS) for bacterial killing and fusing mycobacteria-containing phagosomes with lysosomes as well as autophagy and apoptosis208. However, virulent or multi-drug resistant strains can evade these host responses e.g. by preventing phagolysosome fusion and surviving ROS/RNS209.
During Mtb infection, AMs shift their metabolic programme from OXPHOS to aerobic glycolysis, which is regulated by HIF1α and interferon-gamma (IFN-γ). This metabolic shift and subsequent enhanced glycolytic flux in infected AMs is crucial to control infection. Mice lacking HIF1α in the myeloid lineage are more susceptible to infection and show decreased cytokine and antimicrobial effector production210. To support this metabolic reprogramming, key glycolysis genes are upregulated in the early stages of granuloma formation in mice, supporting the shift towards aerobic glycolysis211. Blocking this shift on the other hand results in decreased levels of IL-1β, increased IL-10 and subsequent increased bacterial survival212. Gleeson et al. suggest furthermore that infection-induced glycolysis limits Mtb survival through the induction of IL-1β during infection with drug susceptible Mtb, as absence or inhibition of the IL-1 receptor (IL-1R) negated the effect of aerobic glycolysis212. In contrast, during infection with multi-drug resistant Mtb, AM metabolic reprogramming and induction of glycolysis is regulated through IFN-β as overexpression of Mtb cell wall lipids blocks the IL-1 receptor type 1 pathway213. These findings suggest that infection-induced glycolysis is necessary for control of bacterial intracellular replication though it remains to be investigated whether glycolytic reprogramming could be further induced to support AM defence.
M. Tuberculosis has developed several mechanisms to evade host defence. Using mice deficient of cystathionine-gamma-lyase (CSE), which catalyses the synthesis of hydrogen sulphide (H2S), H2S has been identified as a regulator for central carbon metabolism during Mtb infection in AM. CSE−/− mice had increased flux through the glycolysis and pentose phosphate pathway in AM, while Mtb infected WT mice produced increased levels of H2S, reducing HIF1α levels, glycolysis and host defence214. Howard et al. show that multi-drug resistant Mtb can drive augmented cell wall lipids synthesis, thereby bypassing the IL-1 receptor pathway and resulting in induction of IFN-β signalling, reprogramming host metabolism213. Furthermore, increased expression of MiR-21 in BMDMs and human MDMs upon exposure with Mtb, resulted in decreased glycolytic response and facilitated bacterial survival by targeting phosphofructokinase isoform M (PFK-M) and limiting IL-1β production. IFN-γ however inhibits MiR-21, forcing an isozyme switch in the PFK-M complex and rescuing glycolysis and host defence215.
Several signalling metabolites have been identified to be important for host defence in AMs during Mtb infection. Decreased levels of fumarase result in accumulation of bactericidal fumarate, which can modify metabolites and proteins through succination216. Furthermore, the antimicrobial metabolite itaconate is increased during Mtb infection, although it can be disabled by β-hydroxylacyl CoA lyase in Mtb and used as a nutrient. Wang et al. show that deletion of this enzyme resulted in attenuated Mtb infection in mice217. Similarly, Mtb exploit intracellular iron as a nutrient. This is highlighted by worse TB outcome with increased dietary ingestion of iron and inhibition of Mtb growth when iron is unavailable218,219. Abreu et al. show that heparin reduced hepcidin expression in macrophages infected with Mtb while heparin-treated macrophages had increased expression of ferroportin and subsequent iron export, limiting iron availability for intracellular bacilli206. Mtb furthermore induce ferroptosis, associated with reduced levels of GSH, superoxide and increased free iron. The ferroptosis inhibitor ferrostatin-1 (Fer-1) as well as iron chelation decreased necrotic cell death of Mtb-infected macrophages in vitro, while in vivo treatment with Fer-1 reduced bacterial load220. Mtb can cope in low iron environments however by downregulating their non-essential protein content via specific sRNA221.
Several changes in fatty acid metabolism of Mtb infected AMs were identified recently. Compared to interstitial macrophages during Mtb infection, which are reliant on glycolysis, AMs utilise FA, which is induced by PPAR-α222 and have a lower burden of Mtb infection223. To escape host defence, Mtb has developed a mechanism inhibiting pathways related to autophagy, lysosomal function and FAO in support of replication by inducing microRNA-33 (MiR-33) in the host cell. Silencing of MiR-33 however induced AM lipid catabolism and autophagy and rescued host defence224. Furthermore, amino acid metabolism is altered during Mtb infection. In mice and macaque lungs, indoleamine 2,3-dioxygenase (IDO), which is involved in tryptophan catabolism, was increased during Mtb infection, while inhibition of IDO in a macaque model of TB decreased bacterial burden and pathology, as tryptophan metabolites suppress host immunity225.
While Mtb relies on host lipids as energy source, existing therapies such as targeting PPAR transcription factors or cholesterol synthesis have been successful mainly in animal models226,227,228,229, whereas retrospective human studies, which investigated the effect of statins in diabetic TB patients did not show any results230. As Mtb can also utilise iron as a substrate, another approach is to prevent iron accumulation. Treatment of Mtb infected human MDMs and primary AM with iron chelator Desferrioxamine (DFX) ex vivo induced the expression of glycolytic enzymes and enhanced glycolysis, as well as IL-1βα, thereby supporting host defence231 and offers a novel therapeutic approach, which will need to be investigated in clinical trials. Together, these findings highlight the distinct phenotype of AMs during Mtb infection, which counteracts intracellular infection through aerobic glycolysis, but is also heavily exploited by Mtb bacteria feeding on host lipids and iron.
Targeting metabolism during chronic lung disease
Many potential targets have been identified recently that could rewire macrophage metabolic and phenotypic changes driving chronic lung disease. Since all cells depend on oxidative phosphorylation or cytoplasmic glycolysis to synthesize ATP, there is the potential for unwanted side effects by targeting specific metabolic processes. However, it is becoming increasingly apparent that it is possible to safely target metabolic pathways in patients. For example, dimethyl fumarate, a known regulator of macrophage phenotype, is a first-line-treatment for relapsing-remitting multiple sclerosis232. Indeed, metabolic processes are highly plastic with significant redundancy, modulation of these processes may have the added benefit of selectively targeting cells with high metabolic demands233. Targeted delivery to AMs may add another layer of selectivity, improving efficacy, sustained drug release and evading capture by mucus234. Systems for inhaled AM targeted drug delivery include the use of micro- and nanocarriers, including liposomes, which are phagocytosed by AMs. Rifampicin-loaded microspheres as a therapeutic approach for Mtb have been described235, and have been further refined to allow a one-step assembly for rifampicin containing microspheres236. Recently, aerosolised delivery of siRNA, which post-translationally downregulates gene expression, has been developed to target AMs specifically237, whilst mannose coated microspheres have been developed which exploit the phagocytotic activity of AMs238. Many of these delivery vehicles have been developed to transport antibiotics targeting intracellular AM bacterial infections, which are helpful for treating TB, however other drugs could be incorporated into aerosolised micro- or nano delivery systems. Specifically, treatment with iron chelators, antioxidants and nitrated fatty acids has shown to rewire AM phenotype and improve diverse chronic lung disease; these may be ideal candidates to develop novel, aerosolised vehicle-assisted drug delivery to AMs during chronic lung disease.
Conclusion
In the last decade enormous strides have been made regarding our understanding of how adaptations in metabolic pathways underlie macrophage phenotype and function. AMs are remarkably plastic cells, orchestrating not only pathogen defence and efferocytosis, but also pulmonary tolerance and resolution. It has become increasingly clear that AMs tailor their metabolic profile to fit their local niche generating ROS for pathogen defence, utilising aerobic glycolysis to rapidly generate cytokines, employing the TCA cycle to fuel inflammatory responses and generating metabolites with secondary signalling functions such as citrate, itaconate, succinate and fumarate. Work elucidating the complexities of AM metabolic alterations in the context of CLDs has highlighted many potential therapeutic targets (summarized in Table 1). Indeed, a lack of understanding of shared cellular mechanisms, which underlie CLDs has been a major obstacle in respiratory biology; identification of common AM-metabolic pathways/metabolites which directly influence core features of CLDs would be a significant advance on the route to devising new AM-directed strategies to treat pulmonary diseases which affect millions worldwide.
References
Lloyd, C. M. & Marsland, B. J. Lung homeostasis: influence of age, microbes, and the immune system. Immunity 46, 549–561 (2017).
Byrne, A. J., Mathie, S. A., Gregory, L. G. & Lloyd, C. M. Pulmonary macrophages: key players in the innate defence of the airways. 1189–1196. https://doi.org/10.1136/thoraxjnl-2015-207020 (2015).
Ginhoux, F. & Jung, S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 14, 392–404 (2014).
Galván-Peña, S. & O’Neill, L. A. J. Metabolic reprograming in macrophage polarization. Front. Immunol. 5, 1–6 (2014).
Bossche, J. Van, den, O’Neill, L. A. & Menon, D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 38, 395–406 (2017).
Tzouvelekis, A. Metabolic disorders in chronic lung diseases. Front. Med. 4, 1–9 (2018).
O’Beirne, S. L. et al. Alveolar macrophage immunometabolism and lung function impairment in smoking and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 125899, 1–14 (2019).
Hussell, T. & Bell, T. J. Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 14, 81–93 (2014).
Lambrecht, B. N. Alveolar macrophage in the driver’s seat. Immunity 24, 366–368 (2006).
Koivisto, L., Bi, J., Häkkinen, L. & Larjava, H. Integrin αvβ6: structure, function and role in health and disease. Int. J. Biochem. Cell Biol. 99, 186–196 (2018).
Byrne, A. J., Maher, T. M. & Lloyd, C. M. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol. Med. 22, 303–316 (2016).
Misharin, A. V., Morales-Nebreda, L., Mutlu, G. M., Budinger, G. R. S. & Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 49, 503–510 (2013).
Zaynagetdinov, R. et al. Identification of myeloid cell subsets in murine lungs using flow cytometry. Am. J. Respir. Cell Mol. Biol. 49, 180–189 (2013).
Guth, A. M. et al. Lung environment determines unique phenotype of alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, 936–946 (2009).
Snelgrove, R. J. et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 9, 1074–1083 (2008).
Schulz, C. et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 336, 86–90 (2012).
Guilliams, M. et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 210, 1977–1992 (2013).
Hashimoto, D. et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804 (2013).
van de Laar, L. et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity 44, 755–768 (2016).
Svedberg, F. R. et al. The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nat. Immunol. 20, 571–580 (2019).
Ginhoux, F. & Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449 (2016).
Tan, S. Y. S. & Krasnow, M. A. Developmental origin of lung macrophage diversity. Development 143, 1318–1327 (2016).
Patel, A. A. et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med 214, 1913–1923 (2017).
Shi, C. & Pamer, E. G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11, 762–774 (2011).
Gibbings, S. L. et al. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood 126, 1357–1366 (2015).
Bittmann, I. et al. Cellular chimerism of the lung after transplantation: an interphase cytogenetic study. Am. J. Clin. Pathol. 115, 525–533 (2001).
Eguíluz-Gracia, I. et al. Long-term persistence of human donor alveolar macrophages in lung transplant recipients. Thorax 71, 1006–1011 (2016).
Nayak, D. K. et al. Long-term persistence of donor alveolar macrophages in human lung transplant recipients that influences donor specific immune responses. Am. J. Transpl. 16, 2300–2311 (2017).
Hunninghake, G. W. et al. The human alveolar macrophage. Methods Cell Biol. 21, 95–112 (1980).
Thomas, E. D., Ramberg, R. E., Sale, G. E., Sparkes, R. S. & Golde, D. W. Direct evidence for a bone marrow origin of the alveolar macrophage in man. Science 192, 1016–1018 (1976).
Byrne, A. J. et al. Dynamics of human monocytes and airway macrophages during healthy aging and after transplant. J. Exp. Med 217, 1–11 (2020).
Metcalf, T. U. et al. Human monocyte subsets are transcriptionally and functionally altered in aging in response to pattern recognition receptor agonists. J. Immunol. 199, 1405–1417 (2017).
Hearps, A. C. et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 11, 867–875 (2012).
Seidler, S., Zimmermann, H. W., Bartneck, M., Trautwein, C. & Tacke, F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 11, 30 (2010).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013).
Mills, E. L. et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e13 (2016).
Huang, S. C.-C. et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 15, 846–855 (2014).
Jha, A. K. et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015).
Kelly, B. & O’Neill, L. A. J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25, 771–784 (2015).
Woods, P. S. et al. Tissue-resident alveolar macrophages do not rely on glycolysis for LPS-induced inflammation. Am. J. Respir. Cell Mol. Biol. 62, 243–255 (2020).
Schneider, C. et al. Induction of the nuclear receptor PPAR-γ 3 by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 15, 1026–1037 (2014).
Sano, H. et al. Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J. Immunol. 163, 387–395 (1999).
Trapnell, B. C. & Whitsett, J. A. GM-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol. 64, 775–802 (2002).
Shibata, Y. et al. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 15, 557–567 (2001).
Baker, A. D. et al. Targeted PPARγ deficiency in alveolar macrophages disrupts surfactant catabolism. J. Lipid Res. 51, 1325–1331 (2010).
Suzuki, T. et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J. Exp. Med 205, 2703–2710 (2008).
Tanaka, T. et al. Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB. J. Med. Genet. 48, 205–209 (2011).
Suzuki, T. et al. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur. Respir. J. 37, 201–204 (2011).
Martinez-Moczygemba, M. et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRα gene in the X chromosome pseudoautosomal region 1. J. Exp. Med 205, 2711–2716 (2008).
Haczku, A. Protective role of the lung collectins surfactant protein A and surfactant protein D in airway inflammation. J. Allergy Clin. Immunol. 122, 861–879 (2008). quiz 880–881.
Watford, W. T., Wright, J. R., Hester, C. G., Jiang, H. & Frank, M. M. Surfactant protein A regulates complement activation. J. Immunol. 167, 6593–6600 (2001).
Yamada, C. et al. Surfactant protein A directly interacts with TLR4 and MD-2 and regulates inflammatory cellular response: Importance of supratrimeric oligomerization. J. Biol. Chem. 281, 21771–21780 (2006).
Kato, K. et al. Membrane-tethered MUC1 mucin counter-regulates the phagocytic activity of macrophages. Am. J. Respir. Cell Mol. Biol. 54, 515–523 (2016).
Roy, M. G. et al. Muc5b is required for airway defence. Nature 505, 412–416 (2014).
Silva, M. A. & Bercik, P. Macrophages are related to goblet cell hyperplasia and induce MUC5B but not MUC5AC in human bronchus epithelial cells. Lab. Investig. 92, 937–948 (2012).
Seibold, M. A. et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 364, 1503–1512 (2011).
Surowiec, I. et al. Multi-platform metabolomics assays for human lung lavage fluids in an air pollution exposure study. Anal. Bioanal. Chem. 408, 4751–4764 (2016).
Huffnagle, G. B., Dickson, R. P. & Lukacs, N. W. The respiratory tract microbiome and lung inflammation: A two-way street. Mucosal Immunol. 10, 299–306 (2017).
Invernizzi, R., Lloyd, C. M. & Molyneaux, P. L. Respiratory microbiome and epithelial interactions shape immunity in the lungs. Immunology 160, 171–182 (2020).
Kumpitsch, C., Koskinen, K., Schöpf, V. & Moissl-Eichinger, C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 17, 1–20 (2019).
Bassis, C. M., Tang, A. L., Young, V. B. & Pynnonen, M. A. The nasal cavity microbiota of healthy adults. Microbiome 2, 27 (2014).
Peng, L., Li, Z.-R., Green, R. S., Holzman, I. R. & Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 139, 1619–1625 (2009).
Postler, T. S. & Ghosh, S. Understanding the holobiont: how microbial metabolites affect human health and shape the immune system. Cell Metab. 26, 110–130 (2017).
Man, W. H., De Steenhuijsen Piters, W. A. A. & Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 15, 259–270 (2017).
Loftus, R. M. & Finlay, D. K. Immunometabolism: cellular metabolism turns immune regulator. J. Biol. Chem. 291, 1–10 (2016).
The European Respiratory Society. The burden of lung disease. 2–15 (2010).
Leung, J. M. & Sin, D. D. Asthma-COPD overlap syndrome: pathogenesis, clinical features, and therapeutic targets. 1–14. https://doi.org/10.1136/bmj.j3772 (2017).
Kusko, R. L. et al. Integrated genomics reveals convergent transcriptomic networks underlying chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 194, 948–960 (2016).
Chilosi, M., Poletti, V. & Rossi, A. The pathogenesis of COPD and IPF: distinct horns of the same devil? Respir. Res. 13, 3 (2012).
Kurai, D., Saraya, T., Ishii, H. & Takizawa, H. Virus-induced exacerbations in asthma and COPD. Front. Microbiol. 4, 1–12 (2013).
Kurt, O. K., Zhang, J. & Pinkerton, K. E. Pulmonary health effects of air pollution. Curr. Opin. Pulm. Med. 22, 138–143 (2016).
Byrne, A. J., Jones, C. P., Gowers, K., Rankin, S. M. & Lloyd, C. M. Lung macrophages contribute to house dust mite driven airway remodeling via HIF-1α. PLoS One 8, 1–10 (2013).
Nagakumar, P. et al. Type 2 innate lymphoid cells in induced sputum from children with severe asthma. J. Allergy Clin. Immunol. 137, 624–626 (2016).
Krausgruber, T. et al. IRF5 promotes inflammatory macrophage polarization and T H1-TH17 responses. Nat. Immunol. 12, 231–238 (2011).
Jenkins, S. J. et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288 (2011).
Byrne, A. J. et al. A critical role for IRF5 in regulating allergic airway inflammation. Mucosal Immunol. 10, 716–726 (2017).
Draijer, C. et al. Human asthma is characterized by more IRF5+ M1 and CD206+ M2 macrophages and less IL-10+ M2-like macrophages around airways compared with healthy airways. J. Allergy Clin. Immunol. 140, 280–283.e3 (2017).
Huynh, M. L. N. et al. Defective apoptotic cell phagocytosis attenuates prostaglandin E 2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am. J. Respir. Crit. Care Med. 172, 972–979 (2005).
Lappalainen, U., Whitsett, J. A., Wert, S. E., Tichelaar, J. W. & Bry, K. Interleukin-1β causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol. 32, 311–318 (2005).
Calhoun, W. J., Bush, R. K., Salisbury, S. M. & Stevens, C. A. Enhanced reactive oxygen species metabolism of airspace cells and airway inflammation follow antigen challenge in human asthma. J. Allergy Clin. Immunol. 86, 306–313 (1990).
Lee, I. & Yang, C. Role of NADPH oxidase / ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 84, 581–590 (2012).
Park, H. S., Kim, S. R. & Lee, Y. C. Impact of oxidative stress on lung diseases. Respirology 14, 27–38 (2009).
Harju, T., Soini, Y., Pääkkö, P. & Kinnula, V. L. Up-regulation of heme oxygenase-1 in alveolar macrophages of newly diagnosed asthmatics. Respir. Med. 96, 418–423 (2002).
Al-Khami, A. A. et al. Fuelling the mechanisms of asthma: increased fatty acid oxidation in inflammatory immune cells may represent a novel therapeutic target. Clin. Exp. Allergy 47, 1170–1184 (2017).
Chavis, C., Godard, P., Michel, F. B., Paulet, A. Cde & Damon, M. Sulfidopeptide leukotrienes contribute to human alveolar macrophage activation in asthma. Prostaglandins, Leukot. Essent. Fat. Acids 42, 95–100 (1991).
Damon, M. et al. Increased generation of the arachidonic metabolites LTB4 and 5-HETE by human alveolar macrophages in patients with asthma: effect in vitro of nedocromil sodium. Eur. Respir. J. 2, 202–209 (1989).
Arm, J., O’hickey, S., Spur, B., Dis, T. L.-A. R. R. & 1989, undefined Airway responsiveness to histamine and leukotriene E4 in subjects with aspirin-induced asthma. atsjournals.org at https://www.atsjournals.org/doi/pdf/10.1164/ajrccm/140.1.148.
Mayatepek, E. et al. Synthesis and metabolism of leukotrienes in γ-glutamyl transpeptidase deficiency. J. Lipid Res. 45, 900–904 (2004).
Vercelli, D. Arginase: Marker, effector, or candidate gene for asthma? J. Clin. Invest. 111, 1815–1817 (2003).
Chang, C.-I., Zoghi, B., Liao, J. C. & Kuo, L. The involvement of tyrosine kinases, cyclic AMP/protein kinase A, and p38 mitogen-activated protein kinase in IL-13-mediated arginase I induction in macrophages: its implications in IL-13-inhibited nitric oxide production. J. Immunol. 165, 2134–2141 (2000).
Lai, C. K. W., Phillips, G. D., Jenkins, J. R. & Holgate, S. T. The effect of inhaled 15-(s)-hydroxyeicosatetraenoic acid (15-HETE) on airway calibre and non-specific responsiveness in normal and asthmatic human subjects. Eur. Respir. J. 3, 38–45 (1990).
Bhavsar, P. K. et al. Corticosteroid suppression of lipoxin A4and leukotriene B4from alveolar macrophages in severe asthma. Respir. Res. 11, 1–9 (2010).
Wenzel, S. E., Trudeau, J. B., Westcott, J. Y., Beam, W. R. & Martin, R. J. Single oral dose of prednisone decreases leukotriene B4 production by alveolar macrophages from patients with nocturnal asthma but not control subjects: Relationship to changes in cellular influx and FEV1. J. Allergy Clin. Immunol. 94, 870–881 (1994).
Lee, K. S. et al. A novel thiol compound, N-acetylcysteine amide, attenuates allergic airway disease by regulating activation of NF-κB and hypoxia-inducible factor-1α. Exp. Mol. Med. 39, 756–768 (2007).
Seidel, P. & Roth, M. A potential new therapy for asthma? Mediators Inflamm. 2013, 1–10 (2013).
Gregory, L. G. & Lloyd, C. M. Orchestrating house dust mite-associated allergy in the lung. Trends Immunol. 32, 402–411 (2011).
Cushen, B. & Menzies-Gow, A. Benralizumab: an updated treatment of eosinophilic asthma. Expert Rev. Respir. Med. 14, 435–444 (2020).
Mannino, D. M. & Buist, A. S. Global burden of COPD: risk factors, prevalence, and future trends. Lancet 370, 765–773 (2007).
Lopez, A. D. et al. Chronic obstructive pulmonary disease: current burden and future projections. Eur. Respir. J. 27, 397–412 (2006).
Brusselle, G. G., Joos, G. F. & Bracke, K. R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 378, 1015–1026 (2011).
Ni, L. & Dong, C. Roles of myeloid and lymphoid cells in the pathogenesis of chronic obstructive pulmonary disease. Front. Immunol. 9, 1–12 (2018).
Barnes, P. J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 138, 16–27 (2016).
Shibata, S. et al. Basophils trigger emphysema development in a murine model of COPD through IL-4–mediated generation of MMP-12–producing macrophages. Proc. Natl Acad. Sci. USA 115, 13057–13062 (2018).
Grumelli, S. et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 1, 075–083 (2004).
Russell, R. E. K. et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 26, 602–609 (2002).
Donnelly, L. E. & Barnes, P. J. Defective phagocytosis in airways disease. Chest 141, 1055–1062 (2012).
Grabiec, A. M. & Hussell, T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin. Immunopathol. 38, 409–423 (2016).
Bewley, M. A. et al. Impaired mitochondrial microbicidal responses in chronic obstructive pulmonary disease macrophages. Am. J. Respir. Crit. Care Med. 196, 845–855 (2017).
Rahman, I. & MacNee, W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic. Biol. Med. 21, 669–681 (1996).
Xia, T., Kovochich, M. & Nel, A. E. Impairment of mitochondrial function by particulate matter (PM) and their toxic components: Implications for PM-induced cardiovascular and lung disease. Front. Biosci. 12, 1238–1246 (2007).
Harju, T., Kaarteenaho-Wiik, R., Soini, Y., Sormunen, R. & Kinnula, V. L. Diminished immunoreactivity of γ-glutamylcysteine synthetase in the airways of smokers’ lung. Am. J. Respir. Crit. Care Med. 166, 754–759 (2002).
Cloonan, S. M. et al. The iron-y of iron overload and iron deficiency in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 196, 1103–1112 (2017).
Philippot, Q. et al. Increased iron sequestration in alveolar macrophages in chronic obtructive pulmonary disease. PLoS One 9, 1–9 (2014).
Belchamber, K. B. R. et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur. Respir. J. 54, 1802244 (2019).
Eapen, M. S., Sharma, P. & Sohal, S. S. Mitochondrial dysfunction in macrophages: A key to defective bacterial phagocytosis in COPD. Eur. Respir. J. 54, 1901641 (2019).
O’Beirne, S. L. et al. Alveolar macrophage immunometabolism and lung function impairment in smoking and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 201, 735–738 (2020).
Ichinose, M., Sugiura, H., Yamagata, S., Koarai, A. & Shirato, K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am. J. Respir. Crit. Care Med. 162, 701–706 (2000).
Zhou, Y., Murthy, J. N., Zeng, D., Belardinelli, L. & Blackburn, M. R. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One 5, e9224 (2010).
Russell, K. E. et al. The MIF antagonist ISO-1 attenuates corticosteroid-insensitive inflammation and airways hyperresponsiveness in an ozone-induced model of COPD. PLoS One 11, 1–17 (2016).
Harvey, C. J. et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 3, 78ra32 (2011).
Cloonan, S. M. et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat. Med. 22, 163–174 (2016).
Hodge, S. et al. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am. J. Respir. Cell Mol. Biol. 44, 673–681 (2011).
Wine, J. J. et al. Progress in understanding mucus abnormalities in cystic fibrosis airways. J. Cyst. Fibros. 17, S35–S39 (2018).
Dhooghe, B., Noël, S., Huaux, F. & Leal, T. Lung inflammation in cystic fibrosis: pathogenesis and novel therapies. Clin. Biochem. 47, 539–546 (2014).
Roesch, E. A., Nichols, D. P. & Chmiel, J. F. Inflammation in cystic fibrosis: an update. Pediatr. Pulmonol. 53, S30–S50 (2018).
Nichols, D. P. & Chmiel, J. F. Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 50, S39–S56 (2015).
Brennan, S. et al. Alveolar macrophages and CC chemokines are increased in children with cystic fibrosis. Eur. Respir. J. 34, 655–661 (2009).
Dakin, C. J. et al. Inflammation, infection, and pulmonary function in infants and young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 165, 904–910 (2002).
Meyer, M. et al. Azithromycin reduces exaggerated cytokine production by M1 alveolar macrophages in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 41, 590–602 (2009).
Bruscia, E. M. et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/-mice. Am. J. Respir. Cell Mol. Biol. 40, 295–304 (2009).
Tarique, A. A. et al. CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J. Cyst. Fibros. 16, 475–482 (2017).
Murphy, B. S. et al. Characterization of macrophage activation states in patients with cystic fibrosis. J. Cyst. Fibros. 9, 314–322 (2010).
Wright, A. K. A. et al. Pivotal Advance: Expansion of small sputum macrophages in CF: failure to express MARCO and mannose receptors. J. Leukoc. Biol. 86, 479–489 (2009).
Snodgrass, S. M., Cihil, K. M., Cornuet, P. K., Myerburg, M. M. & Swiatecka-Urban, A. Tgf-β1 inhibits Cftr biogenesis and prevents functional rescue of ΔF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS One 8, e63167 (2013).
Gaggar, A. et al. Series ‘matrix metalloproteinases in lung health and disease’: the role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir. J. 38, 721–727 (2011).
Morris, D. et al. Glutathione and infection. Biochim. Biophys. Acta - Gen. Subj. 1830, 3329–3349 (2013).
Roum, J. H., Buhl, R., McElvaney, N. G., Borok, Z. & Crystal, R. G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 75, 2419–2424 (1993).
Ghio, A. J. et al. Iron accumulates in the lavage and explanted lungs of cystic fibrosis patients. J. Cyst. Fibros. 12, 390–398 (2013).
Koli, K., Myllärniemi, M., Keski-Oja, J. & Kinnula, V. L. Transforming growth factor-β activation in the lung: Focus on fibrosis and reactive oxygen species. Antioxid. Redox Signal 10, 333–342 (2008).
Gauthier, T. W. et al. Impaired defenses of neonatal mouse alveolar macrophage with cftr deletion are modulated by glutathione and TGFβ1. Physiol. Rep. 5, 1–12 (2017).
Assani, K. et al. Human cystic fibrosis macrophages have defective calcium-dependent protein kinase C activation of the NADPH oxidase, an effect augmented by burkholderia cenocepacia. J. Immunol. 198, 1985–1994 (2017).
Nair, S. et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J. Exp. Med 215, 1035–1045 (2018).
Riquelme, S. A. et al. Pseudomonas aeruginosa utilizes host-derived itaconate to redirect its metabolism to promote biofilm formation. Cell Metab. 31, 1091–1106 (2020).
Riquelme, S. A., Wong Fok Lung, T. & Prince, A. Pulmonary pathogens adapt to immune signaling metabolites in the airway. Front. Immunol. 11, 1–14 (2020).
Worgall, T. S. Lipid metabolism in cystic fibrosis. Curr. Opin. Clin. Nutr. Metab. Care 12, 105–109 (2009).
Andersson, C., Zaman, M. M., Jones, A. B. & Freedman, S. D. Alterations in immune response and PPAR/LXR regulation in cystic fibrosis macrophages. J. Cyst. Fibros. 7, 68–78 (2008).
Ringholz, F. C. et al. Reduced 15-lipoxygenase 2 and lipoxin A4/leukotriene B4 ratio in children with cystic fibrosis. Eur. Respir. J. 44, 394–404 (2014).
Eickmeier, O. et al. Pro-resolving lipid mediator Resolvin D1 serves as a marker of lung disease in cystic fibrosis. PLoS One 12, 1–12 (2017).
Lara-Reyna, S. et al. Metabolic reprograming of cystic fibrosis macrophages via the IRE1α arm of the unfolded protein response results in exacerbated inflammation. Front. Immunol. 10, 1789 (2019).
Roum, J. H. et al. Glutathione aerosol suppresses lung epithelial surface inflammatory cell-derived oxidants in cystic fibrosis. J. Appl. Physiol. 87, 438–443 (1999).
Tazi, M. F. et al. Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy 12, 2026–2037 (2016).
Ferrari, E. et al. Cysteamine re-establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis-relevant F508del-CFTR mutation. Cell Death Dis. 8, 1–11 (2017).
Coste, T. C. et al. An overview of monitoring and supplementation of omega 3 fatty acids in cystic fibrosis. Clin. Biochem. 40, 511–520 (2007).
Al-Turkmani, M. R., Freedman, S. D. & Laposata, M. Fatty acid alterations and n-3 fatty acid supplementation in cystic fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 77, 309–318 (2007).
Biervliet, S. Van et al. Oral DHA supplementation in ΔF508 homozygous cystic fibrosis patients. Prostaglandins Leukot. Essent. Fat. Acids 78, 109–115 (2008).
Lloyd-Still, J. D. et al. Bioavailability and safety of a high dose of docosahexaenoic acid triacylglycerol of algal origin in cystic fibrosis patients: a randomized, controlled study. Nutrition 22, 36–46 (2006).
Vizia, B. D. E. et al. Effect of an 8-month treatment with ω-3 fatty acids (eicosapentaenoic and docosahexaenoic) in patients with cystic fibrosis. J. Parenter. Enter. Nutr. 27, 52–57 (2003).
Beharry, S. et al. Long-term docosahexaenoic acid therapy in a congenic murine model of cystic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G839–G848 (2007).
Raghu, G. Idiopathic pulmonary fibrosis: Guidelines for diagnosis and clinical management have advanced from consensus-based in 2000 to evidence-based in 2011. Eur. Respir. J. 37, 743–746 (2011).
Selman, M. & Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis: An integral model. Am. J. Respir. Crit. Care Med. 189, 1161–1172 (2014).
Zhang, L. et al. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 19, 1–10 (2018).
Mahalanobish, S., Saha, S., Dutta, S. & Sil, P. C. Matrix metalloproteinase: An upcoming therapeutic approach for idiopathic pulmonary fibrosis. Pharmacol. Res. 152, 104591 (2020).
Willems, S. et al. Multiplex protein profiling of bronchoalveolar lavage in idiopathic pulmonary fibrosis and hypersensitivity pneumonitis. Ann. Thorac. Med. 8, 38–45 (2013).
Dancer, R. C. A., Wood, A. M. & Thickett, D. R. Metalloproteinases in idiopathic pulmonary fibrosis. Eur. Respir. J. 38, 1461–1467 (2011).
Tsitoura, E. et al. Accumulation of damaged mitochondria in alveolar macrophages with reduced OXPHOS related gene expression in IPF. Respir. Res. 20, 1–16 (2019).
Xie, N. et al. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L834–L844 (2017).
El-Chemaly, S. et al. Glucose transporter-1 distribution in fibrotic lung disease: Association with [18F]-2-fluoro-2-deoxyglucose-PET scan uptake, inflammation, and neovascularization. Chest 143, 1685–1691 (2013).
Lewis, C. A. et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 55, 253–263 (2014).
Babior, B. M. NADPH oxidase. Curr. Opin. Immunol. 16, 42–47 (2004).
Mills, E. & O’Neill, L. A. J. Succinate: a metabolic signal in inflammation. Trends Cell Biol. 24, 313–320 (2014).
Cordes, T. et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J. Biol. Chem. 291, 14274–14284 (2016).
Mills, E. L. et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 (2018).
Lampropoulou, V. et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166 (2016).
Ogger, P. P. et al. Itaconate controls the severity of pulmonary fibrosis. Sci. Immunol. 5, eabc1884 (2020).
Gonzalez-Gonzalez, F. J., Chandel, N. S., Jain, M. & Budinger, G. R. S. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl. Res. 190, 61–68 (2017).
Soares, M. P. & Hamza, I. Macrophages and iron metabolism. Immunity 44, 492–504 (2016).
Bedard, K. & Krause, K. H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 87, 245–313 (2007).
Murthy, S. et al. Modulation of reactive oxygen species by Rac1 or catalase prevents asbestos-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, 846–855 (2009).
Saci, A., Cantley, L. C. & Carpenter, C. L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 42, 50–61 (2011).
Fung, E. et al. Delta-like 4 induces Notch signaling in macrophages: implications for inflammation. Circulation 115, 2948–2956 (2007).
Saleh, D., Barnes, P. J. & Giaid, A. Increased production of the potent oxidant peroxynitrite in the lungs of patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 155, 1763–1769 (1997).
Yamazaki, C. et al. Production of superoxide and nitric oxide by alveolar macrophages in the bleomycin-induced interstitial pneumonia mice model. Jpn. J. Pharmacol. 78, 69–73 (1998).
Gu, L. et al. Mitochondrial calcium uniporter regulates PGC-1α expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol. 26, 101307 (2019).
Puxeddu, E. et al. Iron laden macrophages in idiopathic pulmonary fibrosis: the telltale of occult alveolar hemorrhage? Pulm. Pharmacol. Ther. 28, 35–40 (2014).
Lee, J. et al. Bronchoalveolar lavage (BAL) cells in idiopathic pulmonary fibrosis express a complex pro-inflammatory, pro-repair, angiogenic activation pattern, likely associated with macrophage iron accumulation. PLoS One 13, 1–15 (2018).
Allden, S. J. et al. The transferrin receptor CD71 delineates functionally distinct airway macrophage subsets during idiopathic pulmonary fibrosis.Am J. Respir. Crit. Care Med. 200, 209–219 (2019).
Oldham, J. M. et al. TOLLIP, MUC5B, and the response to N-acetylcysteine among individuals with idiopathic pulmonary fibrosis.Am J. Respir. Crit. Care Med. 192, 1475–1482 (2015).
Raghu, G., Anstrom, K. J., King, T. E., Lasky, J. A. & Martinez, F. J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 366, 1968–1977 (2012).
Behr, J. et al. Safety and tolerability of acetylcysteine and pirfenidone combination therapy in idiopathic pulmonary fibrosis: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 4, 445–453 (2016).
Santos, C. C., dos, Gopal, B. & Verma, S. Metformin: an old dog with a new trick? Cell Metab. 28, 334–336 (2018).
Sato, N. et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir. Res. 17, 1–12 (2016).
Rangarajan, S. et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 24, 1121 (2018).
Spagnolo, P. et al. Metformin does not affect clinically relevant outcomes in patients with idiopathic pulmonary fibrosis. Respiration 96, 314–322 (2018).
Hecker, L. et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 6, 231ra47 (2014).
Reddy, A. T., Lakshmi, S. P., Zhang, Y. & Reddy, R. C. Nitrated fatty acids reverse pulmonary fibrosis by dedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEB J. 28, 5299–5310 (2014).
Li, H. et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet 2019, 8–11 (2020).
Giamarellos-Bourboulis, E. J. et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe https://doi.org/10.1016/j.chom.2020.04.009 (2020).
Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 (2020).
Liao, M. et al. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv https://doi.org/10.1101/2020.02.23.20026690 (2020).
Yoshikawa, T., Hill, T., Li, K., Peters, C. J. & Tseng, C.-T. K. Severe acute respiratory syndrome (SARS) coronavirus-induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J. Virol. 83, 3039–3048 (2009).
McGonagle, D., Sharif, K., O’Regan, A. & Bridgewood, C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun. Rev. 19, 102537 (2020).
Oliver, B. G. G. et al. Rhinovirus exposure impairs immune responses to bacterial products in human alveolar macrophages. Thorax 63, 519–525 (2008).
Perrone, L. A., Plowden, J. K., García-Sastre, A., Katz, J. M. & Tumpey, T. M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 4, e1000115 (2008).
Rodgers, B. & Mims, C. A. Interaction of influenza virus with mouse macrophages. Infect. Immun. 31, 751–757 (1981).
Wells, M. A., Albrecht, P., Daniel, S. & Ennis, F. A. Host defense mechanisms against influenza virus: interaction of influenza virus with murine macrophages in vitro. Infect. Immun. 22, 758–762 (1978).
Abreu, R., Essler, L., Loy, A., Quinn, F. & Giri, P. Heparin inhibits intracellular Mycobacterium tuberculosis bacterial replication by reducing iron levels in human macrophages. Sci. Rep. 8, 1–12 (2018).
Etna, M. P., Giacomini, E., Severa, M. & Coccia, E. M. Pro-and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis. Semin. Immunol. 26, 543–551 (2014).
Ehrt, S. & Schnappinger, D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol 11, 1170–1178 (2009).
Xu, S. et al. Intracellular trafficking in mycobacterium tuberculosis and mycobacterium avium-infected macrophages. J. Immunol. 153, 2568–2578 (1994).
Braverman, J., Sogi, K. M., Benjamin, D., Nomura, D. K. & Stanley, S. A. HIF-1α Is an essential mediator of IFN-γ–dependent immunity to mycobacterium tuberculosis. J. Immunol. 197, 1287–1297 (2016).
Shi, L. et al. Infection with mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci. Rep. 5, 1–13 (2015).
Gleeson, L. E. et al. Cutting edge: mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J. Immunol. 196, 2444–2449 (2016).
Howard, N. C. et al. Mycobacterium tuberculosis carrying a rifampicin drug resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Nat. Microbiol. 3, 1099–1108 (2018).
Rahman, M. A. et al. Hydrogen sulfide dysregulates the immune response by suppressing central carbon metabolism to promote tuberculosis. Proc. Natl Acad. Sci. USA 117, 6663–6674 (2020).
Hackett, E. E. et al. Mycobacterium tuberculosis limits host glycolysis and IL-1β by restriction of PFK-M via MicroRNA-21. Cell Rep. 30, 124–136.e4 (2020).
Ruecker, N. et al. Fumarase deficiency causes protein and metabolite succination and intoxicates mycobacterium tuberculosis. Cell Chem. Biol. 24, 306–315 (2017).
Wang, H. et al. An essential bifunctional enzyme in Mycobacterium tuberculosis for itaconate dissimilation and leucine catabolism. Proc. Natl Acad. Sci. USA. 116, 15907–15913 (2019).
Boelaert, J. R., Vandecasteele, S. J., Appelberg, R. & Gordeuk, V. R. The effect of the host’s iron status on tuberculosis. J. Infect. Dis. 195, 1745–1753 (2007).
Olakanmi, O., Schlesinger, L. S. & Britigan, B. E. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J. Leukoc. Biol. 81, 195–204 (2007).
Amaral, E. P. et al. A major role for ferroptosis in mycobacterium tuberculosis–induced cell death and tissue necrosis. J. Exp. Med 216, 556–570 (2019).
Gerrick, E. R. et al. Small RNA profiling in mycobacterium tuberculosis identifies mrsi as necessary for an anticipatory iron sparing response. Proc. Natl Acad. Sci. USA 115, 6464–6469 (2018).
Kim, Y. S. et al. PPAR-α activation mediates innate host defense through induction of TFEB and lipid catabolism. J. Immunol. 198, 3283–3295 (2017).
Huang, L., Nazarova, E. V., Tan, S., Liu, Y. & Russell, D. G. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med 215, 1135–1152 (2018).
Ouimet, M. et al. Mycobacterium tuberculosis induces the MIR-33 locus to reprogram autophagy and host lipid metabolism. Nat. Immunol. 17, 677–686 (2016).
Gautam, U. S. et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA 115, E62–E71 (2018).
Almeida, P. E. et al. Mycobacterium bovis bacillus Calmette-Guérin infection induces TLR2-dependent peroxisome proliferator-activated receptor γ expression and activation: functions in inflammation, lipid metabolism, and pathogenesis. J. Immunol. 183, 1337–1345 (2009).
Rajaram, M. V. S. et al. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor γ linking mannose receptor recognition to regulation of immune responses. J. Immunol. 185, 929–942 (2010).
Korf, H. et al. Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J. Clin. Invest. 119, 1626–1637 (2009).
Parihar, S. P. et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J. Infect. Dis. 209, 754–763 (2014).
Kang, Y. A. et al. The effects of statin use on the development of tuberculosis among patients with diabetes mellitus. Int. J. Tuberc. Lung Dis. 18, 717–724 (2014).
Phelan, J. J. et al. Desferrioxamine supports metabolic function in primary human macrophages infected with mycobacterium tuberculosis. Front. Immunol. 11, 1–18 (2020).
Carlström, K. E. et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat. Commun. 10, 3081 (2019).
Pålsson-McDermott, E. M. & O’Neill, L. A. J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. https://doi.org/10.1038/s41422-020-0291-z (2020).
Makled, S., Boraie, N. & Nafee, N. Nanoparticle-mediated macrophage targeting—a new inhalation therapy tackling tuberculosis. Drug Deliv. Transl. Res. https://doi.org/10.1007/s13346-020-00815-3 (2020).
Suarez, S. et al. Respirable PLGA microspheres containing rifampicin for the treatment of tuberculosis: Screening in an infectious disease model. Pharm. Res. 18, 1315–1319 (2001).
Ohashi, K., Kabasawa, T., Ozeki, T. & Okada, H. One-step preparation of rifampicin/poly(lactic-co-glycolic acid) nanoparticle-containing mannitol microspheres using a four-fluid nozzle spray drier for inhalation therapy of tuberculosis. J. Control. Release 135, 19–24 (2009).
Dua, K. et al. The potential of siRNA based drug delivery in respiratory disorders: Recent advances and progress. Drug Dev. Res. 80, 714–730 (2019).
Chen, J. et al. Glycan targeted polymeric antibiotic prodrugs for alveolar macrophage infections. Biomaterials 195, 38–50 (2019).
Henkel, F. D. R. et al. House dust mite drives proinflammatory eicosanoid reprogramming and macrophage effector functions. Allergy Eur. J. Allergy Clin. Immunol. 74, 1090–1101 (2019).
Ye, Q. et al. Decreased expression of haem oxygenase-1 by alveolar macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 31, 1030–1036 (2008).
Larson-Casey, J. L., Deshane, J. S., Ryan, A. J., Thannickal, V. J. & Carter, A. B. Macrophage Akt1 Kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 44, 582–596 (2016).
Stüve, P. et al. De novo fatty acid synthesis during mycobacterial infection is a prerequisite for the function of highly proliferative T cells, but not for dendritic cells or macrophages. Front. Immunol. 9, 1–22 (2018).
Koeken, V. A. C. M. et al. Role of glutamine metabolism in host defense against mycobacterium tuberculosis infection. J. Infect. Dis. 219, 1662–1670 (2019).
Qualls, J. E. & Murray, P. J. Immunometabolism within the tuberculosis granuloma: amino acids, hypoxia, and cellular respiration. Semin. Immunopathol. 38, 139–152 (2016).
Vrieling, F. et al. Analyzing the impact of mycobacterium tuberculosis infection on primary human macrophages by combined exploratory and targeted metabolomics. Sci. Rep. 10, 1–13 (2020).
Acknowledgements
We thank Clare M. Lloyd for critically reading this review and providing valuable feedback. P.P.O. is supported by a Studienstiftung des Deutschen Volkes fellowship, A.J.B. is supported by a Joan Bending, Evelyn Bending, Mervyn Stephens + Olive Stephens Memorial fellowship (AUK-SNF-2017-381).
Author information
Authors and Affiliations
Contributions
P.P.O. and A.J.B. conceived of and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ogger, P.P., Byrne, A.J. Macrophage metabolic reprogramming during chronic lung disease. Mucosal Immunol 14, 282–295 (2021). https://doi.org/10.1038/s41385-020-00356-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-020-00356-5
This article is cited by
-
Role of the protease-activated receptor-2 (PAR2) in the exacerbation of house dust mite-induced murine allergic lung disease by multi-walled carbon nanotubes
Particle and Fibre Toxicology (2023)
-
Macrophages in immunoregulation and therapeutics
Signal Transduction and Targeted Therapy (2023)
-
Gut colonisation with multidrug-resistant Klebsiella pneumoniae worsens Pseudomonas aeruginosa lung infection
Nature Communications (2023)
-
Caspase-8 activation by cigarette smoke induces pro-inflammatory cell death of human macrophages exposed to lipopolysaccharide
Cell Death & Disease (2023)
-
Cigarette smoke exposed airway epithelial cell-derived EVs promote pro-inflammatory macrophage activation in alpha-1 antitrypsin deficiency
Respiratory Research (2022)
