
Abstract
Reactive oxygen species (ROS), initially categorized as toxic by-products of aerobic metabolism, have often been called a double-edged sword. ROS are considered indispensable when host defense and redox signaling is concerned and a threat in inflammatory or degenerative diseases. This generalization does not take in account the diversity of oxygen metabolites being generated, their physicochemical characteristics and their production by distinct enzymes in space and time. NOX/DUOX NADPH oxidases are the only enzymes solely dedicated to ROS production and the prime ROS producer for intracellular and intercellular communication due to their widespread expression and intricate regulation. Here we discuss new insights of how NADPH oxidases act via ROS as multifaceted regulators of the intestinal barrier in homeostasis, infectious disease and intestinal inflammation. A closer look at monogenic VEOIBD and commensals as ROS source supports the view of H2O2 as key beneficial messenger in the barrier ecosystem.
Similar content being viewed by others

Introduction
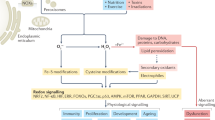
What are ROS? The term ROS is often indiscriminately used.1 Some of the molecules combined under this terminology are reactive species (e.g., superoxide anion radical (O2•−), hydroxyl radical (HO•)), but others are non-radicals (e.g., hydrogen peroxide (H2O2), singlet oxygen (1O2)) (Fig. 1). The reactivity and half-life of these species is very different, with HO• being highly reactive with a very short-half-life (10−9s), followed by the instable O2•− (10−5 s) and the relatively stable (10−2–10−3 s) but weak oxidant H2O2.2,3 The anionic charge of O2•− prevents diffusion through membranes, permitting only oxidation of adjacent targets (e.g., [Fe-S] clusters), while H2O2 is diffusible and reacts preferentially with thiol-containing compounds, initiating redox signaling. Often conversion of oxygen-derived species takes place. Spontaneous and superoxide dismutase-catalyzed dismutation of O2•− forms H2O2, which will be reduced to HO• by the metal-catalyzed Fenton reaction, the Haber-Weiss reaction or Fe2+ autooxidation. In fact, the toxicity attributed to H2O2 is dependent on the presence of ferrous iron (Fe2+) or similarly reactive metal ions, or on conversion to hypohalous or pseudohypohalous acids when peroxidases and halides or pseudohalides are present. The decomposition of H2O2 to water and oxygen is very slow, but the reaction can be accelerated by catalysts such as catalase. The continuous formation, conversion and breakdown of oxygen-derived species assure a rapid, dynamic response to changes in the cellular environment. Intricate regulation of enzymes generating O2•− and H2O2 (termed here “primary ROS”) provides another layer of control, which will induce and limit the output. In some circumstances using the term “ROS” is unavoidable, mainly due to our inability to exactly measure the species generated in a spatiotemporal manner or to correctly identify or discriminate between species responsible for a certain biological event. Sometimes a mixture of oxygen intermediates is produced as several ROS-producing enzymes are stimulated or interact with each other. Therefore, ROS sources cannot be considered isolated enzymatic systems, and biological processes may involve ROS-induced ROS generation. Co-regulation of ROS sources and ROS being an essential part of cellular functions can give rise to various compensation strategies, a challenge when stably deleting a ROS-producing enzyme experimentally. In conclusion, ROS as messengers and signaling molecules are highly versatile due to their diverse reactivity and their localized or long range activity.

Reactive oxygen/nitrogen species production and elimination. Oxygen (O2) is reduced to superoxide (O2•−), which can be either reduced to hydrogen peroxide (H2O2) by superoxide dismutase (SOD) or converted to hydroxyl radical (HO•). H2O2 can undergo the Fenton reaction and is transformed into HO• or is reduced to water (H2O) by catalase (CAT) or the glutathione (GSH)/glutathione peroxidase (GPx) system. In presence of chloride ions (Cl−) H2O2 can be converted to hypochlorous acid (HOCl) by neutrophil myeloperoxidase (MPO) and this reaction can further generate HO•. Nitric oxide synthase (NOS) catalyzes the oxidation of l-arginine to l-citrulline, releasing nitric oxide radicals (NO•) that form the potent oxidant peroxynitrite (ONOO−) following reaction with O2•−.
ROS have been associated with host defense, inflammation and tissue damage for over 40 years,4,5 and in those early reports neutrophil-derived superoxide was considered the harmful radical, which could be rendered harmless by converting it to non-radical H2O2 by superoxide dismutase. As secondary ROS (e.g., HO•) or adduct products (e.g., HOCl, ONOO−) exist, the context is important when anticipating the impact of ROS on any pathophysiological condition. Oxidative tissue damage in terms of modified proteins, lipids or DNA can be assessed, albeit only in a descriptive manner. In contrast, the term oxidative stress is often used without any quantifiable, spatiotemporal information and without knowledge if removal, repair or replacement of oxidative modifications is taking place. To address the biological functions of “ROS” in health or disease, detection of the particular oxygen-derived species (if univocally possible) or of oxidative modifications, and their abrogation with antioxidants or unspecific inhibitors will not adequately provide answers about causality or targeted therapeutic intervention.
More promising will be identifying the enzymatic source at the onset. The tissue and cell expression patterns and the primary oxygen species produced are known for many ROS-generating enzymes. Matching the ROS source with the cellular context and environmental input will determine the biological outcome. For example, the phagocyte NADPH oxidase NOX2 generates large quantities of superoxide into the very small phagosomal space after uptake of microorganisms. Superoxide will be converted to H2O2 and then further by myeloperoxidase and chloride to highly reactive hypochlorous acid (HOCl).6,7 But NOX2 can also be recruited and activated at the neutrophil plasma membrane by other stimuli (e.g., bacterial peptides, very large particles), which will lead to the generation of extracellular H2O2. The first scenario serves as highly toxic antimicrobial defense, while in the second situation extracellular H2O2 concentrations will be much lower and can serve outside-in or long range signaling. In dendritic cells NOX2 activity modulates the phagosomal pH, contributing to antigen presentation to both CD4+ and CD8+ cells and to defense against intracellular E. coli.8,9,10 In other cell types such as endothelial cells the same NOX2 enzyme is localized in lipid rafts or redox active endosomes (redoxosomes) and participates in signal transduction.11 This review focusses on NADPH oxidases as these are the only mammalian enzymes solely dedicated to primary ROS generation, but changes in localization and/or the cellular environment will affect similarly the type of oxygen intermediates generated and their biological function if O2•− and H2O2 are mainly by-products of an enzymatic reaction. Primary ROS generated by these enzymes, in particular by the mitochondrial electron transport chain, participate in immune sensing, redox signaling and stress-induced responses.12,13,14,15
NADPH oxidases
NADPH oxidases form multimeric complexes that generate superoxide or H2O2 in a tightly controlled manner by transferring electrons from NADPH via FAD and across two nonequivalent hemes to molecular oxygen, the electron acceptor. The complete electron shuttle is contained within the six transmembrane spanning NOX domain, which is integral part of all seven NADPH oxidase family members (NOX1–5, DUOX1–2). Additional regulatory features are required for oxidase activity. All oxidases except NOX5 require the dimerization of the NOX/DUOX module with an additional integral membrane protein for maturation, stabilization and localization, leading to membrane-bound heterodimers (NOX1–4/p22phox; DUOX1–2/DUOXA1–2). For NOX1–3 enzymes translocation of cytosolic proteins and their assembly with NOX1–3/p22phox heterodimers is essential for catalytic activity. Many signaling and dissociation/association steps are necessary for partial (see neutrophil priming16) or full enzyme assembly, which include stimulus-induced posttranslational modifications, protein-protein and protein-lipid interactions, as well as GDP to GTP exchange on the RhoGTPases RAC1 or RAC2. NOX5 and DUOX1–2 enzymes require calcium flux and binding of calcium to their EF hand domains for superoxide (NOX5) or H2O2 (DUOX1–2) generation, while NOX4-p22phox heterodimers are seemingly constitutively active. How the one-electron reduction of molecular oxygen can lead to release of H2O2 instead of superoxide by NOX4 and DUOX enzymes is yet unresolved as the additional extracellular peroxidase-like homology domain in mammalian DUOX lacks peroxidase activity. For brevity, we will discuss here only NADPH oxidases firmly connected to ROS generation in the gastrointestinal tract (GIT), namely NOX1 and DUOX2 expressed in the intestinal epithelium and NOX2 in innate immune cells of the lamina propria (LP) (Fig. 2). Other oxidases may play a role in intestinal homeostasis and/or disease states such as NOX4 expressed in intestinal fibroblasts and smooth muscle cells or DUOX1 in lymphocytes, but their contributions to GIT (patho)physiology are not yet defined (see reviews17,18).

Schematic representation of NADPH oxidases present in the intestine. NOX1 is a multi-subunit complex with a catalytic core composed of two transmembrane proteins: p22phox and NOX1. In order to convert molecular oxygen to superoxide, NOX1 heterodimerizes with p22phox, two cytosolic subunits termed NOXA1 and NOXO1, and activated RAC1 GTPase. The NOX2 heterodimer is composed of p22phox and gp91phox (also called NOX2). The NOX2 complex is typically inactive in resting cells, but when stimulated the cytosolic subunits p40phox, p47phox and p67phox and activated RAC assemble with NOX2-p22phox to reduce oxygen to superoxide by electron shuttling. DUOX2 (also known as thyroid oxidase) is an EF hand motif containing protein that forms a membrane-bound heterodimer with its maturation factor DUOXA2 and produces H2O2 in a Ca2+-dependent manner.
The regulatory mechanisms and host defense functions of NOX2 in human neutrophils are well characterized due seminal discoveries in the late 1980/early 1990,19,20,21,22,23,24 which were driven by the genetic and biochemical analysis of neutrophils obtained from patients with chronic granulomatous disease (CGD) (see reviews25,26). CGD, an inherited immunodeficiency, is characterized by recurrent bacterial and fungal infections, often with inflammatory complications. 40–50% of CGD patients develop gastrointestinal symptoms reminiscent of Crohn’s disease (CD). The disorder is caused by loss-of-function variants in genes encoding for any of the proteins forming the assembled NOX2 complex (CYBB (NOX2), CYBA (p22phox), NCF1 (p47phox), NCF2 (p67phox), NCF4 (p40phox), RAC2), leading to markedly reduced to absent superoxide generation.27,28 -X-linked CYBB (70% of CGD patients) and autosomal recessive NCF1 (20%) are commonly affected by inactivating mutations, deletions and frame-shifts. The extent of residual superoxide production predicts disease severity and long-term survival.29 In mice the hyperinflammation phenotype characterized by increased levels of IL-1β, IL-6 and TNFα was linked to loss of Nox2 activity in macrophages and dendritic cells.30 The only cure for CGD is allogeneic hematopoietic stem cell transplantation which also eliminates the gastrointestinal symptoms,31 albeit gene editing technology has recently shown great promise.32
The epithelial NADPH oxidases NOX1 and DUOX1–2 were discovered in 1999 to early 2000.33,34,35,36,37 NOX1 is mainly expressed in epithelial cells of the colon and rectum,38 but can also be found in other cell types such as smooth muscle cells and endothelial cells. NOX1 complex formation follows the NOX2 paradigm, but NOX1 utilizes primarily the p47phox and p67phox homologs NOXO1 and NOXA1 for assembly.39,40,41 In contrast to the tightly controlled, stepwise translocation of cytosolic p47phox and p67phox to the membrane-bound NOX2 heterodimer, both NOXO1 and NOXA1 seem to be membrane-associated, permitting partial NOX1 assembly and activation. This basal level of superoxide generation will be then further enhanced by upregulation of components and by recruitment and accumulation of the assembled NOX1 enzyme at particular cellular locations. In inflammatory conditions TNFα-induced NOXO1 upregulation via AP-1-dependent transcription led to increased superoxide generation.42 Bacterial or endogenous ligands stimulated NOX1 upregulation and/or catalytic activity by triggering Toll-like receptor (TLR2, TLR4, TLR5) or N-formyl peptide receptor (FPR)-mediated pathways.43,44,45,46 In colon cancer cells and colonic biopsies derived from healthy subjects the NOX1-p22phox complex translocated from inner membrane compartments to ruffles and sites of bacterial attachment and invasion when exposed to pathogenic intestinal bacteria.47,48 Increased NOX1 overexpression correlates with oncogenic RAS expression and has been linked to colon cancer cell proliferation and migration.49,50,51
DUOX1–2 enzymes are mainly expressed in the thyroid and mucosal epithelia (lung, nasal, intestinal). In contrast to the pulmonary epithelium DUOX1 expression is very low in the intestine, while DUOX2 can be found all along the GIT with peak expression in the ileum.52,53 In colon or lung epithelia DUOX2 and DUOXA2 will be transcriptionally upregulated (10–40-fold and up to 200-fold, respectively) as part of the host response to pathogenic bacteria and viruses.54,55,56 Cytokines including IFNγ or the combination of IFNβ and TNFα upregulated DUOX2–DUOXA2 expression via STAT1 or STAT2/IRF9 signaling in airway epithelial cells or cancer cell lines.55,57,58 In the murine intestine Duox2 expression is primarily regulated by the microbiota with mucosa-associated segmented filamentous bacteria (SFB) being the predominant inducer of ileal Duox2 expression.53 Experiments using mice deficient in various bacterial signaling transducers (Trif−/−, Myd88−/−, p50−/−, p65fl/fl Vil-cre) combined with pharmacological inhibitors identified two distinct pathways for Duox2 upregulation, involving Trif/p50/p65 signaling in the ileum and a Myd88/p38MAPK pathway in the colon.59 Host defense may require an ileal NOD2–DUOX2 axis as the NOD2 agonist muramyl dipeptide stimulated ROS generation in Caco-2 cells in a partially DUOX2-dependent manner.60
Frequently several NADPH oxidases will be expressed in the same cell type, which can result in coordinated regulation. In the colonic epithelium of Citrobacter rodentium-infected mice activation of Nox1 signaling to Ask1 and p38MAPK induced upregulation of Duox2/Duoxa2 via the transcription factor Atf2. The signaling module Nox1–Ask1 was also observed in vascular smooth muscle cells,61 while the importance of the p38–Atf2 axis is conserved in Drosophila Duox regulation.62 NADPH oxidases can also use H2O2 for functional communication in organisms. An early event in the zebrafish wounding response is H2O2 release by Duox2, which acted as a gradient for leukocyte recruitment.63 Neutrophil sensing of this tissue-scale gradient involved the Lyn tyrosine kinase.64 At the wounding site neutrophils provided Nox2-mediated host defense and used concomitant release of myeloperoxidase for downregulation of the H2O2 gradient.65
NADPH oxidases in inflammatory bowel diseases
The association of acute inflammation with an increased abundance of ROS (oxidative stress) has been the basis for devising antioxidant approaches or inhibitor strategies targeting ROS sources to ameliorate disease. Although ROS have been connected to disease development/progression or are considered a hallmark of disease severity for some disorders, the antioxidant approach has not been very successful in IBD. The notion of a redox imbalance in colonic tissues (e.g., GSH↓, SOD↑, CAT↓) due to ROS overproduction is mostly based on detection of oxidative modifications of lipids, proteins, or DNA, or on ROS measurements with probes that cannot distinguish various ROS/RNS species or give any indication of the relevant ROS source.66,67,68,69,70,71,72,73 Most of these studies have been descriptive without any identification of the responsible enzyme or the time period of activity. Mainly mitochondrial dysfunction with increased O2•− leakage or the oxidative burst of activated neutrophils74 have been implicated in ROS-induced damage. Hyperactivated neutrophils with increased secondary ROS production (e.g., HOCl) and release of proteases and extracellular traps can elicit tissue damage, but in CD chemotaxis is often defective, leading to reduced neutrophil numbers in the intestine and incomplete removal of bacteria.75 In fact, neutrophils are increasingly recognized for their role in resolution of inflammation.76,77,78,79 A better understanding about the spatiotemporal generation of ROS, the chemical composition of the reactive species and their potential to cause damage, and if oxidative damage is predictive for disease severity and/or slow recovery is needed. The upregulation of a ROS-generating enzyme at a certain stage of disease cannot per se be correlated to oxidative damage. Upregulation of NOX1 and particularly of DUOX2–DUOXA2 in CD, ulcerative colitis (UC), pouchitis and irritable bowel syndrome has been discovered in genome-wide transcription profiling, RNA sequencing or array expression analysis, and by protein detection,38,60,80,81,82,83,84 but information about functional complex assembly, posttranslational modifications and catalytic activity in space and time is not available. Most of this vital information cannot be obtained when analyzing patient biopsies and NOX/DUOX isoform selective inhibitors do not exist yet. It is important to consider that increased expression of catalytically active DUOX2 may be protective by augmenting host defense and mucosal healing responses. The low level of H2O2 output by DUOX2 will only lead to tissue damage if conversion to secondary oxygen metabolites occurs. Interestingly, functional annotation enrichment analysis of the DUOX2 co-expression signature in ileal CD indicated upregulation of myeloid cell, lipopolysaccharides, and wounding responses, all considered beneficial for host defense and restitution responses.81
The oxidative stress—IBD correlation is also challenged by the discovery of IBD patients with inactivating rare, inherited or de novo mutations in NADPH oxidases.85 Abolishing superoxide production in innate immune cells due to loss-of-function variants in NOX2 complex components is causally linked with CD-like disease, and CGD patients are sometimes erroneously classified as CD until liver abscesses or lung infections develop. To gain more knowledge about genetic risk factors in IBD in the absence of a primary immunodeficiency, exome or genome sequencing has progressed rapidly. The genomic profile of patients with severe phenotypes who fall into the categories very early onset IBD (VEOIBD) (≤6 years) or infantile IBD (≤2 years) is now almost routinely determined for diagnostic purposes.86 These patients present with IBD-like inflammation or pancolitis, and their underlying genetic defect is, in contrast to adult IBD, considered monogenic. In 2015 the first inactivating missense variants of NOX1 and DUOX2 were identified in five VEOIBD patients.87 Expression levels and intracellular localization of the altered proteins was comparable to wild-type NOX1, but their catalytic function was reduced, thereby impeding ROS-mediated defense against bacterial invasion. Eight more children (age 5–10 years) with IBD pathology harboring NOX1 missense variants have now been identified. The catalytic activity of these NOX1 variants was reduced to a varying degree.88 Recently the first inherited compound heterozygous missense DUOX2 variants were identified in a VEOIBD patient.89 In this case reduced H2O2 generation was accompanied by a defect in DUOX2 expression in colon biopsies of the patient. One of the alleles harbored a mutation close to the first transmembrane domain of DUOX2 which impeded plasma membrane translocation in cell-based studies. Misfolding of the protein in the endoplasmatic reticulum may prevent correct assembly of the DUOX2–DUOXA2 complex even when the other allele is structurally not affected. In adult IBD, a monoallelic DUOX2 missense variant was found to be associated with CD in some members of a cohort of Ashkenazi Jewish individuals.90 Not only reduced function of epithelial NADPH oxidases was associated with VEOIBD, mutations in NOX2 complex components (CYBB, CYBA, NCF1, NCF2 and NCF4) were also identified in VEOIBD patients.91 These mutations weakened primarily protein-protein interactions of cytosolic components that are required for efficient assembly of the complex, and were characterized by decreased, but not abolished superoxide production. VEOIBD patients carrying these NOX2 complex variants did not have CGD, as ROS levels were exceeding the threshold for full disease onset.29 By contrast, it is not yet understood why identified DUOX2 variants are either associated with VEOIBD or with congenital hypothyroidism, but not with both disorders (see review27).
In conclusion, seven of the ~70 genes associated with VEOIBD92,93 belong to the family of NADPH oxidases and their essential partner proteins. All of the NOX1, DUOX2 and NOX2 complex associated variants show deficits in ROS generation, while gain-of-function mutations have not been found. Thus, certain severe VEOIBD phenotypes are associated with a lack of ROS at the intestinal barrier, and not with ROS overproduction.
NADPH oxidase deficiency in colitis mouse models
Experimental models of IBD are indispensable to improve our understanding of intestinal pathophysiology and host-microbiota interactions. Several mouse models that replicate various aspects of intestinal inflammation are commonly used.94,95 Chemically induced (e.g., DSS, TNBS) or infectious (e.g., Citrobacter, Salmonella) colitis are the main methodologies in inbred mouse strains, but it is important to note that clear differences exist between mice and humans in terms of intestinal anatomy, immunology and microbial communities.96,97,98 Further, comparing animal studies between laboratories are complicated by differences in housing conditions such as prevalence of specific and opportunistic pathogens including ecto- and endoparasites, and in the provided food/water regimen. Mice with genetically induced primary immunodeficiencies are particularly sensitive to unusual infections. For example, Cybb−/− (gp91phox, Nox2 knockout) mice acquire spontaneous infections with Staphylococcus xylosus, usually considered a commensal skin bacterium.99 Modeling infectious colitis in mice is hampered by the colonization resistance of the murine microbiota toward human pathogens and the absence of pathogen attachment sites and/or niche environments required for efficient colonization and/or gastrointestinal pathology. The common use of oral antibiotic treatment prior to Salmonella Typhimurium infection of mice will reduce microbiota diversity and colonization resistance, and will alter mucus- and epithelium-associated protective mechanisms,100,101 which excludes modeling of the native host-microbiota-pathogen communication. Table 1
illustrates infectious and chemically induced enteropathy models in mice deficient in ROS generation due to deletion of Nox1, Nox2, Nox2 complex components, Duoxa or p22phox. The absence of innate immune cell-derived superoxide in Nox2 knockout mice (Cybb−/−) did not exacerbate C. rodentium-induced colitis54,102 as long as the colonization resistance of the microbiota was preserved,54 while mice deficient in the Nox2 cytosolic component p47phox (Ncf1−/−) were more susceptible,103 possibly a sign of differentially altered microbiota in these two mouse strains. Knockout of Nox2 or of the Nox2 cytosolic component p40phox (Ncf4−/−) significantly reduced the clearance of S. Typhimurium, while increasing innate immune cell infiltration and pro-inflammatory cytokines.104,105 Mice with loss of Duox1 and Duox2 activity (Duoxa−/−) showed increased H. felis colonization in the stomach106 and augmented bacterial translocation in the small intestine.53 The ileal gene expression profile of mice with Duox1–2 inactivation was comparable to genetic changes observed in the non-inflamed ileal tissue of CD patients with strictly colonic inflammation.53,106 Of note, Duoxa−/− mice require continuous oral L-thyroxine supplementation to maintain sufficient thyroid hormone levels, which may alter the microbiota composition.106 Development of Cyba−/− and Cybafl/fl Vil-cre (p22phox, p22phox ∆IEC) mice revealed the importance of barrier-associated ROS in downregulating C. rodentium colonization and virulence, either by epithelial Nox-Duox2 signaling or in absence of Nox activity by compensatory overgrowth with H2O2-producing lactobacilli.54 Cybb−/− mice were highly susceptible to chemically induced colitis (TNBS) with intense neutrophil involvement that was manifested in hyperinflammation and decreased survival rates due to compromised defense toward invading microbiota.77 In contrast, Nox1 deficiency in mice did not alter TNBS colitis outcome or seemed even protective.107,108 Several studies used DSS colitis as a model to interrogate the relationship between host epithelial damage, microbiota infiltration and resolution of inflammation. Global or epithelial Nox1 deletion or global Duox inactivation were inconsequential,53,107,109 while reports differ widely, ranging from protective to detrimental, when superoxide production was reduced by deletion of Nox2 complex components (Table 1). For example, while Nox2 deficiency was protective in DSS colitis, deficiency in the Nox2 complex components p40phox or p47phox resulted in increased susceptibility in the same colitis model.103,110,111 Disease activity in DSS colitis is strongly influenced by the microbiota composition and the mucus phenotype.112 Both of these parameters are research facility and rodent diet dependent, which may explain these divergent results. But how can we reconcile the colitis risk of VEOIBD patients with NOX1 or DUOX2 variants with the absence of spontaneous or induced pathophysiology in Nox1 and Duoxa knockout mice? It is conceivable that deleting Nox1 or inactivating Duox may trigger compensation by another NADPH oxidase or by a different ROS source. Alternatively, a second risk factor absent in mice, either genetic or environmental (diet) may be present in humans. Differences in the Nox1/Duox2 expression profile in intestinal epithelial cell subsets in mice versus humans and its influence on barrier function may modify the disease outcome. Developing mouse models mimicking epithelial NADPH oxidase deficiency in patients reproducibly will be a key challenge for the near future
Role of epithelial oxidases at the intestinal barrier
The intestinal epithelial barrier together with the microorganisms comprising the microbiota can be considered one interconnected ecosystem. While spatially separated, constant communication between host and microbiota takes place. This crosstalk is mediated by gradients of chemical signals (e.g., oxygen, H2O2, pH, ATP, lactate), immune effectors (e.g., defensins, antimicrobial peptides (AMPs), lysozyme, IgA, RegIIIγ, cytokines, TLR/NLR agonists) and nutrient-derived compounds (e.g., SCFA, oligosaccharides, fibers). H2O2 is particularly suited as means of communication due to its relative stability and diffusibility (Fig. 3). Sensing of bacteria leads to NOX1/DUOX2-mediated release of H2O247,53,113 which can diffuse across bacterial membranes and interfere with bacterial phosphotyrosine signaling47,48,114 and virulence determinants,54 or can re-enter colonic epithelial cells via aquaporin channels, altering intracellular signaling.115 Likewise, some commensals colonizing in the loose mucus layer, in particular lactobacilli and some bifidobacteria, generate H2O2 and are thus uniquely equipped to withstand an oxidative environment.116,117 Bacterial-derived H2O2 modifies niche community structure and reduces pathogen virulence and survival, but will also alter signaling in host epithelial cells.118 Reciprocal interactions between host and microbiota maintain intestinal homeostasis, and compensatory or restorative mechanisms will commence as soon as the equilibrium is disrupted. This interconnectivity provided by H2O2 signaling needs to be considered when attributing biological functions to NOX/DUOX enzymes.

Multi-directional H2O2 signaling in the intestinal mucosa. In the intestine ROS can be released by the immune system, the epithelium and the microbiota. ROS generated by phagocytes in the immune system have antimicrobial and pro-inflammatory functions against invading pathogens. ROS generated by the epithelium contribute to host–microbe interactions and tissue repair mechanisms following an inflammatory response. Commensals such as lactobacilli contribute to the H2O2 environment at the barrier, supporting protective and restorative processes.
Maintenance of barrier function
The intestinal barrier is a complex multilayer system that can be separated into physical and chemical barrier. The physical barrier is formed by a single layer of epithelial cells sealed by tight junctions, adherens junctions and desmosomes, and includes the mucus layer(s), while components of the chemical/immunological barrier are secretory immunoglobulin A, pro-inflammatory mediators, antimicrobial peptides, cytokines and H2O2. Epithelial barrier dysfunction with increased paracellular permeability, redistribution of junction-associated proteins (claudins, occludin, JAM-A) and cytoskeletal dysregulation (increased myosin light chain (MLC) kinase expression and MLC phosphorylation) has been observed in IBD patients119,120,121 and may lead to consistent low grade inflammation. H2O2 has been linked to disruption of junction complexes either by exposing Caco-2 cell monolayers to high concentrations of H2O2 (36 μM–5 mM H2O2/1–2 h)122,123 or indirectly by exposure of cells to TNFα, IL-1β or toxins, which will upregulate NADPH oxidase expression and induce ROS generation.124,125 Lactobacilli such as L. plantarum ATCC 8014 or L. rhamnosus OLL2838 counteracted by a yet unidentified mechanism the TNFα-induced decrease in transepithelial electrical resistance (TEER) and reduced paracellular permeability in several colitis models.126 As H2O2 production by lactobacilli is comparable to the H2O2 output by epithelial NADPH oxidases, these studies suggest that physiological H2O2 concentrations will not disrupt junction complexes in epithelia. In accord, activity of Nox1 or Duox2 is not essential for maintaining TEER, as paracellular permeability of the intestinal epithelium, evaluated with fluorescein-conjugated dextran, was not altered in Nox1−/− or Duoxa−/− mice.53,107
The epithelial monolayer is covered for additional protection by two-layered (colon) or single-layered (small intestine) mucus, which is secreted and continuously renewed by goblet cells. Mucus in the colon and small intestine is mainly composed of MUC2 mucin polymers.127,128 In the colon the inner dense mucus layer separates the microbiota from the host epithelium, thereby rendering the apical epithelial surface free of bacteria. The outer loose mucus layer provides a distinct niche for various commensal bacteria, which either utilize O-glycans as carbon source (mucolytic B. thetaiotaomicron, A. muciniphila, R. torques) or prefer to attach and proliferate in this niche (E. coli, Lactobacillus, butyrate-producing Clostridium).129 In the absence of mucus (Muc2 deficiency) or increased mucus penetrability bacteria will invade into the crypts and interact with the epithelium, leading to inflammation, tight junction opening and bacterial dissemination. This is the case in IBD patients who present with thinner mucus (UC), goblet cell hyperplasia, altered MUC isoform expression or changes in the O-glycosylation profile.130,131,132,133 Recent studies connected intestinal epithelial NADPH oxidases to mucin upregulation and/or secretion. Exposure of Caco-2 cell monolayers to H2O2 (1–5 μM) or stimulation with epidermal growth factor (EGF) increased MUC5AC and MUC3 transcription, which was connected to DUOX2 as downstream target of the EGF receptor.134 This is reminiscent to observations in the airways where EGFR and IL-13 signaling promoted mucin gene induction (MUC5AC, CLCA1) via DUOX1, leading to mucus cell metaplasia and mucus hypersecretion.135,136 Patel and coworkers linked ROS generation by an unspecified Nox enzyme to goblet cell endocytosis, autophagosomes and regulation of mucin secretion.137 Goblet cells in colonic spheroids derived from mice harboring a Cyba mutant (Cybanmf333/nmf333) or lacking the Nox2 complex component p47phox showed enhanced mucin granule accumulation, and an ex vivo bolus of 300 μM H2O2 restored mucin secretion in these spheroids. These experiments would suggest that Nox2, an NADPH oxidase required for LC3-associated phagocytosis (LAP)138,139 is involved in Muc2 secretion in mice. Expression of a functional Nox2 complex was detected in colon epithelial cells,140,141 but has not yet been confirmed in goblet cells. Others reported similarly the coupling of endocytosis, autophagy, and ROS in driving goblet cell exocytosis and mucin secretion in murine colonic explants.142 Pharmacological inhibition of signaling mediators placed the generation of ROS downstream of both, TLR-Myd88 signaling and endocytosis, and upstream of Nlrp6 inflammasome activation without identifying the ROS source. Signals triggering continuous mucin secretion are not only dependent on host goblet cell function, but also on signals emanating from the microbiota. Differences in the health status of animal facilities, breeding strategies and/or diet may account for diverging GIT phenotypes in mice, for example in Nlrp6−/− mice.142,143,144 Identifying the key ROS source in goblet cells will require careful examination of various Nox/Duox deficient mouse strains under controlled conditions. It will be also important to understand how the neutrophil oxidative burst and the subsequent local hypoxia77 will influence goblet cell function. Potential strategies to strengthen the mucus layer of UC patients include supplementation with lactobacilli, which increase mucus thickness in mice,145 in part likely by generating H2O2. This approach will necessitate identifying appropriate Lactobacillus strains as changes in the mucus composition of UC patients reduce the capacity for probiotic colonization.133
Modulation of the microbiota
NADPH oxidases are intimately involved in crosstalk with the microbiota and vice versa. When NADPH oxidases are partially or completely inactivated, changes in the H2O2 gradient reaching commensal bacteria in the loose mucus layer will modify the bacterial community structure by altering gene transcription and intrabacterial signaling which will also affect interbacterial communication. In the epithelium, reduction of superoxide (Nox1) and/or H2O2 (Nox1, Duox2) will disrupt intracellular pathways, leading to changes in secretion of cytokines or other immune mediators, which will further modify the microbiota composition in the vicinity of the ROS source. Hence, changes in the microbiota composition of mice (and VEOIBD patients) with decreased ROS generation due to compromised NADPH oxidase function can be expected, but comparative microbiota analyses are still scarce. Nox1 is mainly expressed in colon and rectum, but can be upregulated 2-fold in jejunum and ileum by microbiota introduction into germ-free mice, while Duox2–Duoxa2 expression increases at the same time 7-15-fold.146 High ileal Duox2 expression was induced by eubiotic SPF microbiota, segmented filamentous bacteria, Salmonella and human dysbiotic microbiota.53,59 Translating these observations to humans, one could argue that the upregulation of DUOX2 in IBD patients is a response to microbiota breaching the physical barrier and a regulatory mechanism for limiting bacterial translocation. The effect of inactivating Nox1–4 globally or solely in the intestinal epithelium (p22phox KO or ∆IEC) on the composition of the cecal microbiota was determined by 16 S rRNA sequencing using Cyba−/− and Cybafl/fl Vil-cre mice.54 The abundance of facultative anaerobic bacteria, and in particular of certain Firmicutes known for protective, anti-inflammatory effects in gastrointestinal disease such as Lactobacillus, Bifidobacterium and Butyricicoccus147,148 was significantly increased in both mouse strains. Analysis of 3 day-old Cyba−/− pups showed highly increased L. reuteri and L. murinus when compared to wild-type littermates. Continuous heterozygous breeding and cohousing revealed dominance of the wild-type microbiota, while separation and analysis at generation F2/F3 stabilized the trend to overgrowth with probiotic bacteria. These experiments reveal that a) the microbiota composition is mainly altered by Nox inactivation in the epithelial compartment and b) the GIT ecosystem will compensate for insufficient ROS by expanding niche colonization for H2O2-generating probiotics. Deleting p22phox will inactivate all four Nox enzymes, leading to a decline in ROS that will not be matched by Nox1 deletion. In accord, lactobacilli were not upregulated in Nox1−/− mice (unpublished, and149). Changes in the microbiota composition were also observed in fecal pellets obtained from Ncf1−/− mice.103 Bacteroidetes, Proteobacteria, and Verrucomicrobiales (A. muciniphila) dominated when p47phox was deleted, while Tenericutes were less abundant when compared to non-littermate wild-type mice. Similar to p22phox knockout mice, cohousing or littermate breeding homogenized the microbiota. In both cases the microbiome signature established at birth played an important role in protection (C. rodentium colitis in Cyba−/− and Cybafl/fl Vil-cre mice) or exacerbation (DSS colitis in Ncf1−/− mice) of disease. While more work is needed to understand how reduced or even increased ROS will alter bacterial community structure and concomitantly disease onset/outcome, these initial studies suggest that the resulting protective or pathogenic microbiota will be dynamically adjusted when exposed to more resilient communities or other microbiota modifiers.
Host defense
Neutrophils and other phagocytic cells generate considerable amounts of superoxide in response to sensing, adherence or phagocytosis of microorganisms. The high concentration of superoxide (~10 nmol O2•−/min/106 cells) and secondary ROS in the enclosed compartment of the innate immune cell phagosome provides an ideal bactericidal environment. The importance of the phagocyte NADPH oxidase NOX2 in this oxidative burst is well established, although the exact “killing” mechanisms are still debated.6,7,150,151 It is assumed that chlorination, oxidation of [Fe-S] clusters, and crosslinking of bacterial (and host) proteins occur together with changes in the phagosome membrane potential, which permits maintaining the neutral pH required for activation of neutrophil proteases (e.g., elastase, cathepsin G). A similar host defense function has been discussed for NADPH oxidases located at apical membranes of mucosal epithelia. In analogy to the NOX2/MPO/Cl− system in neutrophils, DUOX-generated H2O2 oxidizes in conjunction with lactoperoxidase (LPO) thiocyanate to hypothiocyanite (OSCN−). Particularly in the airways this DUOX/LPO/SCN−system has been proposed as microbicidal defense mechanism.152 In mice Lpo is mainly expressed in mature epithelial cells of colon and rectum, but not in the ileum153 where basal Duox2 expression is highest. Increased colonic H2O2 levels in infections (Nox1, Duox2, lactobacilli) would place Lpo at a suitable location for OSCN− production. In the small intestine nitric oxide production by Nos2 may lead to formation of bactericidal ONOO− in the presence of superoxide. The available concentration of intestinal thiocyanate or peroxynitrite is unknown, and the large surface and peristalsis of the GIT will likely not permit reaching bactericidal or bacteriostatic concentrations of secondary reactive oxygen metabolites.
On the other hand, in Drosophila and Anopheles gambiae duox provides antimicrobial host protection.62,154,155 This leads to the question: what kind of host protection, if any, may Nox1/Duox2 activity (~3–30 nmol H2O2/h/106 cells) provide in the mammalian intestine? Overexpression of NOX1 or DUOX2 in epithelial cells reduced adherence and invasion of Salmonella, Campylobacter, EPEC and Listeria.47,48,60,156 When ROS were reduced by silencing of endogenous NOX1, by pharmacological inhibitors or by expressing loss-of-function NOX1 or DUOX2 mutants, pathogen adherence and invasion increased.47,87 Co-incubation of human biopsies and intestinal cells with pathogens stimulated extracellular H2O2 generation and translocation of the NOX1-p22phox complex and of DUOX2 to the entry site of invading pathogens and to sites of active membrane ruffling.47,48 The fate of internalized pathogens in these non-phagocytic cells was not determined as receptors used for uptake and the intracellular behavior of these pathogens differ. In general, autophagy will restrict the growth of intracellular pathogens that cannot counteract or evade this process.157 An unspecified NOX enzyme was associated with the epithelial autophagy machinery in Salmonella-infected Henle intestinal epithelial cells, while in murine goblet cells Nox-mediated ROS production was dependent on the presence of Atg5 and served as link between autophagic and endocytic pathways essential for mucin secretion.137 In differentiated tracheobronchial epithelial cells downregulation of ATG5 decreased apical DUOX1 localization.158 Together with the role of NOX2 in LAP and of NOX4 in regulating autophagy in the cardiovascular system, evidence is mounting for oxidase involvement upstream or downstream of autophagy.139,159
Gastrointestinal pathogens penetrate the dense mucus layer to attach and/or invade the mucosal epithelium. During this process these bacteria will encounter H2O2 released by NOX1/DUOX2 or by lactobacilli, which reduces their virulence in cell culture.47,87,160 But questions if the antivirulence effect of H2O2 can be confirmed in vivo and how low levels of H2O2 will alter pathogen fitness still remained. S. Typhimurium, C. rodentium or Listeria monocytogenes are routinely used to understand how gene deletion in mice will alter colonization and disease outcome. To decrease the colonization resistance of the murine microbiota, antibiotic pretreatment is required for Salmonella infections, while the two other pathogens will colonize wild-type mice with undisturbed microbiota. Colonization by Salmonella in streptomycin-pretreated Nox1−/− or Duoxa−/− mice was not altered, but early systemic dissemination was increased in the absence of Duox activity.53,161 Nox1–4 inactivation in the mucosa due to tissue-specific loss of p22phox was accompanied by compensatory Lactobacillus overgrowth (see microbiota).54 This increase in lactobacilli was required to provide protection against C. rodentium and L. monocytogenes in p22phox-deficient mice. Significant reduction of C. rodentium colonization was accompanied by markedly reduced pro-inflammatory mediators and the virtual absence of crypt pathology, LP immune cell infiltration or other disease markers. Supplementation of wild-type mice with L. johnsonii WT or a mutant deleted in the H2O2-producing NADH-dependent flavin reductase of this commensal (L. johnsonii ∆nfr)118,162 confirmed that H2O2 was critical for host protection. An important point to consider is the significant decrease of epithelial ROS in Cybafl/fl Vil−cre or Cyba−/− mice as even infection-induced upregulation of epithelial Duox2 was curtailed by Nox inactivation, revealing an emerging functional hierarchy in oxidase signaling.54
Cyba-deficient mice and infection studies in intestinal cells, biopsies and a rabbit loop model uncovered novel antivirulence mechanisms triggered by nanomolar H2O2. GIT-induced increased transcription of the LEE pathogenicity island of C. rodentium collected at the peak of disease from p22phox knockout mice or from wild-type mice supplemented with L. johnsonii WT (but not with L. johnsonii ∆nfr) was prevented in elevated H2O2 conditions. In vitro exposure of C. rodentium or EPEC to an environment with continuous flow of H2O2 (~4–10 nmol H2O2/h/106 cells at 5% O2) mirrored these in vivo observations.54 A second mechanism for reducing virulence of pathogens is interference with intrabacterial signaling. In low-oxygen environments H2O2 can cross bacterial membranes and downregulate the bacterial tyrosine signaling network which is essential for numerous metabolic and biosynthetic functions.47,48 Key processes involved in bacterial virulence such as polysaccharide biosynthesis for capsule and biofilm production are under the control of tyrosine kinases and phosphatases.163 Intrabacterial conversion of H2O2 into highly reactive oxygen metabolites together with bacterial peroxidase activity induced oxidative tyrosine dephosphorylation and irreversible DOPA modification of bacterial enzymes.48 Disruption of bacterial tyrosine signaling will weaken bacterial fitness, and decreased encapsulation will facilitate opsonization and phagocytosis by innate immune cells.
Restitution and Mucosal Healing
Restitution of the epithelium following injury involves induced and coordinated proliferation and migration of intestinal epithelial cells, with ROS providing a key signaling mediator for efficient wound healing. Accelerated healing can be stimulated from the extracellular side by exposure to low concentrations of H2O2 (0.15μmole to 1.25μmole H2O2/wound)164,165 or NADPH oxidase-mediated redox signaling.166,167 Outside-in signaling of H2O2 is facilitated by aquaporins61,115,168 and does not always trigger the same intracellular pathways as NOX-mediated signaling. Most NOX enzymes produce initially superoxide (O2•−) and are often part of signaling platforms such as caveolae or lipid rafts. Nonetheless, a review of the literature suggests that redox-mediated effects target usually certain critical signaling mediators, which are connected to the actin cytoskeleton and integrin engagement, both important for cell motility. Increased phosphorylation and activation of focal adhesion kinase (FAK) was mediated by H2O2-inducible Src kinase activation in microvascular endothelial cells, while N-formyl peptide receptor 1-mediated activation of NOX1 induced FAK phosphorylation in colon epithelial cells by inactivation of redox-sensitive low molecular weight (LMW) phosphatases (PTP-PEST, SHP2, PTEN).109,164,169,170 NOX1-mediated inactivation of LMW-PTP modulated the phosphorylation levels of p190RhoGAP and induced transient overactivation of the RhoGTPase RHOA. Redox regulation of a pathway emanating from active RHOA to ROCK and p38MAPK was required for integrin turnover and a switch from random to directional migration of HT29-D4 colon epithelial cells.171 In contrast, in K-Ras transformed kidney cells a Rac1-Nox1 pathway decreased RhoA activity by targeting p190RhoGAP phosphorylation.172 Cell locomotion was altered in smooth muscle cells through Nox1, which was required for the filament severing activity of cofilin by reducing its phosphorylation on Ser-3.173
Dermal wound healing and angiogenesis was reduced in Ncf1−/− mice, presumably due to decreased ROS production by recruited macrophages. This defect in wound closure was corrected by application of H2O2 at a low concentration.164 Similarly, L. rhamnosus GG applied to colonic wounds stimulated proliferation and migration of epithelial cells and enhanced recovery in a DSS colitis model.170 DSS-mediated crypt damage permits access and direct contact of lactobacilli with epithelial cells, and as a result these commensals can stimulate directly Nox1 activity, albeit lactobacillus-generated H2O2 will also be present. Interestingly, Lactobacillus-triggered Nox1 signaling was observed in mouse colon with an intact dense mucus layer, which will require secreted factors such as H2O2, peptides or organic compounds as stimulators of Nox1 activity. Endogenous pro-resolving mediators (ANXA1, Ac2-26, resolvins, lipoxins) promote also colonic wound repair by triggering FPR-Nox1 signaling.174 Taken together, inducing ROS or supplying physiological levels of H2O2 to the intestinal barrier ecosystem accelerates mucosal healing and restitution.
Conclusions and future directions
The general view of ROS as culprits of redox imbalance, damaging oxidative modifications, cell death and chronic inflammation is slowly being revised, not only by recognizing primary ROS as essential signaling molecules, but also by acknowledging diffusible H2O2 for its protective and restorative potential. Wound closure studies have provided an accessible and reproducible tool to determine the efficacy and safety profile of H2O2, which cannot yet be matched on internal mucosal surfaces.164,165 Lactobacilli are used for various intestinal disorders, but assessing qualitatively and quantitatively their mode of action (H2O2, bacteriocins, organic compounds) in a particular stage of disease has only been achieved in rare cases. Analytical methods for measuring individual ROS and RNS selectively in the GIT are needed to gain a better understanding of the relationship between oxygen, ROS and nitric oxide metabolites. Generation of ROS is dependent on the oxygen concentration along the intestine (pO2 3–20 mm Hg),175,176,177 which can vary considerably between the fed/fasted state and homeostasis/inflammation. ROS, RNS and oxygen are interconnected in homeostasis, infection and inflammatory disease178,179,180,181 and any changes in their chemistry and/or availability will induce shifts in the microbiota composition. In order to understand how dysbiotic microbiota develops and how to restore eubiosis quantifiable approaches will be vital. Oxygen consumption will be very high in active inflammatory disease due to the neutrophil oxidative burst, increasing hypoxia in the epithelium.77,182 On the other hand, antibiotic treatment, infection and inflammation are accompanied by the bloom of facultative anaerobic Enterobacteriaceae.178,180 Loss of the majority of Nox/Duox activity (Cyba−/− mice) which will likely increase the availability of oxygen, resulted in overgrowth of facultative anaerobic lactobacilli which will consume oxygen by generating H2O2.54 The abundance of Enterobacteriaceae was not significantly increased in these mice, presumably as nitrate was not abundant and/or reduced by lactobacilli to nitrite.181 Reconciling the chemical environment with quantitative microbiome profiling along the GIT will provide a rational approach for microbiota modification.
Cyba−/− mice mimic AR22CGD patients with loss-of-function CYBA variants. These mutations are extremely rare (6% of total CGD cases; prevalence CGD worldwide 1/220,000), but a high rate of consanguinity in some countries (e.g., Turkey, Tunisia, Iran) can result in 40–50% of CGD patients carrying CYBA variants.183 Most of these patients have severe infections in childhood with an overrepresentation of gastrointestinal infections, which may reflect poor sanitary conditions and not necessarily increased susceptibility to GIT infections due to NOX1–4 inactivation. The protective microbiota that developed in Cyba−/− mice was eliminated by exposure to normal SPF mouse microbiota or to a Western-style diet (low fiber, medium fat, high monosaccharides), suggesting that Lactobacillus overgrowth may not persist in humans. Most CGD patients require aggressive antibacterial or antifungal medication independently of their GIT disease status. In contrast, VEOIBD patients with inactivating NOX1 or DUOX2 variants are usually very young and treatment-naïve when they present with intestinal inflammation. The importance of NOX1 and DUOX2 in host defense and microbiota composition suggests that increased bacterial load, shifts in the microbiota composition, translocation of bacteria into the LP and impaired bacterial clearance will be present in these patients. Other IBD risk factors are also involved in immune dysfunction and some of them participate in signaling pathways (e.g., NOD2, ATG16L1, CARD9 and INAVA) that connect to NADPH oxidases. A recent study with five VEOIBD patients showed encouraging results by targeting gram-positive bacteria with non-absorbable antibiotics,184 supporting the view that restoring eubiosis by modifying bacterial communities with antibiotics or other means to enhance diversity and resilience of beneficial species may become a promising therapeutic option.147,185
References
Egea, J. et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 13, 94–162 (2017).
Pryor, W. A. Oxy-radicals and related species: their formation, lifetimes, and reactions. Ann. Rev. Physiol. 48, 657–667 (1986).
Sies, H. Strategies of antioxidant defense. Eur. J. Biochem. 215, 213–219 (1993).
McCord, J. M. Free radicals and inflammation: protection of synovial fluid by superoxide dismutase. Science 185, 529–531 (1974).
Weissmann, G., Smolen, J. E. & Korchak, H. M. Release of inflammatory mediators from stimulated neutrophils. N. Engl. J. Med. 303, 27–34 (1980).
Rada, B. & Leto, T. L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib. Microbiol. 15, 164–187 (2008).
Winterbourn, C. C. & Kettle, A. J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 18, 642–660 (2013).
Savina, A. et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126, 205–218 (2006).
Vulcano, M. et al. Toll receptor-mediated regulation of NADPH oxidase in human dendritic cells. J. Immunol. 173, 5749–5756 (2004).
Kotsias, F., Hoffmann, E., Amigorena, S. & Savina, A. Reactive oxygen species production in the phagosome: impact on antigen presentation in dendritic cells. Antioxid. Redox Signal. 18, 714–729 (2013).
Oakley, F. D., Abbott, D., Li, Q. & Engelhardt, J. F. Signaling components of redox active endosomes: the redoxosomes. Antioxid. Redox Signal. 11, 1313–1333 (2009).
Chandel, N. S. Mitochondria as signaling organelles. BMC Biol. 12, 34 (2014).
West, A. P., Shadel, G. S. & Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402 (2011).
Cao, S. S. & Kaufman, R. J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 21, 396–413 (2014).
Berger, E. et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 7, 13171 (2016).
El-Benna, J. et al. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol. Rev. 273, 180–193 (2016).
Bedard, K. & Krause, K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 (2007).
Lambeth, J. D. & Neish, A. S. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Ann. Rev. Pathol. 9, 119–145 (2014).
Babior, B. M. The respiratory burst of phagocytes. J. Clin. Invest. 73, 599–601 (1984).
Segal, A. W. Absence of both cytochrome b-245 subunits from neutrophils in X-linked chronic granulomatous disease. Nature 326, 88–91 (1987).
Dinauer, M. C., Orkin, S. H., Brown, R., Jesaitis, A. J. & Parkos, C. A. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature 327, 717–720 (1987).
Nunoi, H., Rotrosen, D., Gallin, J. I. & Malech, H. L. Two forms of autosomal chronic granulomatous disease lack distinct neutrophil cytosol factors. Science 242, 1298–1301 (1988).
Volpp, B. D., Nauseef, W. M. & Clark, R. A. Two cytosolic neutrophil oxidase components absent in autosomal chronic granulomatous disease. Science 242, 1295–1297 (1988).
Knaus, U. G., Heyworth, P. G., Evans, T., Curnutte, J. T. & Bokoch, G. M. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 254, 1512–1515 (1991).
Babior, B. M. NADPH oxidase. Curr. Opin. Immunol. 16, 42–47 (2004).
Groemping, Y. & Rittinger, K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem. J. 386, 401–416 (2005).
O’Neill, S., Brault, J., Stasia, M. J. & Knaus, U. G. Genetic disorders coupled to ROS deficiency. Redox Biol. 6, 135–156 (2015).
van den Berg, J. M. et al. Chronic granulomatous disease: the European experience. PLoS ONE 4, e5234 (2009).
Kuhns, D. B. et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N. Engl. J. Med 363, 2600–2610 (2010).
Deffert, C. et al. Hyperinflammation of chronic granulomatous disease is abolished by NOX2 reconstitution in macrophages and dendritic cells. J. Pathol. 228, 341–350 (2012).
Holland, S. M. Chronic granulomatous disease. Hematol. Oncol. Clin. North Am. 27, 89–99 (2013).
De Ravin, S. S. et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med 9, 372 (2017).
Lambeth, J. D., Cheng, G., Arnold, R. S. & Edens, W. A. Novel homologs of gp91phox. Trends Biochem. Sci. 25, 459–461 (2000).
Banfi, B. et al. A mammalian H+channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science 287, 138–142 (2000).
Dupuy, C. et al. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J. Biol. Chem. 274, 37265–37269 (1999).
De Deken, X. et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 275, 23227–23233 (2000).
Kawahara, T., Quinn, M. T. & Lambeth, J. D. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol. Biol. 7, 109 (2007).
Szanto, I. et al. Expression of NOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J. Pathol. 207, 164–176 (2005).
Ueyama, T., Geiszt, M. & Leto, T. L. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol. Cell. Biol. 26, 2160–2174 (2006).
Takeya, R. et al. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J. Biol. Chem. 278, 25234–25246 (2003).
Banfi, B., Clark, R. A., Steger, K. & Krause, K. H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 278, 3510–3513 (2003).
Kuwano, Y. et al. Tumor necrosis factor alpha activates transcription of the NADPH oxidase organizer 1 (NOXO1) gene and upregulates superoxide production in colon epithelial cells. Free Radic. Biol. Med. 45, 1642–1652 (2008).
Lee, J. H. et al. Interaction of NADPH oxidase 1 with Toll-like receptor 2 induces migration of smooth muscle cells. Cardiovasc. Res. 99, 483–493 (2013).
O’Leary, D. P. et al. TLR-4 signalling accelerates colon cancer cell adhesion via NF-kappaB mediated transcriptional up-regulation of Nox-1. PLoS ONE 7, e44176 (2012).
Lee, H. K. et al. RGS2 is a negative regulator of STAT3-mediated Nox1 expression. Cell Signal. 24, 803–809 (2012).
Kawahara, T. et al. Role of nicotinamide adenine dinucleotide phosphate oxidase 1 in oxidative burst response to Toll-like receptor 5 signaling in large intestinal epithelial cells. J. Immunol. 172, 3051–3058 (2004).
Corcionivoschi, N. et al. Mucosal reactive oxygen species decrease virulence by disrupting campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe 12, 47–59 (2012).
Alvarez, L. A. et al. NADPH oxidase-derived H2O2 subverts pathogen signaling by oxidative phosphotyrosine conversion to PB-DOPA. Proc. Natl Acad. Sci. USA 113, 10406–10411 (2016).
Laurent, E. et al. Nox1 is over-expressed in human colon cancers and correlates with activating mutations in K-Ras. Int. J. Cancer 123, 100–107 (2008).
Sadok, A. et al. Nox1-dependent superoxide production controls colon adenocarcinoma cell migration. Biochim. Biophys. Acta 1783, 23–33 (2008).
Juhasz, A. et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 292, 7866–7887 (2017).
El Hassani, R. A. et al. Dual oxidase2 is expressed all along the digestive tract. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G933–942 (2005).
Grasberger, H. et al. Increased expression of DUOX2 is an epithelial response to mucosal dysbiosis required for immune homeostasis in mouse intestine. Gastroenterology 149, 1849–1859 (2015).
Pircalabioru, G. et al. Defensive mutualism rescues NADPH oxidase inactivation in gut infection. Cell Host Microbe 19, 651–663 (2016).
Fink, K. et al. IFNbeta/TNFalpha synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH oxidase-mediated airway antiviral response. Cell Res. 23, 673–690 (2013).
Strengert, M. et al. Mucosal reactive oxygen species are required for antiviral response: role of Duox in influenza a virus infection. Antioxid. Redox Signal. 20, 2695–2709 (2014).
Harper, R. W. et al. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 579, 4911–4917 (2005).
Wu, Y. et al. Up-regulation and sustained activation of Stat1 are essential for interferon-gamma (IFN-gamma)-induced dual oxidase 2 (Duox2) and dual oxidase A2 (DuoxA2) expression in human pancreatic cancer cell lines. J. Biol. Chem. 286, 12245–12256 (2011).
Sommer, F. & Backhed, F. The gut microbiota engages different signaling pathways to induce Duox2 expression in the ileum and colon epithelium. Mucosal Immunol. 8, 372–379 (2015).
Lipinski, S. et al. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J. Cell Sci. 122, 3522–3530 (2009).
Al Ghouleh, I. et al. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc. Res. 97, 134–142 (2013).
Ha, E. M. et al. Coordination of multiple dual oxidase-regulatory pathways in responses to commensal and infectious microbes in drosophila gut. Nat. Immunol. 10, 949–957 (2009).
Niethammer, P., Grabher, C., Look, A. T. & Mitchison, T. J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459, 996–999 (2009).
Yoo, S. K., Starnes, T. W., Deng, Q. & Huttenlocher, A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature 480, 109–112 (2011).
Pase, L. et al. Neutrophil-delivered myeloperoxidase dampens the hydrogen peroxide burst after tissue wounding in zebrafish. Curr. Biol. 22, 1818–1824 (2012).
Lee, I. A., Bae, E. A., Hyun, Y. J. & Kim, D. H. Dextran sulfate sodium and 2,4,6-trinitrobenzene sulfonic acid induce lipid peroxidation by the proliferation of intestinal gram-negative bacteria in mice. J. Inflamm. (Lond) 7, 7 (2010).
McKenzie, S. J., Baker, M. S., Buffinton, G. D. & Doe, W. F. Evidence of oxidant-induced injury to epithelial cells during inflammatory bowel disease. J. Clin. Invest. 98, 136–141 (1996).
Nair, J. et al. Increased etheno-DNA adducts in affected tissues of patients suffering from Crohn’s disease, ulcerative colitis, and chronic pancreatitis. Antioxid. Redox Signal. 8, 1003–1010 (2006).
Keshavarzian, A. et al. Increases in free radicals and cytoskeletal protein oxidation and nitration in the colon of patients with inflammatory bowel disease. Gut 52, 720–728 (2003).
Tardieu, D. et al. Dextran sulfate enhances the level of an oxidative DNA damage biomarker, 8-oxo-7,8-dihydro-2’-deoxyguanosine, in rat colonic mucosa. Cancer Lett. 134, 1–5 (1998).
Kruidenier, L., Kuiper, I., Lamers, C. B. & Verspaget, H. W. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J. Pathol. 201, 28–36 (2003).
Beltran, B. et al. Mitochondrial dysfunction, persistent oxidative damage, and catalase inhibition in immune cells of naive and treated Crohn’s disease. Inflamm. Bowel. Dis. 16, 76–86 (2010).
Oldenburg, B., van Kats-Renaud, H., Koningsberger, J. C., van Berge Henegouwen, G. P. & van Asbeck, B. S. Chemiluminescence in inflammatory bowel disease patients: a parameter of inflammatory activity. Clin. Chim. Acta 310, 151–156 (2001).
Naito, Y., Takagi, T. & Yoshikawa, T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J. Gastroenterol. 42, 787–798 (2007).
Wera O., Lancellotti P., Oury C. The dual role of neutrophils in inflammatory bowel diseases. J. Clin. Med. 5, pii E118 (2016).
Parkos, C. A. Neutrophil-epithelial interactions: a double-edged sword. Am. J. Pathol. 186, 1404–1416 (2016).
Campbell, E. L. et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40, 66–77 (2014).
Denning, T. L. & Parkos, C. A. Neutrophils enlist IL-22 to restore order in the gut. Proc. Natl Acad. Sci. USA 110, 12509–12510 (2013).
Sumagin, R. et al. Neutrophil interactions with epithelial-expressed ICAM-1 enhances intestinal mucosal wound healing. Mucosal Immunol. 9, 1151–1162 (2016).
MacFie, T. S. et al. DUOX2 and DUOXA2 form the predominant enzyme system capable of producing the reactive oxygen species H2O2 in active ulcerative colitis and are modulated by 5-aminosalicylic acid. Inflamm. Bowel. Dis. 20, 514–524 (2014).
Haberman, Y. et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 124, 3617–3633 (2014).
Mirza, A. H. et al. Transcriptomic landscape of lncRNAs in inflammatory bowel disease. Genome Med. 7, 39 (2015).
Csillag, C. et al. Expression of the genes dual oxidase 2, lipocalin 2 and regenerating islet-derived 1 alpha in Crohn’s disease. Scand. J. Gastroenterol. 42, 454–463 (2007).
Aerssens, J. et al. Alterations in mucosal immunity identified in the colon of patients with irritable bowel syndrome. Clin. Gastroenterol. Hepatol. 6, 194–205 (2008).
Aviello, G. & Knaus, U. G. ROS in gastrointestinal inflammation: rescue or sabotage? Br. J. Pharmacol. 174, 1704–1718 (2017).
Uhlig, H. H. & Muise, A. M. Clinical genomics in inflammatory bowel disease. Trends Genet. 33, 629–641 (2017).
Hayes, P. et al. Defects in NADPH oxidase genes NOX1 and DUOX2 in very early onset inflammatory bowel disease. Cell Mol. Gastroenterol. Hepatol. 1, 489–502 (2015).
Schwerd T., et al. NOX1 loss-of-function genetic variants in patients with inflammatory bowel disease. Mucosal Immunol. 2017.
Parlato, M. et al. First identification of biallelic inherited DUOX2 inactivating mutations as a cause of very early onset inflammatory bowel disease. Gastroenterology 153, 609–611 (2017). e603.
Levine, A. P., Pontikos, N., Schiff, E. R., Jostins, L. & Speed, D., Consortium NIBDG. et al. Genetic Complexity of Crohn’s Disease in Two Large Ashkenazi Jewish Families. Gastroenterology 151, 698–709 (2016).
Dhillon, S. S. et al. Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology 147, 680–689 (2014). e682.
Uhlig, H. H. et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 147, 990–1007 (2014). e1003.
Turner, D. & Muise, A. M. Very Early Onset IBD: How Very Different ‘on Average’? J. Crohns Colitis 11, 517–518 (2017).
Mizoguchi, A. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Transl. Sci. 105, 263–320 (2012).
Kiesler, P., Fuss, I. J. & Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell Mol. Gastroenterol. Hepatol. 1, 154–170 (2015).
Nguyen, T. L., Vieira-Silva, S., Liston, A. & Raes, J. How informative is the mouse for human gut microbiota research? Dis. Models Mech. 8, 1–16 (2015).
Gibbons, D. L. & Spencer, J. Mouse and human intestinal immunity: same ballpark, different players; different rules, same score. Mucosal Immunol. 4, 148–157 (2011).
Gu, S. et al. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE 8, e74957 (2013).
Gozalo, A. S. et al. Spontaneous Staphylococcus xylosus infection in mice deficient in NADPH oxidase and comparison with other laboratory mouse strains. J. Am. Assoc. Lab. Anim. Sci. 49, 480–486 (2010).
Lichtman, J. S. et al. Host-Microbiota Interactions in the Pathogenesis of Antibiotic-Associated Diseases. Cell Rep. 14, 1049–1061 (2016).
Schubert, A. M., Sinani, H. & Schloss, P. D. Antibiotic-induced alterations of the murine gut microbiota and subsequent effects on colonization resistance against Clostridium difficile. mBio 6, e00974 (2015).
Fattouh, R. et al. Rac2-deficiency leads to exacerbated and protracted colitis in response to Citrobacter rodentium infection. PLoS. One. 8, e61629 (2013).
Falcone, E. L. et al. Colitis susceptibility inp47(phox-/-) mice is mediated by the microbiome. Microbiome 4, 13 (2016).
Li, Y. et al. p40 (phox) -Deficient Mice Exhibit Impaired Bacterial Clearance and Enhanced Pro-inflammatory Responses during Salmonella enterica serovar Typhimurium Infection. Front. Immunol. 8, 1270 (2017).
Felmy, B. et al. NADPH oxidase deficient mice develop colitis and bacteremia upon infection with normally avirulent, TTSS-1- and TTSS-2-deficient Salmonella Typhimurium. PLoS ONE 8, e77204 (2013).
Grasberger, H., El-Zaatari, M., Dang, D. T. & Merchant, J. L. Dual oxidases control release of hydrogen peroxide by the gastric epithelium to prevent Helicobacter felis infection and inflammation in mice. Gastroenterology 145, 1045–1054 (2013).
Treton, X. et al. Combined NADPH oxidase 1 and interleukin 10 deficiency induces chronic endoplasmic reticulum stress and causes ulcerative colitis-like disease in mice. PLoS ONE 9, e101669 (2014).
Yokota, H. et al. NOX1/NADPH oxidase expressed in colonic macrophages contributes to the pathogenesis of colonic inflammation in trinitrobenzene sulfonic acid-induced murine colitis. J. Pharmacol. Exp. Ther. 360, 192–200 (2017).
Leoni, G. et al. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J. Clin. Invest. 123, 443–454 (2013).
Rodrigues-Sousa, T. et al. Deficient production of reactive oxygen species leads to severe chronic DSS-induced colitis in Ncf1/p47phox-mutant mice. PLoS ONE 9, e97532 (2014).
Conway, K. L. et al. p40phox expression regulates neutrophil recruitment and function during the resolution phase of intestinal inflammation. J. Immunol. 189, 3631–3640 (2012).
Jakobsson, H. E. et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 16, 164–177 (2015).
Jones, R. M. et al. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J. 32, 3017–3028 (2013).
Imlay, J. A. Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 77, 755–776 (2008).
Thiagarajah, J. R., Chang, J., Goettel, J. A., Verkman, A. S. & Lencer, W. I. Aquaporin-3 mediates hydrogen peroxide-dependent responses to environmental stress in colonic epithelia. Proc. Natl Acad. Sci. USA 114, 568–573 (2017).
Ito, A. et al. The screening of hydrogen peroxide-producing lactic acid bacteria and their application to inactivating psychrotrophic food-borne pathogens. Curr. Microbiol. 47, 231–236 (2003).
Chooruk A., Piwat S., Teanpaisan R. Antioxidant activity of various oral Lactobacillus strains. J. Appl. Microbiol. 2017.
Knaus, U. G. et al. Pathogen control at the intestinal mucosa - H2O2 to the rescue. Gut Microbes 8, 67–74 (2017).
Zeissig, S. et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56, 61–72 (2007).
Vetrano, S. et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology 135, 173–184 (2008).
Blair, S. A., Kane, S. V., Clayburgh, D. R. & Turner, J. R. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Invest. 86, 191–201 (2006).
Katsube, T., Tsuji, H. & Onoda, M. Nitric oxide attenuates hydrogen peroxide-induced barrier disruption and protein tyrosine phosphorylation in monolayers of intestinal epithelial cell. Biochim. Biophys. Acta 1773, 794–803 (2007).
Rao, R. K., Baker, R. D., Baker, S. S., Gupta, A. & Holycross, M. Oxidant-induced disruption of intestinal epithelial barrier function: role of protein tyrosine phosphorylation. Am. J. Physiol. 273, G812–823 (1997).
Tesoriere, L., Attanzio, A., Allegra, M., Gentile, C. & Livrea, M. A. Indicaxanthin inhibits NADPH oxidase (NOX)-1 activation and NF-kappaB-dependent release of inflammatory mediators and prevents the increase of epithelial permeability in IL-1beta-exposed Caco-2 cells. Br. J. Nutr. 111, 415–423 (2014).
Choi, Y. J. et al. Polychlorinated biphenyls disrupt intestinal integrity via NADPH oxidase-induced alterations of tight junction protein expression. Environ. Health Perspect. 118, 976–981 (2010).
Ahrne, S. & Hagslatt, M. L. Effect of lactobacilli on paracellular permeability in the gut. Nutrients 3, 104–117 (2011).
Johansson, M. E., Sjovall, H. & Hansson, G. C. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 10, 352–361 (2013).
Birchenough, G. M., Johansson, M. E., Gustafsson, J. K., Bergstrom, J. H. & Hansson, G. C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 8, 712–719 (2015).
Li, H. et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nat. Commun. 6, 8292 (2015).
Theodoratou, E. et al. The role of glycosylation in IBD. Nat. Rev. Gastroenterol. Hepatol. 11, 588–600 (2014).
Larsson, J. M. et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel. Dis. 17, 2299–2307 (2011).
Johansson, M. E. et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 63, 281–291 (2014).
Vigsnaes, L. K. et al. Microbiotas from UC patients display altered metabolism and reduced ability of LAB to colonize mucus. Sci. Rep. 3, 1110 (2013).
Damiano, S. et al. Dual oxidase 2 generated reactive oxygen species selectively mediate the induction of mucins by epidermal growth factor in enterocytes. Int. J. Biochem. Cell. Biol. 60, 8–18 (2015).
Shao, M. X. & Nadel, J. A. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc. Natl. Acad. Sci. USA 102, 767–772 (2005).
Habibovic, A. et al. DUOX1 mediates persistent epithelial EGFR activation, mucous cell metaplasia, and airway remodeling during allergic asthma. JCI Insight 1, e88811 (2016).
Patel, K. K. et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. Embo. J. 32, 3130–3144 (2013).
Huang, J. et al. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl Acad. Sci. USA 106, 6226–6231 (2009).
Martinez, J. et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17, 893–906 (2015).
Regmi, S. C., Park, S. Y., Ku, S. K. & Kim, J. A. Serotonin regulates innate immune responses of colon epithelial cells through Nox2-derived reactive oxygen species. Free Radic. Biol. Med. 69, 377–389 (2014).
Banskota, S. et al. Serotonin disturbs colon epithelial tolerance of commensal E. coli by increasing NOX2-derived superoxide. Free Radic. Biol. Med. 106, 196–207 (2017).
Birchenough, G. M., Nystrom, E. E., Johansson, M. E. & Hansson, G. C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542 (2016).
Wlodarska, M. et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059 (2014).
Mamantopoulos, M. et al. Nlrp6- and ASC-dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity 47, 339–348 (2017). e334.
Ahl, D. et al. Lactobacillus reuteri increases mucus thickness and ameliorates dextran sulphate sodium-induced colitis in mice. Acta Physiol. 217, 300–310 (2016).
Larsson, E. et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 61, 1124–1131 (2012).
Vieira, A. T., Fukumori, C. & Ferreira, C. M. New insights into therapeutic strategies for gut microbiota modulation in inflammatory diseases. Clin. Transl. Immunol. 5, e87 (2016).
Eeckhaut, V. et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 62, 1745–1752 (2013).
Matziouridou C., et al iNOS- and NOX1-dependent ROS production maintains bacterial homeostasis in the ileum of mice. Mucosal Immunol. 2017.
Green, J. N., Kettle, A. J. & Winterbourn, C. C. Protein chlorination in neutrophil phagosomes and correlation with bacterial killing. Free Radic. Biol. Med. 77, 49–56 (2014).
Levine A. P., Segal A. W. The NADPH oxidase and microbial killing by neutrophils, with a particular emphasis on the proposed antimicrobial role of myeloperoxidase within the phagocytic vacuole. Microbiol. Spectr. 2016; 4.
Gerson, C. et al. The lactoperoxidase system functions in bacterial clearance of airways. Am. J. Respir. Cell Mol. Biol. 22, 665–671 (2000).
Kim, B. W., Esworthy, R. S., Hahn, M. A., Pfeifer, G. P. & Chu, F. F. Expression of lactoperoxidase in differentiated mouse colon epithelial cells. Free Radic. Biol. Med. 52, 1569–1576 (2012).
Bae, Y. S., Choi, M. K. & Lee, W. J. Dual oxidase in mucosal immunity and host-microbe homeostasis. Trends Immunol. 31, 278–287 (2010).
Kumar, S., Molina-Cruz, A., Gupta, L., Rodrigues, J. & Barillas-Mury, C. A peroxidase/dual oxidase system modulates midgut epithelial immunity in Anopheles gambiae. Science 327, 1644–1648 (2010).
Botteaux, A., Hoste, C., Dumont, J. E., Van Sande, J. & Allaoui, A. Potential role of Noxes in the protection of mucosae: H(2)O(2) as a bacterial repellent. Microbes Infect. 11, 537–544 (2009).
Benjamin, J. L., Sumpter, R. Jr, Levine, B. & Hooper, L. V. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 13, 723–734 (2013).
Dickinson, J. D. et al. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol. 14, 272–284 (2018).
Forte, M., Palmerio, S., Yee, D., Frati, G. & Sciarretta, S. Functional role of Nox4 in autophagy. Adv. Exp. Med. Biol. 982, 307–326 (2017).
Dumont, J. E. et al. H2O2, signal, substrate, mutagen and chemorepellent from physiology to biochemistry and disease. Bull. Mem. Acad. R. Med. Belg. 165, 231–234 (2010).
Chu, F. F., Esworthy, R. S., Doroshow, J. H. & Shen, B. NADPH oxidase-1 deficiency offers little protection in Salmonella typhimurium-induced typhlitis in mice. World J. Gastroenterol. 22, 10158–10165 (2016).
Hertzberger, R. et al. H(2)O(2) production in species of the Lactobacillus acidophilus group: a central role for a novel NADH-dependent flavin reductase. Appl. Environ. Microbiol. 80, 2229–2239 (2014).
Grangeasse, C., Nessler, S. & Mijakovic, I. Bacterial tyrosine kinases: evolution, biological function and structural insights. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2640–2655 (2012).
Roy, S., Khanna, S., Nallu, K., Hunt, T. K. & Sen, C. K. Dermal wound healing is subject to redox control. Mol. Ther. 13, 211–220 (2006).
Loo, A. E. et al. Effects of hydrogen peroxide on wound healing in mice in relation to oxidative damage. PLoS ONE 7, e49215 (2012).
Chan, E. C., Jiang, F., Peshavariya, H. M. & Dusting, G. J. Regulation of cell proliferation by NADPH oxidase-mediated signaling: potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol. Ther. 122, 97–108 (2009).
Fu, X. J. et al. NADPH oxidase 1 and its derived reactive oxygen species mediated tissue injury and repair. Oxid. Med. Cell Longev. 2014, 282854 (2014).
Bienert, G. P. & Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 1840, 1596–1604 (2014).
Swanson, P. A. et al. Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc. Natl Acad. Sci. USA 108, 8803–8808 (2011).
Alam, A. et al. Redox signaling regulates commensal-mediated mucosal homeostasis and restitution and requires formyl peptide receptor 1. Mucosal Immunol. 7, 645–655 (2014).
Sadok, A. et al. NADPH oxidase 1 controls the persistence of directed cell migration by a RhoA-dependent switch of α2/α3 integrins. Mol. Cell. Biol. 29, 3915–3928 (2009).
Shinohara, M. et al. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J. Biol. Chem. 282, 17640–17648 (2007).
Lee, M. Y. et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 29, 480–487 (2009).
Leoni, G. & Nusrat, A. Annexin A1: shifting the balance towards resolution and repair. Biol. Chem. 397, 971–979 (2016).
Albenberg, L. et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 147, 1055–1063 (2014). e1058.
Goda, F. et al. In vivo oximetry using EPR and India ink. Magn. Res. Med. 33, 237–245 (1995).
He, G. et al. Noninvasive measurement of anatomic structure and intraluminal oxygenation in the gastrointestinal tract of living mice with spatial and spectral EPR imaging. Proc. Natl Acad. Sci. USA 96, 4586–4591 (1999).
Rivera-Chavez, F., Lopez, C. A. & Baumler, A. J. Oxygen as a driver of gut dysbiosis. Free Radic. Biol. Med. 105, 93–101 (2017).
Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 7, 1256–1261 (2013).
Zeng, M. Y., Inohara, N. & Nunez, G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 10, 18–26 (2017).
Tiso, M. & Schechter, A. N. Nitrate reduction to nitrite, nitric oxide and ammonia by gut bacteria under physiological conditions. PLoS ONE 10, e0119712 (2015).
Taylor, C. T. & Colgan, S. P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 17, 774–785 (2017).
Stasia, M. J. CYBA encodingp22(phox), the cytochrome b558 alpha polypeptide: gene structure, expression, role and physiopathology. Gene 586, 27–35 (2016).
Lev-Tzion, R. et al. Oral vancomycin and gentamicin for treatment of very early onset inflammatory bowel disease. Digestion 95, 310–313 (2017).
Simpson, H. L., Campbell, B. J. & Rhodes, J. M. IBD: microbiota manipulation through diet and modified bacteria. Dig. Dis. 32, 18–25 (2014).
Esworthy, R. S. et al. Nox1 causes ileocolitis in mice deficient in glutathione peroxidase-1 and -2. Free Radic. Biol. Med. 68, 315–325 (2014).
Kato, M. et al. The ROS-generating oxidase Nox1 is required for epithelial restitution following colitis. Exp. Anim. 65, 197–205 (2016).
Krieglstein, C. F. et al. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J. Exp. Med. 194, 1207–1218 (2001).
Bao, S., Carr, E. D., Xu, Y. H. & Hunt, N. H. Gp91(phox) contributes to the development of experimental inflammatory bowel disease. Immunol. Cell Biol. 89, 853–860 (2011).
Woo, H., Okamoto, S., Guiney, D., Gunn, J. S. & Fierer, J. A model of Salmonella colitis with features of diarrhea in SLC11A1 wild-type mice. PLoS ONE 3, e1603 (2008).
Chu, F. F. et al. Deficiency in Duox2 activity alleviates ileitis in GPx1- and GPx2-knockout mice without affecting apoptosis incidence in the crypt epithelium. Redox Biol. 11, 144–156 (2017).
Acknowledgements
The work was supported by Science Foundation Ireland (UGK), the European Crohn’s and Colitis Organization (ECCO) (GA) and the Medical Research Council UK (GA).
Author information
Authors and Affiliations
Contributions
Concept and writing (UGK); figures and writing (GA).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aviello, G., Knaus, U.G. NADPH oxidases and ROS signaling in the gastrointestinal tract. Mucosal Immunol 11, 1011–1023 (2018). https://doi.org/10.1038/s41385-018-0021-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-018-0021-8
This article is cited by
-
Leaf microbiome dysbiosis triggered by T2SS-dependent enzyme secretion from opportunistic Xanthomonas pathogens
Nature Microbiology (2024)
-
Gastrointestinal redox homeostasis in ageing
Biogerontology (2023)
-
The role of NADPH oxidase 1 in alcohol-induced oxidative stress injury of intestinal epithelial cells
Cell Biology and Toxicology (2023)
-
Recombinant ACE2 protein protects against acute lung injury induced by SARS-CoV-2 spike RBD protein
Critical Care (2022)
-
Mesenchymal stem cells against intestinal ischemia–reperfusion injury: a systematic review and meta-analysis of preclinical studies
Stem Cell Research & Therapy (2022)
