
Abstract
The gene encoding for Epstein-Barr virus-induced G-protein-coupled receptor 2 (EBI2) is a risk gene for inflammatory bowel disease (IBD). Together with its oxysterol ligand 7α,25-dihydroxycholesterol, EBI2 mediates migration and differentiation of immune cells. However, the role of EBI2 in the colonic immune system remains insufficiently studied. We found increased mRNA expression of EBI2 and oxysterol-synthesizing enzymes (CH25H, CYP7B1) in the inflamed colon of patients with ulcerative colitis and mice with acute or chronic dextran sulfate sodium (DSS) colitis. Accordingly, we detected elevated levels of 25-hydroxylated oxysterols, including 7α,25-dihydroxycholesterol in mice with acute colonic inflammation. Knockout of EBI2 or CH25H did not affect severity of DSS colitis; however, inflammation was decreased in male EBI2−/− mice in the IL-10 colitis model. The colonic immune system comprises mucosal lymphoid structures, which accumulate upon chronic inflammation in IL-10-deficient mice and in chronic DSS colitis. However, EBI2−/− mice formed significantly less colonic lymphoid structures at baseline and showed defects in inflammation-induced accumulation of lymphoid structures. In summary, we report induction of the EBI2-7α,25-dihydroxycholesterol axis in colitis and a role of EBI2 for the accumulation of lymphoid tissue during homeostasis and inflammation. These data implicate the EBI2-7α,25-dihydroxycholesterol axis in IBD pathogenesis.
Similar content being viewed by others

Introduction
Inflammatory bowel diseases (IBD), with the main forms Crohn’s disease (CD) and ulcerative colitis (UC), are chronic inflammatory conditions of the human gut. The pathogenesis of IBD is incompletely understood, but genetic and environmental factors were shown to contribute to disease development and progression. Genome wide association studies (GWAS) have identified more than 240 genetic regions in the human genome affecting the risk for IBD.1,2 The majority of IBD-specific single nucleotide polymorphisms (SNPs) confer an increased risk for both, CD and UC.1 Genes identified by GWAS provide a framework for future scientific studies addressing IBD pathogenesis.
Epstein-Barr virus-induced G-protein-coupled receptor 2 (EBI2, also known as GPR183), is an IBD risk gene identified by GWAS.1 EBI2 exerts a crucial function for the correct activation and maturation of naïve B cells in secondary lymphoid organs.3,4,5 Oxysterols are ligands for EBI2, the most potent being 7α,25-dihydroxycholesterol (7α,25-diHC).6,7 7α,25-diHC acts as a chemoattractant, directing migration of EBI2 expressing B cells, T cells, and DCs.6,7,8,9 A 7α,25-diHC gradient in secondary lymphoid organs seems to be important for correct positioning of immune cells and a rapid and efficient antibody response.10 7α,25-diHC is produced from cholesterol via two hydroxylation steps, at position 25 and 7α, by the enzymes cholesterol 25-hydroxylase (CH25H) and cytochrome P450 family 7 subfamily member B1 (CYP7B1), respectively.
Besides B cells and DCs, EBI2 is also expressed in macrophages and natural killer cells,6 and CH25H and CYP7B1 are expressed in immune cells and many tissues including lymph nodes, lung, and colon.6,11 Therefore, the oxysterol-EBI2 axis might constitute a fundamental mechanism in the regulation of the immune system and tissue homeostasis.
Intestinal immune responses are orchestrated in lymphoid tissue localized directly in the intestinal tract and draining lymph nodes. Local lymphoid tissues show considerable plasticity, varying in organization and cellular composition depending on the segments of the gut and the immune status. The colonic immune system comprises two types of secondary lymphoid organs: large colonic patches (CLP), similar to Peyer’s patches (PP) in the small intestine, and smaller structures referred to as solitary intestinal lymphoid tissue (SILT).12 CLP and PP develop before birth, whereas SILT develop strictly postnatally. SILT comprise a continuum of lymphoid structures ranging from nascent small immature cryptopatches (CP) to mature isolated lymphoid follicles (ILF) containing B cells.
General principles of secondary lymphoid tissue formation are shared between mesenteric lymph nodes and intestinal lymphoid tissues including CLP/PP and SILT (reviewed in ref. 13) Development of these lymphoid tissues includes clustering of lymphoid tissue inducer (LTi) cells, a subclass of innate lymphoid cells (ILC), and requires an intact lymphotoxin signaling pathway. Most previous studies focused on the development of lymphoid tissue in the small intestine; in contrast, formation of SILT in the colon has been studied to a lower extent and specific factors required beyond lymphotoxin remain unknown. While in the small intestine, maturation of SILT depends on CXCL13, RANKL, and CCR6/CCL20, these chemokines are not essential in the colon.12,14 Furthermore, while the gut microbiota stimulates small intestinal SILT, the microbiota dampens SILT formation in the colon, an effect mediated by IL-25 and IL-23.15 Therefore, additional molecular factors besides those cytokines seem to be required, and very recently, a role of EBI2 expressing ILCs for the development of colonic lymphoid structures has been demonstrated.16
In the colon, adaptation to inflammation includes formation of additional lymphoid structures. Induction of newly formed lymphoid structures upon inflammation requires the presence of microbiota and lymphotoxin signaling but was independent from the nuclear hormone receptor ROR-γt,17 suggesting that ILCs and LTi cells are not strictly necessary. SILT were proposed to host a flexible pool of B cells for the formation of an IgA response complementing PP and CLP with a fixed B cell pool size.18 However, the role of SILT in colon inflammation has not been clarified.
Given the function of EBI2 in immune cell migration, we aimed at investigating the role of EBI2 and oxysterols in the pathogenesis of intestinal inflammation and the development of colonic lymphoid tissues. Our results implicate the EBI2-oxysterol axis in colonic SILT development and inflammatory responses in the colon.
Results
Human intestinal lymphocytes express EBI2
To test for surface expression of EBI2 on intestinal immune cells, colon lamina propria mononuclear cells (LPMC) from patients undergoing intestinal surgery were analyzed by flow cytometry. We observed robust EBI2 expression on various subsets of B and T cells. As reported previously, expression levels were highest in memory CD4+ T cells (Fig. 1a) in human peripheral blood mononuclear cells (PBMC).19 EBI2 expression was also observed in PBMCs without significant differences between healthy volunteers and IBD patients (CD and UC, Fig. 1b). Direct comparison of LPMCs with identical cellular subsets of PBMCs revealed significantly higher EBI2 expression in the gut (p = 0.0001).

High EBI2 surface expression in human intestinal lymphocytes and upregulated gene expression of EBI2 and oxysterol-synthesizing enzymes in inflamed intestinal tissue. (a) EBI2 expression on human LPMCs of colon resections shown as mean fluorescence intensity ratio (MFIR) in naïve (CD19+CD27−) and memory (CD19+CD27+) subsets of B cells and in naïve (CD45RA+) and memory (CD45RA−) CD4+ and CD8+ T cells determined by FACS analysis. (b) Percentages of memory CD4+ T cells expressing EBI2 on human PBMCs from healthy volunteers (HV) and CD and UC patients and on human LPMCs of colon resections determined by FACS analysis. (c) Data from a human whole genome microarray (GEO data sets: GDS3268) was analyzed regarding mRNA expression levels of EBI2, CH25H and CYP7B1 in non-inflamed colon tissue of healthy volunteers (n = 63) and non-inflamed (n = 61) and inflamed (n = 62) colon tissue of UC patients. The dotted line represents the mean of healthy tissue (set to 1). (d) mRNA expression levels from rectal biopsies of UC patients, either non-inflamed from patients with quiescent disease activity (n = 24) or inflamed from patients with moderate to severe disease activity (n = 20) from the Swiss IBD cohort study were determined by RT-PCR and normalized to GAPDH using the ΔΔCt method. Data shown as mean ± SEM. Statistical analysis: (a): One-way ANOVA with Dunnetts test; (b-d): Mann–Whitney U test; *p < 0.05, ***p < 0.001, ****p < 0.0001
Upregulation of gene expression of EBI2 and oxysterol-synthesizing enzymes in inflamed tissue of UC patients
We found an inflammation dependent upregulation of the EBI2-oxysterol axis in the gut. Results of a whole human genome microarray performed with total RNA isolated from inflamed and non-inflamed intestinal tissue from UC patients (GEO data sets: GDS3268)20 were analyzed. RNA expression levels of the oxysterol-synthesizing enzymes CH25H, CYP7B1, and CYP27A1 from inflamed tissue of UC patients were significantly higher compared to non-inflamed tissue of UC patients (p < 0.05 and p < 0.001) and tissue of healthy controls (p < 0.001 and p < 0.0001; Fig. 1c and Supplementary Figure S1a). A similar increase was observed for the oxysterol receptor EBI2 (p < 0.001 for inflamed vs. healthy). Expression levels of HSD3B7 remained unchanged in all three sample groups (Supplementary Figure S1a).
To validate these results, we analyzed colon biopsy samples from inflamed and non-inflamed tissue of UC patients from the Swiss IBD cohort study (SIBDCS). Biopsy samples were obtained from patients with moderate to severe or quiescent UC disease activity, respectively (Supplementary Table S1).
SIBDCS samples confirmed an upregulation of CH25H (p < 0.01), CYP7B1 (p < 0.0001), and EBI2 (p < 0.0001) mRNA levels in inflamed tissue (Fig. 1d). mRNA expression levels of genes encoding proinflammatory cytokines (TNF, IFNG, and IL1B) were used to confirm the severity of colonic inflammation (Supplementary Figure S1b). Expression of genes of the EBI2-oxysterol axis also depended on clinical parameters: In a multivariate analysis, EBI2 expression was related to UC activity (modified Truelove and Witts activity index) while expression of CH25H and CYP7B1 was related to endoscopic activity and a history of past or current TNF treatment (p < 0.05; Supplementary Table S2).
Expression levels of CYP7B1, EBI2, and TNF strongly correlated (r ≥ 0.6, p < 0.001 for all correlations) and CYP7B1 expression correlated with CH25H (r = 0.46, p < 0.05), suggesting an upregulation of the EBI2-oxysterol system in active UC in parallel with the critical cytokine TNF (Supplementary Table S3). However, TNF does not seem to directly affect EBI2 expression since treatment of PBMCs with TNF did not affect EBI2 surface expression (Supplementary Figure S1c-d).
Increased gene expression of EBI2 and oxysterol-synthesizing enzymes in murine DSS colitis
To further study the function of the EBI2-oxysterol system in gut inflammation, we induced acute and chronic dextran sulfate sodium (DSS) colitis in mice. In acute colitis, expression levels of Ebi2, Ch25h, and Cyp7b1 were significantly increased (p < 0.001; Fig. 2a). This increase was more pronounced than in human samples (Fig. 1c, d), potentially reflecting the more acute and severe inflammation in DSS colitis. In chronic DSS colitis, robust upregulation of Ebi2 (p < 0.05), Ch25h (p < 0.05), and Cyp7b1 (p < 0.0001) was evident even though the increase was weaker in chronic than in acute inflammation, reminiscent of the human situation (Fig. 2b). No significant changes were observed in Cyp27a1 and Hsd3b7 expression in both acute and chronic colitis (Supplementary Figure S2a, b).

Increased expression levels of oxysterol-synthesizing enzymes CH25H and CYP7B1 accompanied by elevated oxysterol levels in murine DSS colitis. Acute and chronic DSS colitis was induced; on day 8 (acute colitis) or 80 (chronic colitis) mice underwent colonoscopy and were sacrificed to obtain tissue samples. (a) mRNA expression levels of Ch25h, Cyp7b1 and Ebi2 from colon tissue of wild-type mice with acute colitis and (b) chronic colitis and water controls were determined by RT-PCR and normalized to GAPDH using the ΔΔCt method. (c) Oxysterol levels from acute DSS colitis experiments were measured by LC-MS/MS. Upper panel: Oxysterol levels in colon tissue of inflamed wild-type (n = 4) and inflamed CH25H−/− mice (n = 3) with wild-type water controls (n = 3). Lower panel: Oxysterol levels in liver tissue of inflamed wild-type (n = 12) and inflamed CH25H−/− mice (n = 6) and respective water controls (n = 12/ n = 6). (d) Oxysterol levels from chronic DSS colitis. Upper panel: Colon tissue from wild-type mice (DSS: n = 6, water: n = 6). # indicates measurements, which were on the limit of detection (grey). Lower panel: Liver tissue of inflamed wild-type (n = 6) and inflamed CH25H−/− (n = 5) mice and respective water controls (n = 5/ n = 6). Data from acute and chronic DSS colitis are pooled from two independent experiments each. Data shown as mean ± SEM. Statistical analysis: Mann–Whitney U test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Increased oxysterol levels in murine colitis
To determine the functional outcome of altered CH25H and CYP7B1 expression during inflammation, we compared oxysterol concentrations between mice with acute DSS colitis and controls. Eight oxysterol derivatives (hydroxycholesterol: HC; dihydroxylated cholesterols: diHC) were measured by mass spectrometry: 7α,25-diHC, 7β,25-diHC, 25-HC, 7α,27-diHC, 7β,27-diHC, 27-HC, 24S-HC, and 7α,24-diHC (Fig. 2c, d and Supplementary Figure S2c-f).
We found a trend for higher levels of 25-HC, 7α,25-diHC, and 7α,27-diHC in colon tissue of DSS-treated mice with acute colitis compared to untreated controls (Fig. 2c, upper panel) and significantly higher levels of 25-HC and 7α,25-diHC in liver tissue of inflamed versus untreated mice (Fig. 2c, lower panel). In both, colon and liver tissue knockout of CH25H led to lower levels of 25-hydroxylated oxysterols whereas lack of EBI2 did not influence any of the liver oxysterols (Fig. 2c and Supplementary Table S4).
In a multivariate linear regression analysis of liver oxysterol levels (Supplementary Table S4) controlling for DSS, CH25H, and EBI2 genotype, DSS treatment increased the levels of all 25-hydroxylated oxysterols (25-HC, 7α,25-diHC, 7β,25-diHC) and all 24-hydroxylated oxysterols (24S-HC, 7α,24-diHC; p ≤ 0.0015 for all compounds). In contrast, inflammation did not seem to change 7α- or 27-hydroxylation activity. In the same multivariate regression analysis, CH25H knockout significantly decreased all 25-hydroxylated oxysterols (7α,25-diHC, 7β,25-diHC, and 25-HC, p ≤ 0.01 for all compounds). However, as expected, the effect of CH25H knockout was not absolute and high levels of all 25-hydroxylated oxysterols remained, most likely due to the 25-hydroxylation activity of other enzymes including CYP27A1, CYP46A1, and CYP3A4.21 Intestinal and liver gene expression levels of all analyzed oxysterol-related enzymes did not increase predictive value of the analysis (data not shown). A multivariate analysis for colon oxysterol measurements was not possible due to a low number of observations.
Our data thus indicate pronounced changes in liver oxysterol levels upon induction of acute colitis and CH25H knockout. Of note, inflammation increased levels of the EBI2 ligand 7α,25-diHC (p = 0.0015) while CH25H knockout decreased its concentration (p < 0.0001).
In chronic DSS colitis, oxysterol levels were not significantly elevated in colon or liver tissue (Fig. 2d), in agreement with only mild upregulation of oxysterol-producing enzymes (Fig. 2b).
Lack of EBI2 and CH25H does not affect severity of inflammation in the DSS colitis model
Despite the upregulation of the EBI2-oxysterol system in acute and chronic DSS colitis, knockout of neither EBI2 nor CH25H substantially decreased histological or endoscopical scoring of inflammation (Fig. 3a–d, Supplementary Figure S3a–c, and submitted manuscript.22) For CH25H−/− mice we observed a trend towards slightly increased inflammation indicated by significantly higher endoscopic colitis scores in acute DSS colitis (affecting all sub-scores, Supplementary Figure S3b). Of note, in chronic colitis we also observed slightly increased inflammation, but for unclear reasons only the histological score with all sub-scores was affected (submitted manuscript.22) Reboldi et al. reported a 25-HC dependent suppression of Il1b expression and inflammasome activity.23 However, in our experiments, increased inflammation in CH25H−/− mice could not be explained by increased Il1b mRNA expression (Supplementary Figure S3d, e). Similar to wild-type animals, CH25H and EBI2 knockout mice showed increased expression levels of Ch25h, Cyp7b1, and Ebi2 in acute and chronic DSS colitis (data not shown). Taken together, in the DSS-induced colitis model, the activity of EBI2 and CH25H was not essential.

Lack of EBI2 and CH25H does not affect severity of inflammation in the DSS colitis model. (a) Histological scores for acute DSS colitis were determined on HE-stained colon sections (scale bars: 200 µm) in wild-type and EBI2−/− mice (b), and wild-type and CH25H −/− mice (c). (d) Histological scores for chronic DSS colitis in EBI2−/− mice were determined accordingly. Data from acute and chronic DSS colitis are pooled from two independent experiments each. Data shown as mean ± SEM. Statistical analysis: Mann–Whitney U test; *p < 0.05, **p < 0.01
EBI2 promotes colon inflammation in the IL-10 colitis model
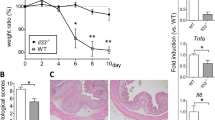
Genetic defects of IL-10 or its receptor have been linked to human IBD,24 hence we also addressed the role of EBI2 in the IL-10 model of spontaneous colitis. For this aim, we generated EBI2−/−IL10−/− mice and compared inflammatory activity to EBI2+/+IL10−/− (in the following named IL10−/−) littermate controls. In 200-days-old mice, the histological colitis score of male EBI2−/−IL10−/− mice was significantly reduced compared to IL10−/− controls (p < 0.01; Fig. 4a, b). Reduced spleen weight in these EBI2−/−IL10−/− animals (p < 0.05) confirmed reduced systemic inflammation in EBI2−/−IL10−/− males (Fig. 4c). Effects of EBI2 on colon inflammation were restricted to male animals, in females no EBI2-dependent changes were detected (Supplementary Figure S4a–c).

EBI2 promotes inflammation in the IL-10 colitis model. EBI2−/−IL10−/− and IL10−/− male mice were sacrificed after onset of rectal prolapse or at the age of 120 or 200 days. Histological scoring (a) and representative images (b) from HE-stained colon sections from 200 days old mice. Scale bars: 200 µm. (c) Spleen weight of animals from (a). (d) Onset of prolapse in EBI2−/−IL10−/− and IL10−/− mice depicted in a survival curve. Each animal represents an independent observation from continuous breeding > 12 months. Data shown as mean ± SEM. Statistical analysis: Mann–Whitney U test; *p < 0.05, **p < 0.01
A minority of IL10−/− animals developed rectal prolapses due to colonic inflammation; however, time to develop prolapse did not differ between EBI2−/−IL10−/− and IL10−/− in male and female animals (Fig. 4d, Supplementary Figure S4c). The genotype also did not affect colon inflammation of animals with prolapse (Fig. 4a).
Testing expression levels of a panel of immune regulatory genes in 200-days-old male EBI2−/−IL10−/− and IL10−/− mice revealed similar expression of most cytokines and T cell regulatory genes except Tbx21 and Il23 for which expression was significantly higher in EBI2−/−IL10−/− mice (Supplementary Figure S5a, b). Expression levels of enzymes regulating the concentration of EBI2 ligands (CH25H, CYP7B1, CYP27A1, and HSD3B7) did not differ significantly between IL10−/− and EBI2−/−IL10−/− mice (Supplementary Figure S5c, d).
EBI2 is required for a normal number of colonic SILT
Since EBI2 affects the localization of immune cells in secondary lymphatic organs, we speculated that EBI2 knockout might also alter the distribution of immune cells in the colon. For quantification of lymphoid structures in whole colons, B cells were visualized by B220 staining in a whole mount approach. The number of B220+ structures was strongly reduced in EBI2−/− mice in comparison to wild-type littermate controls (p < 0.05, Fig. 5a, b). This difference was much stronger than experimental variation (i.e., effects of cages). Stratifying B220+ structures by size revealed a strongly reduced number of small and intermediate B220+ structures (<20’000 μm2: p = 0.004; <100’000 μm2: p < 0.0001) while the number of large B220+ structures (>100’000 μm2) remained unchanged (Fig. 5c). Furthermore, classification into multifollicular or single structures revealed lower numbers for both types in EBI2−/− mice, but with a much stronger reduction for single structures, likely representing isolated lymphoid follicles (ILF) belonging to the group of SILT (Fig. 5d).

Lack of EBI2 leads to a lower number of lymphoid structures in the colon. Colonic lymphoid structures of 12 weeks old female EBI2−/− and wild-type littermate mice were assessed using complementary approaches. (a) B220 B cell staining in a whole mounted colon of wild-type and EBI2−/− mice (B220: white). (b) Quantification of B cell follicles in B220-stained whole mounted colons. (c) Stratification of B220+ structures from (b) by size (area). ## = p < 0.01 and #### = p < 0.0001 comparing structures < 20’000 µm2 and < 100’000 µm2 respectively with a multivariate Poisson regression using model-based t-tests. (d) Classification of B220+ structures from (b) by presence or absence of multiple follicles per structure. (e) HE-stained colon Swiss rolls (left) and representative SILT and CLP structures stained with HE (middle) and B220 and αSMA (right); scale bars: 200 µm. (f) Lymphoid structures were quantified and categorized according to their location in 20 HE-stained colon Swiss roll sections per mouse. Scale bars: 200 µm. Data shown as mean ± SEM. Statistical analysis: Mann–Whitney U test; *p < 0.05
Lymphoid structures can also be identified on HE-stained colon “Swiss rolls”, where SILT are clearly distinguishable from CLP: SILT locate in the lamina propria, CLP between the two muscular layers and the muscularis mucosae12 (Fig. 5e). This approach also allows for the visualization of small lymphoid structures including cryptopatches, largely devoid of B cells. While the number of SILT in EBI2−/− animals was reduced (p < 0.05), the number of large CLP remained unchanged (Fig. 5f).
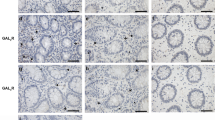
Immunohistochemical stainings of the colon revealed normal structures of both SILT and CLP in EBI2−/− animals with a normal distribution of B and T cells and c-kit+ LTi cells recapitulating the situation of wild-type mice (Fig. 6). Taken together, our data suggest that EBI2 does not seem to be required for maturation of SILT since the few detected lymphoid structures in EBI2−/− animals were indistinguishable from lymphoid structures in wild-types.

Similar lymphoid structures (CLP and SILT) in wild-type and EBI2−/− mice. Cellular characterization of CLP and SILT with immunohistochemical staining of colon Swiss rolls for c-kit (brown) and B220 (pink; please note unspecific staining of alkaline phosphatase at the top of crypts) and with immunofluorescent staining for B220 (green) and CD3 (red). Scale bars: 200 µm, images are representative from at least 3 mice
Development of intestinal lymphoid structures is a complex process involving ROR-γt-expressing ILCs, cytokines, and chemokines. However, colonic expression of RORC and chemokines important for lympho-organogenesis and lymphocyte recruitment including CCL20, CXCL13, and CCL19 were not altered in EBI2−/− compared to wild-type mice (Supplementary Figure S6a) suggesting that the effect of EBI2 is direct and not mediated via altered expression of any of the tested chemokines.
EBI2 deficiency does not affect levels of B cells, IgA, and microbiota composition
To further assess effects of EBI2 knockout on the colonic immune system, we quantified B and T cells by FACS. Overall, the fraction of B and T cells in the colon, mesenteric lymph nodes, and spleen of EBI2−/− animals was similar to wild-types (Supplementary Figure S6b, c). Similarly, the substantial reduction of SILT numbers in EBI2−/− mice did not significantly affect fecal IgA levels (Supplementary Figure S6d). Furthermore, overall microbiota composition of wild-type and EBI2−/− animals was indistinguishable, and the microbiota of EBI2−/− or wild-type animals resembled more strongly the microbiota of wild-type littermates from the same cage than animals of the same genotype housed in a different cage (data not shown). Finally, the fraction of colonic bacteria covered by IgA was similar in wild-type and EBI2−/− animals (Supplementary Figure S6e). Overall, these experiments indicate an intact intestinal adaptive immune system and microbiota composition in the non-inflamed colon of EBI2−/− animals.
EBI2 promotes an increase of the number of colonic lymphoid structures in intestinal inflammation
To test for effects of EBI2 on the formation of lymphoid structures during inflammation, we quantified lymphoid structures in mice with chronic DSS colitis. As previously reported, in wild-type mice with DSS colitis, the number of colonic lymphoid structures was approximately two-fold higher compared to control animals (p < 0.05; Fig. 7a, b). Furthermore, in EBI2−/− animals the number of lymphoid structures did not significantly increase upon DSS treatment (p = 0.07, Fig. 7a) and remained well below levels observed in inflamed wild-type colons even though severity of inflammation was comparable between wild-type and EBI2−/− mice (Fig. 3b, d). In CH25H−/− mice the number of lymphoid structures at baseline was also lower compared to wild-type animals (p < 0.05; Fig. 7b). However, DSS-induced colonic inflammation increased the number of lymphoid structures in CH25H−/− mice almost to wild-type levels. Therefore, CH25H activity seems necessary for normal development of lymphoid structures at baseline but not for an increase in chronic inflammation. No increase in lymphoid structures was observed in acute DSS colitis (Supplementary Figure S7a, b).

EBI2 promotes an increase in the number of colonic lymphoid structures in chronic intestinal inflammation. Lymphoid structures were quantified in representative HE-stained colon sections. (a) Quantification of lymphoid structures in female wild-type (n = 6) and EBI2−/− mice (n = 6) with chronic DSS colitis and water controls (n = 6 each). (b) As in (a), but with wild-type (n = 6) and CH25H−/− mice (n = 5) and water controls (n = 5 / n = 6). (c) Quantification of lymphoid structures in EBI2−/−IL10−/− and IL10−/− male mice at the indicated ages or after occurrence of prolapse. The dotted line indicates the mean number of lymphoid structures in wild-type animals at the age of approximately 200 days. (d) Correlation of the numbers of lymphoid structures with histological scoring of 120 (grey) and 200 days old (black) EBI2−/−IL10−/− and IL10−/− male mice. (e, f) Cellular characterization of lymphoid structures (e): SILT and (f): CLP with immunofluorescent staining of colon Swiss rolls for B220 (green; left panels) or c-kit (green; right panels), CD3 (red) and DAPI (blue) in wild-type and IL10−/− mice at the age of 200 days. Scale bars: 200 µm. Data from chronic DSS colitis (a and b) are pooled from two independent experiments each. Data from IL-10 colitis: each animal represents an independent observation from continuous breeding > 12 months. Data shown as mean ± SEM. Statistical analysis: Mann–Whitney U test; *p < 0.05, **p < 0.01, ****p < 0.0001; correlation analysis: Spearman R
Compared to untreated wild types, the number of lymphoid structures was increased in IL10−/− mice (>three-fold; Fig. 7c and Supplementary Figure S8a), and the number of lymphoid structures strongly correlated with the level of intestinal inflammation in IL10−/− animals (Fig. 7d). However, this was not the case in EBI2−/−IL10−/− mice, which had clearly decreased numbers of colonic lymphoid structures compared to IL10−/− mice (p < 0.05 and p < 0.0001, at day 120 and 200, respectively). Furthermore, inflammation did not significantly increase the number of lymphoid structures in EBI2−/−IL10−/− animals, suggesting that EBI2 is required for efficient accumulation of lymphoid structures during inflammation (Fig. 7c, d and Supplementary Table S5). The cellular composition of gut mucosal lymphoid structures in IL10−/− mice was very similar to the structures seen in wild-type mice (Fig. 7e, f).
In IL10−/− mice, accumulation of lymphoid structures was clearly accompanied by an increase in IgA levels in fecal colon extracts. However, this effect was much lower in EBI2−/− animals (Supplementary Figure S8b-c), due to a reduced number of SILTs and/ or other effects of missing EBI2 function. Taken together, our data indicate an essential role of EBI2 in the accumulation of lymphoid structures upon colonic inflammation.
Discussion
This study assessed the role of EBI2 and oxysterols for the development of colonic lymphoid tissue and human and murine colitis. Key observations of our study include: i) The EBI2-7α,25-diHC axis is upregulated in colitis, indicated by mRNA measurements of EBI2 and oxysterol-synthesizing enzymes in human and murine colon samples and oxysterol levels in mice. ii) EBI2 is required for efficient formation of solitary intestinal lymphoid tissue (SILT) in the mouse colon. iii) EBI2 is required for accumulation of lymphoid tissue in chronic mouse colitis. iv) EBI2 increases the severity of colitis in the IL-10 colitis model but not in acute or chronic DSS colitis.
We show an activation of the EBI2-oxysterol axis in colon inflammation using mRNA and direct oxysterol measurements. Analogous to previous studies, our results demonstrate that inflammation increased CYP7B1 and CH25H mRNA expression and levels of all 25-hydroxylated oxysterols tested, including the EBI2 ligand 7α,25-diHC.11,25,26,27,28 Inflammation did not affect concentrations of most 7α-hydroxylated oxysterols, even though Cyp7b1 mRNA levels were induced 10-fold. Effects of inflammation on enzyme expression and oxysterols levels were less pronounced in chronic DSS colitis. No oxysterol measurements for the IL-10 colitis models are available, which is a limitation of our study. Yet, very recently an independent study observed a similar increase of oxysterol levels both in other mouse models of colitis and in human colitis cohorts.29
Knockout of the 7α,25-diHC receptor EBI2 decreased the number of colonic lymphoid structures approximately five-fold, as also demonstrated very recently.16 Further, the number of lymphoid structures was reduced in CH25H−/− animals, independently confirming a role of the EBI2-7α,25-diHC axis in colonic lymphoid tissue formation. SILT in EBI2−/− and wild-type animals were morphologically indistinguishable, suggesting that EBI2 promotes induction but is not essential for subsequent maturation of SILT. Further, our results indicate an intact IgA response with intact bacterial IgA coating in homeostasis in EBI2−/− mice with a reduced number of SILTs.
Our results agree with the recent paper by Emgård et al., describing EBI2-dependent lymphoid tissue formation during homeostasis.16 Emgård et al. showed EBI2 expression by ILC with a LTi phenotype. LTi cells migrate towards a 7α,25-diHC gradient in vitro and EBI2 knockout reduced the number of cryptopatches and ILF (i.e., immature and mature SILT). Emgård et al. also demonstrated expression of CH25H and CYP7B1 by CD34-Podoplanin+ fibroblasts as a likely source for 7α,25-diHC production, which would attract EBI2+ LTi-cells.16
There is agreement that chronic inflammation increases the number of intestinal lymphoid tissues in the DSS colitis model.17,30,31 However, in a carefully performed study by Olivier et al., three large colonic lymphoid structures were induced in inflammation in three distinct locations in the colon.31 In contrast, in a study by Lochner et al, up to 30 lymphoid structures were found in inflamed colonic tissue,17 similar to our experiments. The reason for this discrepancy remains unclear but might be related to differences in the intestinal microbiota or different scoring systems used in these studies.
Our study shows that accumulation of lymphoid structures in chronic DSS colitis strongly depends on EBI2 activity. Similarly, in IL-10 colitis, the level of inflammation increased the number of colonic lymphoid structures and EBI2 knockout abolished accumulation of lymphoid structures in inflammation (Supplementary Table S5). In contrast, CH25H knockout did not significantly affect the number of lymphoid structures in inflammation, suggesting that CH25H activity is not limiting for immune cell recruitment. In inflammation, we found an increase of the other EBI2 ligand 7α,27-diHC produced by CYP27A1 in colon tissue (Figure 2c). 7α,27-diHC might replace 7α,25-diHC as a chemoattractant in intestinal inflammation, as 7α,27-diHC has been shown to act as an efficient EBI2 ligand for the positioning of dendritic cells in the spleen.32 However, we cannot completely rule out that other enzymes with 25-hydroxylation activity can replace CH25H. In any case, future studies with CH25H/ CYP27A1 double knockouts are warranted. In previous studies, several requirements for induction of lymphoid tissue in inflammation, including a role of lymphotoxin, IL-22, IL-23, and CXCL13 have been described.17,33,34 Our study identifies EBI2 as a further molecule promoting the accumulation of lymphoid tissue in chronic inflammation.
Even though lymphoid structures uniformly accompany colonic inflammation, it is unclear whether their accumulation promotes colon inflammation or whether it is a reaction to chronic inflammation, potentially enabling downregulation of inflammation, or induction of tolerance.35 Some experimental evidence for proinflammatory effects of SILT in murine colitis has been published: ROR-γt deficient mice displayed more colonic SILT and more severe DSS colitis compared to wild–type. Vice versa, reduction of the number of SILT by lymphotoxin neutralization decreased colitis severity.17 A similar correlation between intestinal lymphoid tissue accumulation and severity of inflammation was observed in the TNFΔARE model. Interference by anti-CCR7 treatment further increased lymphoid tissue formation and severity of inflammation.36
Our results argue against an essential proinflammatory role of lymphoid structures in the chronic DSS and IL-10 colitis model: Severity of inflammation in chronic DSS colitis was identical in EBI2 wild-type and knockout animals. Further, EBI2 knockout did not reduce inflammation in female IL10−/− animals even though the number of lymphoid structures was decreased to a similar level as in males.
In contrast to DSS colitis, EBI2 knockout reduces inflammation in IL-10 colitis; however, EBI2 deficiency only reduced inflammation in male mice. This gender dependent dimorphism of EBI2−/−IL10−/− animals has parallels in the literature. Other sexual dimorphisms regarding colitis severity have been related to various factors including microbiota37 and colon cellular infiltrate38 or direct of effects of estradiol.39 Further, in a recent study with the murine TNFΔARE model, protection from ileitis was observed in male, but not female mice with a specific pathogen-free flora.40 Strikingly, for a genetic polymorphism within the human IL10 gene, a sexual dimorphism regarding the risk of ulcerative colitis has been described.41 For the enzyme CYP7B1, producing the EBI2 ligand 7α,25-diHC, differential expression in mouse livers (male > females) has been described;42 however, our analysis did not reveal relevant gender specific differences of all oxysterol producing enzymes tested (Supplementary Figure S5c and data not shown). Further, the number of lymphoid structures was similar in both sexes. Taken together, the mechanism for the sexual dimorphism of the EBI2 knockout in IL-10 colitis regarding inflammation cannot be explained by our experiments, which remains a further limitation of our study.
Further, EBI2 knockout only affected IL-10 colitis but not acute or chronic DSS colitis, likely reflecting different mechanisms of inflammation.43 In DSS colitis, pathogenesis comprises destruction of the epithelial barrier and primarily innate immune system activation. In contrast, IL-10 colitis results from lack of immune-modulatory effects of IL-10 on several immune cells.44 To test whether EBI2 knockout would reduce recruitment of proinflammatory T cells, we analyzed mRNA levels of several T cell markers but no clear difference was detected (Supplementary Figure S5a).
ILCs were shown to have high EBI2 expression and ILCs lacking EBI2 failed to localize into colonic lymphoid structures.16,45 Impaired ILC recruitment might explain reduced inflammation in EBI2−/−IL10−/− animals. The role of ILCs in IL-10 colitis has not been formally addressed; however, in a related infectious colitis model with Campylobacter jejuni infecting IL10−/− mice, colonic ILCs were increased and depletion of ILCs abrogated colitis.46 ILCs have also been implicated in other models of colitis.47,48,49 In CD40 colitis, ILCs within colonic cryptopatches increased their motility shortly after induction of inflammation, resulting in ILC accumulation at the tip of the villus.50 The signal driving ILC movements is unclear but 7α,25-diHC produced immediately after onset of inflammation might stimulate EBI2-dependent ILC motility.
Emgård et al. also demonstrated reduced colon inflammation upon EBI2 knockout: In CD40 colitis, EBI2 expressing ILCs and myeloid cells accumulated in inflammatory foci in the colon mucosa and colitis severity were much lower in EBI2−/− animals.16 These data, together with our data, suggest that the dependence of inflammation on EBI2 varies regarding the colitis model. EBI2 dependence would be expected for models with a critical role of ILCs including CD40 colitis,16,48 TRUC colitis (spontaneous colitis in RAG2−/−TBX21−/− animals),49 and some infectious colitis models.47 In contrast, DSS colitis with breakdown of the physical barrier of the colon seems to develop independently of EBI2. ILCs have also been implicated in human IBD since proinflammatory ILCs were found in intestinal samples.51 In addition, a SNP associated with the EBI2 gene increased the risk for both, UC and CD.1
In summary, we describe increased oxysterol synthesis in colon inflammation and a role of the EBI2-7α,25-diHC axis for generation of colonic lymphoid structures in steady state and inflammation. Our results provide further insights to lymphoid tissue development in the colon, which has been substantially less studied than the small intestine. We also report a role of EBI2 in IL-10 colitis, pointing to EBI2 as a potential drug target in IBD since specific EBI2 inhibitors are available.52
Materials and methods
Human samples
LPMCs were isolated from colon resections of patients undergoing intestinal surgery. PBMCs were isolated from healthy volunteers and IBD patients. Intestinal biopsies from IBD patients were obtained from the Swiss IBD Cohort Study (SIBDCS), a large, prospective nation-wide registry. The cohort goals and methodology are described elsewhere.53 Sample and data collection was approved by local ethics committees (BASEC 2017-01868, EK-1316) and all patients provided written informed consent. Data from a whole human genome oligo microarray (GEO data sets: GDS3268)20 were used as a complementary data set.
Flow cytometric analysis of human samples
PBMCs and LPMCs (prepared as described in the supplementary methods) were thawed and resuspended in RPMI-PSG-10% FCS and plated for resting at 37 °C (PBMCs: overnight; LPMCs: 5 h). The cells were stained with LIVE/DEAD Fixable Dead Cell Stain Kit (Life technologies/ Thermo Fisher scientific, Waltham, USA) according to the manufacturer’s instructions. For cell staining the following antibodies were purchased from BioLegend (San Diego, USA): CD8 (SK1), CD4 (SK3), CD45RA (HI100), CD27 (M-T271), CD19 (HIB19), and CCRR6 (G0343E3). The following antibodies were purchased from BD Biosciences (Franklin Lakes, USA): PE-Streptavidin and CD3 (SK7). The isotype control was purchased from R&D Systems (Minnesota, USA): Mouse IgG2a control biotin conjugated (Cat: IC0038). The EBI2 antibody (57C9B5C9) was provided by Andreas Sailer from Novartis and biotinylated. All patient samples were analyzed with an LSR II flow cytometer (BD Biosciences, Franklin Lakes, USA) within the same month. The analysis was performed with FlowJo software (FlowJo LLC).
Animals
All mice were kept in a specific pathogen-free (SPF) facility in individually ventilated cages. EBI2−/− mice (C57BL/6 x C129) were originally purchased from Deltagen (San Mateo, USA) and CH25H-deficient mice (C57/BL6) were provided by David. W. Russell, University of Texas Southwestern.6 Both strains were subsequently back-crossed to C57BL/6 for more than 10 generations.6 In our facility, they were crossed with wild-type C57BL/6 mice and heterozygous offspring was mated to generate knockout and wild-type littermates. All animal experiments were conducted according to Swiss animal well fare law and approved by the local animal welfare authority of Zurich county (Tierversuchskommission Zürich, Zurich, Switzerland, License No. ZH256-2014).
Colitis models
Acute dextran sulfate sodium (DSS) colitis was induced in age- and weight-matched females by administration of 3% DSS (MW: 36-50 kDa; MP Biomedicals/ Thermo Fischer Scientific, Waltham, USA) in the drinking water for 7 days. Mice were sacrificed after colonoscopy on day 8. To induce chronic colitis, animals underwent 4 DSS cycles, consisting of 2% DSS administration for 7 days followed by 10 days of normal drinking water, each. Mice were sacrificed after colonoscopy 3 weeks after the last DSS cycle. Colonoscopy was performed as described previously and scored using the murine endoscopic index of colitis severity (MEICS) scoring system.54
IL10−/− mice (C57BL/6) were crossed with EBI2−/− mice; animals heterozygous for EBI2 (EBI2+/-IL10−/−) were used to generate EBI2−/−IL10−/− and EBI2+/+IL10−/− littermates. IL10−/− animals are highly susceptible to develop spontaneous colitis. As described in previous studies,55 1% DSS for 4 days in drinking water at the age of 90 days was used to trigger inflammation, as the spontaneous development of colitis is dependent on the gut microbiota and is drastically reduced under SPF conditions. Mice were sacrificed at 120 or 200 days of age or upon development of rectal prolapse.
Histological score
Colons were dissected, fixed in 4% formalin, embedded in paraffin, and cut into 5-μm sections. Deparaffinized sections were stained with hematoxylin and eosin (HE). Histological scoring for acute and chronic DSS colitis was performed as described;56 a slightly adapted score was used for IL10−/− colitis (Supplementary Table S6).
Oxysterol measurements
Extraction of oxysterols from murine liver samples
Frozen colon or liver samples from colitis experiments were pulverized using a CryoPrepTM CP02 (Covaris, Woburn, USA), weighed and lysed in homogenization buffer (0.9% sodium chloride) using a Qiagen TissueLyser II (Qiagen, Venlo, NL) at 4 °C. Oxysterols were extracted from the lysate by two liquid–liquid extractions using a methanol:dichloromethane (1:1) mix and a homogenization buffer:dichloromethane (1:2) mix, respectively. The organic phases from both extractions were pooled and dried under nitrogen. The residue was reconstituted with ethanol containing 0.1% formic acid and filtered before analysis on the liquid chromatography tandem-mass spectrometry (LC-MS/MS) system.
LC-MS/MS analysis
The LC-MS/MS analysis was performed on a Dionex UltiMate 3000 RS with HPG Pump (Thermo Scientific, Waltham, USA), coupled with a Sciex Triple QuadTM 5500 mass spectrometer (AB Sciex, Zug, CH).
Detailed protocols of oxysterol extraction and LC-MS/MS analysis are provided in the supplementary methods.
Quantification of lymphoid structures
Whole mount
Colons were removed intact, flushed with cold PBS, opened along the mesenteric border, and mounted, lumen facing up. Colons were then incubated two times for 10 min with warmed HBSS containing 2 mM EDTA at 37 °C on a shaker to remove epithelial cells. After washing with PBS, colons were fixed with 4% Paraformaldehyde (PFA) for 1 h at 4 °C. Colons were washed five times with TBST (1 M Tris (pH 7.2), 1 M NaCl, 0.2% Triton X-100) and blocked with TBST containing 2% rat serum for 1 h at 4 °C. Colons were incubated with Cy3-conjugated anti-mouse B220 antibody (clone TIB146; provided by Oliver Pabst: Institute for Molecular Medicine, RWTH Aachen University, Aachen, Germany) in the above solution overnight at 4 °C. The next day, colons were washed with TBST and mounted on glass slides. B220+ cell clusters were quantified under the microscope.
Swiss rolls
Colons were dissected and opened along the mesenteric border. Swiss rolls were prepared with the luminal side facing outwards and embedded in optimum cutting temperature (OCT) compound, and frozen in liquid nitrogen. Quantification of lymphoid structures was performed in 20 HE-stained cryosections (6 µm) per colon, ~100 µm apart (the protocol for immunofluorescent stainings is provided in the supplementary methods). Zeiss Axio Scan.Z1 with ZEN blue Software (Zeiss, Oberkochen, Germany) was used to scan and analyze sections. SILT and CLP were defined according to their size and localization; CLP: composed of large lymphoid follicles between the two external muscular layers and the muscularis mucosae, SILT: smaller clusters of lymphoid cells in the lamina propria.
Lymphoid structures in animals from colitis experiments were enumerated using one HE-stained section of the rolled-up colon per mouse.
If not indicated otherwise, a Zeiss Axio Imager Z2 Microscope with Axio Vision Software (Zeiss, Oberkochen, Germany) was used.
Statistical analysis
If not indicated otherwise, Mann–Whitney U test was performed with GraphPad Prism software (GraphPad, San Diego, USA). For the multivariate linear regression analysis, appropriate modules of Matlab R2017b were used. To compare the number of B220+ lymphoid structures of different sizes (Fig. 5c) we used R studio to generate a multivariate Poisson regression relating log(average counts) with the genotype using model-based t-tests. Data are presented as mean ± SEM. P-values less than 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
References
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017).
Gatto, D., Paus, D., Basten, A., Mackay, C. R. & Brink, R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity 31, 259–269 (2009).
Gatto, D., Wood, K. & Brink, R. EBI2 operates independently of but in cooperation with CXCR5 and CCR7 to direct B cell migration and organization in follicles and the germinal center. J. Immunol. 187, 4621–4628 (2011).
Pereira, J. P., Kelly, L. M., Xu, Y. & Cyster, J. G. EBI2 mediates B cell segregation between the outer and centre follicle. Nature 460, 1122–1126 (2009).
Hannedouche, S. et al. Oxysterols direct immune cell migration via EBI2. Nature 475, 524–527 (2011).
Liu, C. et al. Oxysterols direct B-cell migration through EBI2. Nature 475, 519–523 (2011).
Gatto, D. et al. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. Nat. Immunol. 14, 876 (2013).
Li, J., Lu, E., Yi, T. & Cyster, J. G. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature 533, 110–114 (2016).
Yi, T. et al. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity 37, 535–548 (2012).
Bauman, D. R. et al. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl Acad. Sci. USA 106, 16764–16769 (2009).
Baptista, A. P. et al. Colonic patch and colonic SILT development are independent and differentially regulated events. Mucosal Immunol. 6, 511–521 (2013).
Buettner, M. & Lochner, M. Development and function of secondary and tertiary lymphoid organs in the small intestine and the colon. Front. Immunol. 7, 342 (2016).
Knoop, K. A., Butler, B. R., Kumar, N., Newberry, R. D. & Williams, I. R. Distinct developmental requirements for isolated lymphoid follicle formation in the small and large intestine: RANKL is essential only in the small intestine. Am. J. Pathol. 179, 1861–1871 (2011).
Donaldson, D. S., Bradford, B. M., Artis, D. & Mabbott, N. A. Reciprocal regulation of lymphoid tissue development in the large intestine by IL-25 and IL-23. Mucosal Immunol. 8, 582–595 (2015).
Emgard, J. et al. Oxysterol sensing through the receptor GPR183 promotes the lymphoid-tissue-inducing function of innate lymphoid cells and colonic inflammation. Immunity 48, 120–132 e128 (2018).
Lochner, M. et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J. Exp. Med. 208, 125–134 (2011).
Knoop, K. A. & Newberry, R. D. Isolated lymphoid follicles are dynamic reservoirs for the induction of intestinal IgA. Front. Immunol. 3, 84 (2012).
Clottu, A. S. et al. EBI2 expression and function: robust in memory lymphocytes and increased by natalizumab in multiple sclerosis. Cell Rep. 18, 213–224 (2017).
Noble, C. L. et al. Regional variation in gene expression in the healthy colon is dysregulated in ulcerative colitis. Gut 57, 1398–1405 (2008).
Honda, A. et al. Cholesterol 25-hydroxylation activity of CYP3A. J. Lipid Res. 52, 1509–1516 (2011).
Raselli T., et al. The oxysterol synthesizing enzyme CH25H contributes to the development of intestinal fibrosis. https://www.biorxiv.org/content/early/2018/09/23/424820 .
Reboldi, A. et al. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 345, 679–684 (2014).
Kotlarz, D. et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology 143, 347–355 (2012).
Diczfalusy, U. et al. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J. Lipid Res. 50, 2258–2264 (2009).
Xiang, Y. et al. Identification of cholesterol 25-hydroxylase as a novel host restriction factor and a part of the primary innate immune responses against hepatitis C virus infection. J. Virol. 89, 6805–6816 (2015).
Wu, J. et al. LC/MS/MS profiling of tissue oxysterols and its application in dextran sodium sulphate induced mouse colitis models. Curr. Top. Med. Chem. 17, 2781–2790 (2017).
Blanc, M. et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118 (2013).
Guillemot-Legris O., et al. Colitis alters oxysterol metabolism and is affected by 4beta-hydroxycholesterol administration. J. Crohns Colitis 2018. https://doi.org/10.1093/ecco-jcc/jjy157. Epub ahead of print.
Okayasu, I. et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98, 694–702 (1990).
Olivier, B. J. et al. Vagal innervation is required for the formation of tertiary lymphoid tissue in colitis. Eur. J. Immunol. 46, 2467–2480 (2016).
Lu, E., Dang, E. V., McDonald, J. G. & Cyster, J. G. Distinct oxysterol requirements for positioning naive and activated dendritic cells in the spleen. Sci. Immunol. 2, 10 (2017).
Ota, N. et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat. Immunol. 12, 941–948 (2011).
Marchesi, F. et al. CXCL13 expression in the gut promotes accumulation of IL-22-producing lymphoid tissue-inducer cells, and formation of isolated lymphoid follicles. Mucosal Immunol. 2, 486–494 (2009).
McNamee, E. N. & Rivera-Nieves, J. Ectopic tertiary lymphoid tissue in inflammatory bowel disease: protective or provocateur? Front. Immunol. 7, 308 (2016).
McNamee, E. N. et al. Ectopic lymphoid tissue alters the chemokine gradient, increases lymphocyte retention and exacerbates murine ileitis. Gut 62, 53–62 (2013).
Kozik, A. J., Nakatsu, C. H., Chun, H. & Jones-Hall, Y. L. Age, sex, and TNF associated differences in the gut microbiota of mice and their impact on acute TNBS colitis. Exp. Mol. Pathol. 103, 311–319 (2017).
Gao, Y., Postovalova, E. A., Makarova, O. V., Dobrynina, M. T. & Mikhailova, L. P. Sex-related differences in the morphology and subpopulation composition of colon lymphocytes in experimental acute colitis. Bull. Exp. Biol. Med 165, 503–507 (2018).
Babickova, J. et al. Sex differences in experimentally induced colitis in mice: a role for estrogens. Inflammation 38, 1996–2006 (2015).
Schaubeck, M. et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 65, 225–237 (2016).
Tedde, A. et al. Interleukin-10 promoter polymorphisms influence susceptibility to ulcerative colitis in a gender-specific manner. Scand. J. Gastroenterol. 43, 712–718 (2008).
Li-Hawkins, J., Lund, E. G., Bronson, A. D. & Russell, D. W. Expression cloning of an oxysterol 7alpha-hydroxylase selective for 24-hydroxycholesterol. J. Biol. Chem. 275, 16543–16549 (2000).
Jones-Hall, Y. L. & Grisham, M. B. Immunopathological characterization of selected mouse models of inflammatory bowel disease: Comparison to human disease. Pathophysiology 21, 267–288 (2014).
Keubler, L. M., Buettner, M., Hager, C. & Bleich, A. A multihit model: colitis lessons from the interleukin-10-deficient mouse. Inflamm. Bowel Dis. 21, 1967–1975 (2015).
Chu, C. et al. Anti-microbial functions of group 3 innate lymphoid cells in gut-associated lymphoid tissues are regulated by G-protein-coupled receptor 183. Cell Rep. 23, 3750–3758 (2018).
Malik, A. et al. Contrasting immune responses mediate Campylobacter jejuni-induced colitis and autoimmunity. Mucosal Immunol. 7, 802–817 (2014).
Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Vonarbourg, C. et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat( + ) innate lymphocytes. Immunity 33, 736–751 (2010).
Powell, N. et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor + innate lymphoid cells. Immunity 37, 674–684 (2012).
Pearson, C. et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. elife 5, e10066 (2016).
Geremia, A. et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 208, 1127–1133 (2011).
Gessier, F. et al. Identification and characterization of small molecule modulators of the Epstein-Barr virus-induced gene 2 (EBI2) receptor. J. Med. Chem. 57, 3358–3368 (2014).
Pittet, V. et al. Cohort profile: the Swiss Inflammatory Bowel Disease Cohort Study (SIBDCS). Int. J. Epidemiol. 38, 922–931 (2009).
Becker, C., Fantini, M. C. & Neurath, M. F. High resolution colonoscopy in live mice. Nat. Protoc. 1, 2900–2904 (2006).
Michael, S. et al. Quantitative phenotyping of inflammatory bowel disease in the IL-10-deficient mouse by use of noninvasive magnetic resonance imaging. Inflamm. Bowel Dis. 19, 185–193 (2013).
Hausmann, M. et al. In vivo treatment with the herbal phenylethanoid acteoside ameliorates intestinal inflammation in dextran sulphate sodium-induced colitis. Clin. Exp. Immunol. 148, 373–381 (2007).
Acknowledgements
The authors would like to thank Silvia Lang for technical support, Jean-Benoît Rossel, Nicolas Fournier, and Delphine Golay for help with SIBDC data and biosamples, Brian Lang for help with the multivariate linear regression analysis, and the scientific committee of SIBDC for generous support.
Author information
Authors and Affiliations
Contributions
A.W., G.R. and B.M. conceived, designed, and supervised the study. A.W., T.R., G.S., F.R., C.P., A.M., R.R., M.R.S., L.H., I.F.W., K.A. and S.L. performed experiments and/or were involved in data analysis. M.H., G.K. and N.P. performed oxysterol measurements. O.P. performed analysis of microbial IgA coating. A.W. and B.M. wrote the paper. M.R.S., A.W.S., I.F.W., O.P., M.H., C.P., M.S. and G.R. critically revised the manuscript for important intellectual content. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
This work was supported by grants from the Swiss National Science Foundation to BM (grant No. 32473B_156525), CP (PP00P3_157476), FR (MD-PhD fellowship) and GR (for the Swiss IBD Cohort, grant no. 33CS30_148422) and a grant from the Hartmann-Müller Foundation to BM. AWS is an employee of Novartis Pharma AG and does hold stock and stock options in this company. The remaining authors declare no competing interests.
Additional information
Disclosures:This work was supported by grants from the Swiss National Science Foundation to BM (grant No. 32473B_156525), CP (PP00P3_157476), FR (MD-PhD fellowship) and GR (for the Swiss IBD Cohort, grant no. 33CS30_148422) and a grant from the Hartmann-Müller Foundation to BM. AWS is an employee of Novartis Pharma AG and does hold stock and stock options in this company. The remaining authors have nothing to disclose.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wyss, A., Raselli, T., Perkins, N. et al. The EBI2-oxysterol axis promotes the development of intestinal lymphoid structures and colitis. Mucosal Immunol 12, 733–745 (2019). https://doi.org/10.1038/s41385-019-0140-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-019-0140-x
This article is cited by
-
Development of non-alcoholic steatohepatitis is associated with gut microbiota but not with oxysterol enzymes CH25H, EBI2, or CYP7B1 in mice
BMC Microbiology (2024)
-
Gut-inhabiting Clostridia build human GPCR ligands by conjugating neurotransmitters with diet- and human-derived fatty acids
Nature Microbiology (2021)
-
Cholesterol 25-hydroxylase protects against experimental colitis in mice by modulating epithelial gut barrier function
Scientific Reports (2020)
