
Abstract
The gabapentinoids, gabapentin, and pregabalin, target the α2δ subunits of voltage-gated calcium channels. Initially licensed for pain and seizures, they have become widely prescribed drugs. Many of these uses are off-label for psychiatric indications, and there is increasing concern about their safety, so it is particularly important to have good evidence to justify this usage. We conducted a systematic review and meta-analysis of the evidence for three of their common psychiatric uses: bipolar disorder, anxiety, and insomnia. Fifty-five double-blind randomised controlled trials (RCTs) and 15 open-label studies were identified. For bipolar disorder, four double-blind RCTs investigating gabapentin, and no double-blind RCTs investigating pregabalin, were identified. A quantitative synthesis could not be performed due to heterogeneity in the study population, design and outcome measures. Across the anxiety spectrum, a consistent but not universal effect in favour of gabapentinoids compared to placebo was seen (standardised mean difference [SMD] ranging between -2.25 and -0.25). Notably, pregabalin (SMD -0.55, 95% CI -0.92 to -0.18) and gabapentin (SMD -0.92, 95% CI -1.32 to -0.52) were more effective than placebo in reducing preoperative anxiety. In insomnia, results were inconclusive. We conclude that there is moderate evidence of the efficacy of gabapentinoids in anxiety states, but minimal evidence in bipolar disorder and insomnia and they should be used for these disorders only with strong justification. This recommendation applies despite the attractive pharmacological and genetic rationale for targeting voltage-gated calcium channels.
Similar content being viewed by others

Introduction
The gabapentinoids comprise gabapentin and pregabalin. Gabapentin is licensed for use in the USA for the treatment of focal seizures and post-herpetic neuralgia [1] and in the UK for focal seizures and peripheral neuropathic pain [2]. Pregabalin has similar indications, as well as for fibromyalgia in the USA and generalised anxiety disorder (GAD) in the UK [1, 2]. Since their introduction in 1993, these drugs have become two of the most commonly prescribed medications [3], with a significant proportion likely to have been off-label. An early study of 105 Medicaid patients showed that 95% of prescriptions for gabapentin were for off-label indications, at least 10% of which were for psychiatric disorders [4]. In the UK, at least half of all gabapentinoid prescriptions are off-label, and one in five are co-prescribed with opioids [5]. A recent survey using the US-based TriNetX electronic health records network [6] showed that gabapentin had been prescribed at least once in 13.6% of patients with bipolar disorder (BD), 11.5% with anxiety disorders, and 12.7% with insomnia disorder; for pregabalin, the figures were 2.9%, 2.6%, 3.0% respectively (PJH, unpublished observations).
Despite the widespread off-label use of gabapentin in BD, the evidence for its efficacy is unclear [7]. Gabapentin was not found to be effective over placebo in a comprehensive network meta-analysis of pharmacologic treatments in acute mania [8]. Systematic reviews of gabapentin treatment in psychiatric and/or substance use disorders showed inconclusive evidence for efficacy in BD, but possible efficacy for some anxiety disorders [9, 10]. These studies did not examine pregabalin, did not attempt a quantitative synthesis, and only included published studies.
Beyond its licensed use for GAD in the UK, pregabalin has been used in the management of other anxiety disorders [11] and acute anxiety states such as preoperative anxiety [12]. The efficacy of pregabalin is well established in GAD [13] but is unclear in other anxiety diagnoses such as social anxiety disorder [14]. A review of pregabalin for preoperative anxiolysis did not attempt a quantitative synthesis [15]. Gabapentinoids may also be of benefit in the treatment of insomnia in some conditions [16], but again the evidence is unclear.
Overall, these studies show that there is limited evidence for the efficacy of gabapentinoids in several of the disorders for which they are used. Moreover, these drugs have potential harms, including a range of serious side-effects, and evidence of misuse and addiction potential [17,18,19,20], leading to their re-categorisation as controlled drugs in the UK in 2019. In the United States, pregabalin (but not gabapentin) has been a federally controlled drug since market availability because of its potential for dependence and abuse [21].
A priori, gabapentinoids represent attractive candidate molecules for psychiatric indications because of their mode of action. Despite the nomenclature, they act primarily by inhibiting neuronal voltage-gated calcium channel (VGCC) currents, by binding to the α2δ auxiliary subunit that regulates channel trafficking and function [22]. Though genes encoding α2δ subunits have not thus far been associated with psychiatric phenotypes, other VGCC genes – particularly CACNA1C, that encode the α-1 subunit of the CaV1.2 L-type VGCC subtype – show robust, transdiagnostic associations with multiple disorders, including BD [23,24,25,26]. Furthermore, the prescription of calcium channel blockers, which block the α-1 subunit of L-type VGCCs, is associated with reduced hospitalisations in patients with severe mental illness [27]. More broadly, dysregulation of cellular calcium is observed in patients with BD and some other psychiatric disorders [28,29,30].
Given the potential harms of gabapentinoids, and their widespread off-label prescribing, it is important to assess rigorously the evidence base for their use. Thus, we have carried out a systematic review and meta-analysis of the efficacy, acceptability and tolerability of gabapentin and pregabalin in the treatment of BD, anxiety and insomnia. To complement this analysis, we discuss further the rationale for their use in psychiatric indications.
Methods
Search strategy
To identify published studies, we searched the Cochrane Central Register of Controlled Trials (CENTRAL), EMBASE, MEDLINE, MEDLINE In-Process and PsycINFO from their inception to 4th August 2020 (see published protocol [7] and Supplementary Appendix for full information about search terms). For unpublished studies, we searched international trial registries (ClinicalTrials.gov; the International Clinical Trials Registry Platform [ICTRP], www.who.int/clinical-trials-registry-platform/the-ictrp-search-portal) and the websites of the following regulatory agencies: the European Medicines Agency, the United States Food and Drug Administration, the Medicines and Healthcare Products Regulatory Agency in the UK, the Medicines Evaluation Board, The Medical Products Agency, the Pharmaceuticals and Medical Devices Agency, and the Therapeutic Goods Administration in Australia. Reference lists of included studies were hand-checked for relevant papers and systematic reviews.
Selection criteria
To assess efficacy and acceptability (dropout rate), we included double-blind, randomised controlled trials (DB-RCT) comparing gabapentin or pregabalin, in any dose, frequency, route of administration or setting, with placebo or any other active pharmacological treatment. RCTs investigating adjunctive gabapentin/pregabalin to pre-existing treatment and trials allowing rescue medications were included if pre-existing treatments or rescue medications were evenly distributed in the experimental and comparator intervention arms. To assess tolerability (adverse effects), we included randomised and non-randomised studies, irrespective of blinding. Crossover trials were only included if data from the first period, prior to crossover, were available. Cluster randomised trials were excluded. Patients of any age, sex, ethnicity, and clinical setting were included, but we excluded studies with patients with serious medical illnesses.
For BD, we included studies that assessed both acute treatment (follow-up of 3 weeks for manic and mixed episodes; 8 weeks for depression) and long-term treatment (>12 weeks). We included studies with patients of any BD subtype based on standardised diagnostic criteria, i.e., the International Classification of Diseases (ICD) and the Diagnostic and Statistical Manual of Mental Disorders (DSM). Studies were excluded if a diagnosis of BD was defined using cut-off scores on screening questionnaires, or if patients reported a concurrent Axis I disorder (excluding comorbid anxiety or insomnia) [7].
For anxiety, we included studies of any anxiety disorder as defined in ICD or DSM. We also included studies investigating preoperative anxiety. Studies were excluded if patients reported a concurrent Axis I disorder (excluding BD or insomnia) [7].
For insomnia, we included studies investigating sleep or insomnia, including healthy populations and those with a defined sleep disorder [7].
Three authors (LZA, JSWH, AA) independently screened titles and abstracts generated by the search strategies. Full-text articles were reviewed for inclusion and the relevant information was extracted from included trials. Where both published and unpublished data were available, data from unpublished sources were prioritised in the case of any discrepancies. We contacted authors for additional data if necessary. Graphical data were extracted using WebPlotDigitizer version 4.4 (https://automeris.io/WebPlotDigitizer). The risk of bias for each study was independently assessed by four authors (LZA, JSWH, NA, and AA) using the Cochrane Risk of Bias tool [31]. Any disagreements were resolved by consensus with another team member.
Outcomes
Primary outcome
For the acute treatment of BD, our primary outcome was the efficacy of gabapentin or pregabalin as measured by the following: (i) number of hospital admissions during the study period, (ii) length of hospital admission, (iii) change on validated manic or depressive symptom rating scales from baseline, (iv) change on validated psychotic symptom rating scales from baseline, (v) response to treatment (i.e., at least 50% improvement on any validated rating scale), and (vi) time to cessation of additional treatment for manic/depressive symptoms. For the long-term treatment of BD, our primary outcome was efficacy measured by the following: (i) time to recurrence of any mood episodes, (ii) number of recurrences of any mood episodes during the trial period, and (iii) number of recurrences of manic, mixed, or depressive episodes. For the acute and long-term treatment of anxiety, our primary outcome was efficacy as measured by a change in validated and standardised anxiety rating scales. For the acute and long-term treatment of insomnia, our primary outcome was efficacy measured by the following: (i) objectively measured or self-reported sleep time, (ii) self-reported sleep quality, and (iii) sleep onset latency.
Secondary outcomes
Secondary outcomes included the acceptability and tolerability of gabapentin or pregabalin in acute/long-term treatment of BD, anxiety, and insomnia. Acceptability was measured as follows: (i) participants dropping out of treatment due to any cause, (ii) participants dropping out of treatment due to adverse events, and (iii) participants dropping out of treatment due to inefficacy. Tolerability was measured as follows: (i) the number of participants experiencing at least one side effect and (ii) the number of participants experiencing a pre-specified list of side effects in the British National Formulary [2].
Data analysis
Continuous outcomes were calculated as mean differences (MDs) or standardised MDs (SMDs) with 95% confidence intervals (CI). For studies where the continuous outcome of interest was reported in median/interquartile range (IQR)/range, the method by Wan and colleagues [32] was used to convert values to mean/standard deviation (SD). To deal with the multiplicity of continuous outcome measures, we selected the most relevant outcome guided by previous literature relevant to the diagnostic category, e.g., in GAD, we used the baseline-to-endpoint change in the Hamilton Anxiety Rating Scale (HAM-A) where possible [13]. In preoperative anxiety studies, we used the primary outcome measure of preoperative anxiety recorded at 1–2 h post-drug administration and closest to the time of the induction of anaesthesia. For dichotomous outcomes, risk ratios (RRs) were calculated with 95% CI. For studies with multiple treatment arms of the same type of interventional drug, the mean/SDs were combined following methods described in the Cochrane Handbook (https://training.cochrane.org/handbook/current) and elsewhere [33].
Random effects meta-analyses were conducted, as appropriate, within each disorder [7] as well as across the spectrum of anxiety-related disorders. The grouping of this anxiety spectrum was decided post-hoc to include any disorder classified as an ‘anxiety disorder’ under DSM or ICD classifications, as well as acute anxiety states. Heterogeneity between studies was assessed by visual inspection of forest plots and I2 statistics. Cases of significant heterogeneity were investigated through subgroup analyses of dose. The following sensitivity analyses were undertaken: (i) excluding trials of gabapentin/pregabalin as adjunctive treatment, (ii) excluding trials involving patients with psychiatric comorbidities, (iii) excluding trials allowing rescue medications and (iv) excluding trials where the outcome measure of interest was originally reported in median/IQR/range. Funnel plots were visually inspected for asymmetry (small study effect) in meta-analyses containing at least 10 studies. Statistical analyses were conducted using Review Manager [34] and R [35]. Data that could not be meta-analysed are presented narratively in a table. The certainty of the evidence was estimated using the Grading of Recommendations, Assessment, Development and Evaluation (GRADE) framework [36]. This study is registered with PROSPERO (registration number CRD42016041802), and we followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [37].
Results
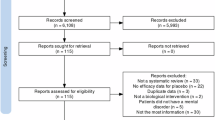
Out of 4268 records initially identified, 70 studies were selected for inclusion: 55 DB-RCTs and 15 open-label studies (Fig. 1). One open-label study [38] included an observational component. The risk of bias was unclear for the majority of studies in many domains and a significant proportion of trials were at high risk in terms of attrition bias (Supplementary Appendix, Figs. 1 and 2). A full description of the results is reported in the Supplementary Appendix (Figs. 3–25 and Tables 1–6).

Flowchart of included and excluded studies.
Bipolar disorder (BD)
Four DB-RCTs investigating the efficacy of gabapentin in BD were identified. 101 patients were randomised to receive gabapentin, 81 to placebo, 30 to lamotrigine and 19 to carbamazepine. The mean age of all randomised patients was 37.5 years and 64.1% were female.
Acute treatment
Three studies compared gabapentin with three different comparators for the acute treatment of BD across heterogeneous patient groups with manic/hypomanic, depressed and/or mixed symptoms [39,40,41] (Table 1). Each study assessed the efficacy of gabapentin using a different outcome measure. Quantitative analysis was not performed due to clinical and methodological heterogeneity.
Gabapentin was significantly more effective than lamotrigine and carbamazepine in reducing depressive symptoms on the Minnesota Multiphasic Personality Inventory-2 (MMPI-2) depression subscale (50%, 33.5%, and 13.6% reduction, respectively), but there were no group differences in improvements on the MMPI-2 mania subscale [39]. A cross-over study [40] observed response rates of 50% for lamotrigine, 33% for gabapentin, and 18% for placebo on the Clinical Global Impressions-Bipolar Version (CGI-BP) during the first phase, but without clear statistical significance. A third study, testing adjunctive gabapentin vs. placebo, reported a significantly greater improvement in the total Young Mania Rating Scale (YMRS) score in the placebo group but no significant between-group difference in the Hamilton Rating Scale for Depression (HAM-D) change score [41].
Long-term treatment
One long-term treatment study of 25 patients with BD in clinical remission (13 on gabapentin, 12 on placebo) [42] showed a significant benefit of gabapentin versus placebo on CGI-BP change scores (gabapentin: -2.1, placebo: -0.6) (Table 1, Supplementary Appendix Tables 1 and 2).
Anxiety disorders or states
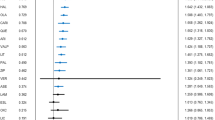
Forty-two DB-RCTs investigating anxiety disorders/states (GAD; social anxiety disorder, SAD; preoperative anxiety; post-traumatic stress disorder, PTSD; obsessive-compulsive disorder, OCD; panic disorder, PD) were selected for inclusion. 3539 patients were randomised to receive pregabalin, 525 to gabapentin, 2280 to placebo, and 937 to active comparators. The mean age of all randomised patients was 43.0 years, and 60.4% were female. 5190 patients from eligible anxiety studies were analysed for efficacy. Gabapentinoids were significantly more effective than placebo across a range of disorders within the anxiety spectrum (SMDs between -2.25 and -0.25) (Fig. 2). Findings for PTSD, OCD and PD are also presented narratively below, as only one DB-RCT was identified for each of these disorders.

CI Confidence interval, IV Inverse variance, SD Standard deviation.
The side effect profile of gabapentinoids was comparable across the anxiety studies. Nineteen studies were included to examine the tolerability of pregabalin in GAD, SAD, PTSD, OCD and PD of that 17 reported side-effect outcomes (Supplementary Appendix, Table 6). Two DB-RCTs reported side-effect outcomes of gabapentin in SAD and PD (Supplementary Appendix, Table 6). For studies of preoperative anxiety, assessment of tolerability was limited due to inconsistent reporting of preoperative side effects across trials. Common side effects of gabapentin and pregabalin included drowsiness, dry mouth, dizziness, headache, fatigue and visual disturbance (see Supplementary Appendix, Table 6).
Generalised anxiety disorder (GAD)
Pregabalin was significantly more effective than placebo in treating GAD (SMD -0.37, 95% CI -0.45 to -0.29) (Fig. 2). Sensitivity analyses excluding studies in which pregabalin was used as adjunctive treatment, participants had psychiatric comorbidities, or rescue medications were allowed did not significantly alter the effect estimate or estimate of uncertainty (Supplementary Appendix, Table 3). The RR of all-cause drop-out was comparable for pregabalin and placebo. Compared to placebo, pregabalin significantly reduced the RR of drop-out due to inefficacy (RR 0.44, 95% CI 0.28 to 0.70), but showed a trend increase in RR of drop-out due to adverse events (RR 1.30, 95% CI 0.99–1.71) (Supplementary Appendix, Fig. 21). Primary and secondary outcomes relating to the efficacy and acceptability of pregabalin versus lorazepam and venlafaxine are presented in the supplementary analyses (Supplementary Appendix, Figs. 23 and 24). Visual inspection of the funnel plot did not show evidence of small-study effects (Supplementary Appendix, Fig. 6).
Social anxiety disorder (SAD)
Acute treatment
Pregabalin was significantly more effective than placebo in the acute treatment of SAD (SMD -0.25, 95% CI -0.45 to -0.04) (Fig. 2). There was no significant difference in the RR of all-cause drop-out or drop-out due to adverse events (Supplementary Appendix, Figure 8). Compared to placebo, pregabalin showed a trend reduction in RR of drop-out due to inefficacy (RR 0.39, 95% CI 0.15 to 1.03) (Supplementary Appendix, Figure 22).
Long-term treatment
A 14-week study of gabapentin in SAD [43] showed significant improvement in symptoms compared to placebo across multiple clinician-evaluated and self-report rating scales. A 26-week study [44] investigating pregabalin for relapse prevention found that a fixed dose of 450 mg/day significantly reduced the overall relapse rate compared to placebo (27.5% vs. 43.8%, based on CGI-Improvement/CGI-Severity criteria).
Preoperative anxiety
Compared with placebo, both pregabalin (SMD -0.55, 95% CI -0.92 to -0.18) and gabapentin (SMD -0.92, 95% CI -1.32 to -0.52) were significantly more effective in reducing preoperative anxiety (Fig. 2). Significant heterogeneity was found between these studies, which was investigated post-hoc through subgroup analyses based on empirically guided dose thresholds of dose-dependent efficacy [45, 46]. High doses (>600 mg) of gabapentin were found to be significantly effective in reducing preoperative anxiety (SMD -1.30, 95% CI -1.72 to -0.87), but low doses (600 mg) were not (Fig. 3). A high degree of heterogeneity across studies in the high dose group remained (I2 = 81%), hence the validity of the high dose effect estimate is uncertain. Visual inspection of a funnel plot for gabapentin versus placebo in preoperative anxiety suggested possible small study effects, with greater effect size being observed in high-dose gabapentin studies with smaller samples (Supplementary Appendix, Fig. 5.1). There was no significant subgroup difference between low (≤150 mg) and high (300 mg) doses of pregabalin (Fig. 4). Interestingly, low-dose pregabalin was significantly more effective than placebo (SMD -0.29, 95% CI -0.54 to -0.05), whilst the high dose was not. It is worth noting that the splitting of patients in the placebo arm across the intervention arms, to facilitate dose-related subgroup analysis, resulted in a slightly reduced overall effect estimate (SMD -0.43, 95% CI -0.73 to -0.13) (Fig. 4) compared with the original analysis (Fig. 2), but the overall effect remained significant. Sensitivity analyses did not significantly alter the results (Supplementary Appendix, Tables 4 and 5). It was not feasible to perform meta-analyses for the acceptability of pregabalin and gabapentin in preoperative anxiety studies. Short outcome assessment times (lasting hours rather than weeks) meant that dropouts occurred in very few studies. In such cases, dropouts were due to practicalities of preoperative procedures and not due to participant decisions.

CI Confidence interval, IV Inverse variance, SD Standard deviation.

CI Confidence interval, IV Inverse variance, SD Standard deviation.
PTSD, OCD, and PD
Three DB-RCTs were included, two assessing adjunctive pregabalin for the treatment of PTSD [47] and OCD [48] and one assessing gabapentin for the treatment of PD [49] (also see Supplementary Appendix, Table 2). Quantitative synthesis was not conducted due to clinical and methodological heterogeneity. Adjunctive pregabalin was more effective than placebo in combat-related chronic PTSD (significantly reduced the severity of symptoms on the PTSD checklist military version at 6 weeks, but not HAM-A, HAM-D, or Spitzer Quality of Life Index) and SSRI-resistant OCD (significantly reduced score on the Yale-Brown Obsessive-Compulsive Scale at 12 weeks). However, eight weeks of flexibly dosed gabapentin was not more effective than placebo in patients with PD (comparable baseline-to-endpoint change on the Panic and Agoraphobia Scale).
Insomnia/Sleep disturbance
Eight DB-RCTs assessed the efficacy of gabapentin on sleep-related outcomes. Of two trials with sufficient data to conduct a random-effects meta-analysis [50, 51], 111 patients were randomized to receive gabapentin and 60 to placebo (mean age 44.8 years; 44.9% female). In participants with alcohol dependence and related sleep disturbance, gabapentin did not demonstrate improvement in sleep scales compared to placebo (Supplementary Appendix, Figs. 12 and 13). The RR for all-cause drop-out was comparable for gabapentin and placebo (RR 0.96, 95% CI 0.59–1.57) (Supplementary Appendix, Fig. 25). Dropouts due to adverse events and inefficacy could not be assessed as there was no incidence of dropouts contributing to either outcome. Side effect data are reported in the supplementary material (Supplementary Appendix, Table 6).
Just one DB-RCT [52] assessed the efficacy of pregabalin (n = 121) vs. venlafaxine (n = 125) and placebo (n = 128) on sleep-related outcomes (mean age of all randomised patients 40.8 years, 60.7% female). Pregabalin significantly reduced scores on the Medical outcomes study sleep scale and sleep problems index II compared with placebo at weeks 4 and 8.
Discussion
We conducted a systematic review on the efficacy, acceptability and tolerability of gabapentinoids in BD, anxiety, and insomnia/sleep disturbance. By comparison with prior studies, our systematic review covered more disorders and databases, considered only DB-RCTs for evidence synthesis on efficacy, included unpublished literature, and, as far as possible, conducted quantitative syntheses. Our study shows that there is minimal evidence to support the use of gabapentinoids in BD and insomnia. The moderate effect size was seen across the anxiety spectrum; this was also significant for several individual anxiety states and showed a dose-effect of gabapentin in preoperative anxiolysis.
In BD, the small number of DB-RCTs investigating gabapentin, and the absence of studies investigating pregabalin, highlight the sparse evidence based on that to evaluate the efficacy of gabapentinoids in the treatment of BD. Quantitative synthesis was not performed due to heterogeneity in study population, design, and outcome measures. The evidence is inconclusive and does not support the current use of gabapentinoids in the management of BD.
Our analysis of all anxiety-related studies showed statistically significant low to large effect sizes favoring gabapentinoid use, compared to placebo, across the spectrum of anxiety disorders/states. This transdiagnostic effect is supported by the fact that state, trait, and pathological anxiety are mediated by a common brain network [53, 54] and by the common neural phenotypes observed across anxiety disorders [55]. Our approach necessitated the inclusion of baseline-to-endpoint change scores as well as post-intervention scores, but a study of 21 meta-analyses found that combining post-intervention and change scores produces valid meta-analytical results [56]. The results suggest a dose-dependent effect of preoperative gabapentin, with >600 mg being required for significant acute anxiolysis. These analyses were performed post-hoc to investigate the large between-study heterogeneity and therefore should be considered as hypothesis-generating to guide future studies. The choice of dose thresholds was based on empirical reports of dose-dependent effects in GAD, which is a limitation of this subgroup analysis. For gabapentin, a dose-response pattern has been observed in GAD with remission/mild anxiety on total daily doses of gabapentin ≥900 mg/day and recurrence of severe anxiety, suggesting ineffectiveness, at <600 mg/day [46]. For pregabalin, we used a similar approach based on a reported difference in efficacy between 150 mg/day versus 200–600 mg/day in GAD [45].
Our meta-analysis of studies of alcohol-related insomnia found that gabapentin was not significantly better than placebo in improving self-reported sleep quality. One relevant study [50] of alcohol-related insomnia reported total sleep time and sleep onset latency but did not show a significant benefit of gabapentin over placebo on these outcome measures. Two studies of gabapentin versus placebo in healthy adults with occasional sleep disturbance [57, 58] suggest a limited benefit in this population for sleep quality and total sleep time. Gabapentinoids may improve sleep in patients with a range of clinical conditions, such as GAD- and neuropathic pain-associated insomnia [59], but the extent to which sleep parameters are affected by direct or indirect effects in these conditions is unclear. Whilst studies of pregabalin versus placebo in GAD included in our review reported improvements in insomnia subscales of HAM-A, these were not meta-analyzed due to concerns regarding confounding and the validity of HAM-A subscales in insomnia.
The limited evidence for the efficacy of gabapentinoids in several of the conditions we studied here is complemented by good evidence of their side effects and potential harms. Consistent with previous reports [1], our findings show that gabapentinoids commonly cause side effects associated with central nervous system depression, such as drowsiness and dizziness, which may be associated with an increased risk of accidental physical injuries and road traffic incidents [20]. There is also increasing concern about the addictive potential of gabapentinoids, and the harms when used concurrently with opioids [17, 60]. Another important limitation of the retrieved evidence is the methodological quality and the risk of bias of the included studies. The prominent side-effect profiles of gabapentinoids [61] may be associated with difficulties in maintaining the blinding of patients and participants, and this may have led to the overestimation of effect sizes based on subjective outcomes [62]. Therefore, trial results suggesting gabapentinoid efficacy should be interpreted with caution, particularly when such findings form the basis for their use in unlicensed indications. Against this background, the extensive off-label usage of gabapentinoids appears unwarranted and requires closer scrutiny.
The rationale for α2δ ligands in psychiatry
Neurobiological and pharmacological considerations supporting the candidacy of a drug target should never trump the empirical clinical evidence regarding efficacy and safety. Nevertheless, they may be useful when evaluating the potential for further investigations of the target where evidence is limited.
As outlined earlier, a role for VGCCs in psychiatric disorders is supported by genomic data. The evidence includes both common and rare variants and is most robust for schizophrenia [25] and BD [26], but VGCC associations are observed across a range of psychiatric disorders [63], albeit not (yet) for anxiety disorders [64]. However, the genetic associations to VGCCs are primarily with α-1 and β subunits; we are unaware of robust evidence directly implicating α2δ subunit genes.
Furthermore, the desired nature and direction of the manipulation required for therapeutic benefit remains to be determined. Whilst the existing VGCC data are based on channel blockade [27, 65], alternative and more nuanced approaches are likely to be important. For example, VGCC genes encode multiple isoforms [66] with different properties, including sensitivity to the existing channel blockers [67], and with differential expression between tissues [68]. The impact of rare mutations also hints at the need to modulate, rather than simply block, VGCC function in psychiatric disorders. In the case of CACNA1C, for example, gain-of-function mutations cause Timothy Syndrome, in which autism is a cardinal feature [69], but autism has also been reported as a feature with loss-of-function mutations [70]. The latter findings are consistent with the presence of psychiatry-relevant phenotypes in Cacna1c heterozygous rats [71], which have reduced gene dosage. Taken together, these findings highlight the need for therapeutic agents that are capable of fine-tuning function, perhaps by targeting specific isoforms, or acting homeostatically, in order to maximize clinical benefit and minimize side effects.
Targetting the α2δ subunits, as the gabapentinoids do, provides a potential means to achieve nuanced modulation of VGCC function. α2δ subunits increase the density of VGCCs on the plasma membrane, direct trafficking of these channels to subcellular sites, and enhance function by altering their biophysical properties [72]. Conversely, gabapentin decreases the number of α2δ and α-1 subunits on the cell surface [73] and reduces VGCC currents [74], suggesting an inhibitory role. However, the precise effect of gabapentin on calcium currents depends on the stoichiometry of VGCC auxiliary subunits [74] which, like the other VGCC subunits, differ in abundance between tissues, raising the possibility that gabapentinoids may differentially affect VGCC currents in different cell types. Furthermore, it is possible that the gabapentinoids’ effects on anxiety are mediated by α2δ-dependent, but VGCC-independent, mechanisms [75]. Notably, α2δ-1 interacts with NMDA receptors (NMDARs) to promote dendritic spine maturation and NMDAR trafficking to synaptic sites [76]. Thus, as well as clarifying the clinical effects of the gabapentinoids, it will be of interest to establish the underlying molecular mechanisms in order to illuminate pathophysiology and identify novel therapeutic strategies. Ultimately, a personalized medicine approach may well be appropriate [77], given the genomic and other contributions to VGCC function and involvement in psychiatric disorders [78].
Future directions for clinical research: Targeted treatment of anxiety in BD
Gabapentinoids, as shown here, are broadly efficacious across the anxiety spectrum, and it is likely that this reflects a pharmacologic effect on transdiagnostic anxiety phenotypes mediated by α2δ-dependent mechanisms. Anxiety is the most common co-morbidity in patients with BD [79], reflecting in part a shared genetic predisposition [80]. It is also associated with a greater symptom burden and a range of worse clinical outcomes [81]. However, there are to date no clinical trials investigating the efficacy of gabapentinoids in the treatment of anxiety in BD. Therefore, a future area of research would be to explore the targeted treatment of ‘bipolar anxiety’ using gabapentinoids or modified α2δ ligands.
Conclusions
The results of this systematic review and meta-analysis show that the widespread and often off-label psychiatric prescribing of gabapentinoids is not supported by robust evidence except for some anxiety states. Thus, despite the attractive genetic and pharmacological rationale for their use, caution is indicated, and further evidence of efficacy and safety is required. It may also be possible to develop modified α2δ ligands, targeting particular subtypes or isoforms, with a more beneficial therapeutic profile.
References
Goodman CW, Brett AS. A clinical overview of off-label use of gabapentinoid drugs. JAMA Intern Med. 2019;179:695–701.
Joint Formulary Committee. British National Formulary (online): London: BMJ Group and Pharmaceutical Press http://www.medicinescomplete.com/ [Last accessed on 9th June 2021].
Goodman CW, Brett AS. Gabapentin and pregabalin for pain — is increased prescribing a cause for concern? N. Engl J Med. 2017;377:411–4.
Hamer AM, Haxby DG, McFarland BH, Ketchum K. Gabapentin use in a managed Medicaid population. J Manag Care Spec Pharm. 2002;8:266–71.
Montastruc F, Loo SY, Renoux C. Trends in first gabapentin and pregabalin prescriptions in primary care in the United Kingdom, 1993-2017. JAMA 2018;320:2149–51.
Taquet M, Geddes JR, Husain M, Luciano S, Harrison PJ. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records. Lancet Psychiatry. 2021;8:416–27.
Houghton KT, Forrest A, Awad A, Atkinson LZ, Stockton S, Harrison PJ, et al. Biological rationale and potential clinical use of gabapentin and pregabalin in bipolar disorder, insomnia and anxiety: protocol for a systematic review and meta-analysis. BMJ Open. 2017;7:e013433.
Cipriani A, Barbui C, Salanti G, Rendell J, Brown R, Stockton S, et al. Comparative efficacy and acceptability of antimanic drugs in acute mania: a multiple-treatments meta-analysis. Lancet 2011;378:1306–15.
Berlin RK, Butler PM, Perloff MD. Gabapentin therapy in psychiatric disorders: a systematic review. Prim Care Companion CNS Disord. 2015;17, https://doi.org/10.4088/PCC.15r01821.
Ahmed S, Bachu R, Kotapati P, Adnan M, Ahmed R, Farooq U, et al. Use of gabapentin in the treatment of substance use and psychiatric disorders: a systematic review. Front Psychiatry. 2019;10:228 https://doi.org/10.3389/fpsyt.2019.00228.
Blanco C, Bragdon LB, Schneier FR, Liebowitz MR. The evidence-based pharmacotherapy of social anxiety disorder. Int J Neuropsychopharmacol. 2013;16:235–49.
Nutt D, Mandel F, Baldinetti F. Early onset anxiolytic efficacy after a single dose of pregabalin: double-blind, placebo- and active-comparator controlled evaluation using a dental anxiety model. J Psychopharmacol. 2009;23:867–73.
Slee A, Nazareth I, Bondaronek P, Liu Y, Cheng Z, Freemantle N. Pharmacological treatments for generalised anxiety disorder: a systematic review and network meta-analysis. Lancet 2019;393:768–77.
Williams T, McCaul M, Schwarzer G, Cipriani A, Stein DJ, Ipser J. Pharmacological treatments for social anxiety disorder in adults: a systematic review and network meta-analysis. Acta Neuropsychiatr. 2020;32:169–76.
Torres-González MI, Manzano-Moreno FJ, Vallecillo-Capilla MF, Olmedo-Gaya MV. Preoperative oral pregabalin for anxiety control: a systematic review. Clin Oral Investig. 2020;24:2219–28.
Atkin T, Comai S, Gobbi G. Drugs for insomnia beyond benzodiazepines: pharmacology, clinical applications, and discovery. Pharm Rev. 2018;70:197–245.
Bonnet U, Scherbaum N. How addictive are gabapentin and pregabalin? A systematic review. Eur Neuropsychopharmacol. 2017;27:1185–215.
Morrison EE, Sandilands EA, Webb DJ. Gabapentin and pregabalin: do the benefits outweigh the harms? J R Coll Physicians Edinb 2017;47:310–3.
Goodman CW, Brett AS. Gabapentinoids for pain: potential unintended consequences. Am Fam Physician. 2019;100:672–5.
Molero Y, Larsson H, D’Onofrio BM, Sharp DJ, Fazel S. Associations between gabapentinoids and suicidal behaviour, unintentional overdoses, injuries, road traffic incidents, and violent crime: population based cohort study in Sweden. BMJ. 2019;365:l2147.
Schjerning O, Rosenzweig M, Pottegård A, Damkier P, Nielsen J. Abuse potential of pregabalin: a systematic review. CNS Drugs. 2016;30:9–25.
Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J, et al. Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci USA. 2008;105:3628–33.
Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ, et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol. 2015;134:36–54.
Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 2019;179:1469–82 e11.
Ripke S, Walters JT, O’Donovan MC. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv. 2020:2020.09.12.20192922.
Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JRI, Qiao Z, et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet. 2021;53:817–29.
Hayes JF, Lundin A, Wicks S, Lewis G, Wong ICK, Osborn DPJ, et al. Association of hydroxylmethyl glutaryl coenzyme A reductase inhibitors, L-type calcium channel antagonists, and biguanides with rates of psychiatric hospitalization and self-harm in individuals with serious mental illness. JAMA Psychiatry. 2019;76:382–90.
Harrison PJ, Cipriani A, Harmer CJ, Nobre AC, Saunders K, Goodwin GM, et al. Innovative approaches to bipolar disorder and its treatment. Ann N. Y Acad Sci. 2016;1366:76–89.
Harrison PJ, Hall N, Mould A, Al-Juffali N, Tunbridge EM. Cellular calcium in bipolar disorder: systematic review and meta-analysis. Mol Psychiatry. 2019. https://doi.org/10.1038/s41380-019-0622-y. Epub ahead of print.
Berridge MJ. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013;7:2–13.
Higgins JP, Altman DG, Gotzsche PC, Juni P, Moher D, Oxman AD, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928.
Wan X, Wang W, Liu J, Tong T. Estimating the sample mean and standard deviation from the sample size, median, range and/or interquartile range. BMC Med Res Methodol. 2014;14:135 https://doi.org/10.1186/471-2288-14-135.
Rucker G, Cates CJ, Schwarzer G. Methods for including information from multi-arm trials in pairwise meta-analysis. Res Synth Methods. 2017;8:392–403.
The Cochrane Collaboration. Review Manager (RevMan) [Computer program]. Version 5.4. 2020.
Balduzzi S, Rücker G, Schwarzer G. How to perform a meta-analysis with R: a practical tutorial. Evid Based Ment Health. 2019;22:153–60.
Balshem H, Helfand M, Schunemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64:401–6.
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535.
Schaffer LC, Schaffer CB, Miller AR, Manley JL, Piekut JA, Nordahl TE. An open trial of pregabalin as an acute and maintenance adjunctive treatment for outpatients with treatment resistant bipolar disorder. J Affect Disord. 2013;147:407–10.
Mokhber N, Lane CJ, Azarpazhooh MR, Salari E, Fayazi R, Shakeri MT, et al. Anticonvulsant treatments of dysphoric mania: a trial of gabapentin, lamotrigine and carbamazepine in Iran. Neuropsychiatr Dis Treat. 2008;4:227–34.
Frye MA, Ketter TA, Kimbrell TA, Dunn RT, Speer AM, Osuch EA, et al. A placebo-controlled study of lamotrigine and gabapentin monotherapy in refractory mood disorders. J Clin Psychopharmacol. 2000;20:607–14.
Pande AC, Crockatt JG, Janney CA, Werth JL, Tsaroucha G, Gabapentin Bipolar Disorder Study Group. Gabapentin in bipolar disorder: a placebo-controlled trial of adjunctive therapy. Bipolar Disord. 2000;2:249–55.
Vieta E, Manuel Goikolea J, Martínez-Arán A, Comes M, Verger K, Masramon X, et al. A double-blind, randomized, placebo-controlled, prophylaxis study of adjunctive gabapentin for bipolar disorder. J Clin Psychiatry. 2006;67:473–7.
Pande AC, Davidson JR, Jefferson JW, Janney CA, Katzelnick DJ, Weisler RH, et al. Treatment of social phobia with gabapentin: a placebo-controlled study. J Clin Psychopharmacol. 1999;19:341–8.
Greist JH, Liu-Dumaw M, Schweizer E, Feltner D. Efficacy of pregabalin in preventing relapse in patients with generalized social anxiety disorder: results of a double-blind, placebo-controlled 26-week study. Int Clin Psychopharmacol. 2011;26:243–51.
Baldwin DS, Ajel K, Masdrakis VG, Nowak M, Rafiq R. Pregabalin for the treatment of generalized anxiety disorder: an update. Neuropsychiatr Dis Treat. 2013;9:883–92.
Markota M, Morgan RJ. Treatment of generalized anxiety disorder with gabapentin. Case Rep Psychiatry. 2017;2017:6045017.
Baniasadi M, Hosseini G, Fayyazi Bordbar MR, Rezaei Ardani A, Mostafavi, Toroghi H. Effect of pregabalin augmentation in treatment of patients with combat-related chronic posttraumatic stress disorder: a randomized controlled trial. J Psychiatr Pract. 2014;20:419–27.
Mowla A, Ghaedsharaf M. Pregabalin augmentation for resistant obsessive–compulsive disorder: a double-blind placebo-controlled clinical trial. CNS Spectr. 2020;25:552–6.
Pande AC, Pollack MH, Crockatt J, Greiner M, Chouinard G, Lydiard RB, et al. Placebo-controlled study of gabapentin treatment of panic disorder. J Clin Psychopharmacol. 2000;20:467–71.
Brower KJ, Myra Kim H, Strobbe S, Karam-Hage MA, Consens F, Zucker RA. A randomized double-blind pilot trial of gabapentin versus placebo to treat alcohol dependence and comorbid insomnia. Alcohol Clin Exp Res. 2008;32:1429–38.
Mason BJ, Quello S, Goodell V, Shadan F, Kyle M, Begovic A. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174:70–7.
Bollu V, Bushmakin AG, Cappelleri JC, Chen C-C, Feltner D, Wittchen H-U. Pregabalin reduces sleep disturbance in patients with generalized anxiety disorder via both direct and indirect mechanisms. Eur J Psychiatry. 2010;24:18–27.
Spampinato MV, Wood JN, De Simone V, Grafman J. Neural correlates of anxiety in healthy volunteers: a voxel-based morphometry study. J Neuropsychiatry Clin Neurosci. 2009;21:199–205.
Takagi Y, Sakai Y, Abe Y, Nishida S, Harrison BJ, Martinez-Zalacain I, et al. A common brain network among state, trait, and pathological anxiety from whole-brain functional connectivity. Neuroimage 2018;172:506–16.
Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164:1476–88.
da Costa BR, Nuesch E, Rutjes AW, Johnston BC, Reichenbach S, Trelle S, et al. Combining follow-up and change data is valid in meta-analyses of continuous outcomes: a meta-epidemiological study. J Clin Epidemiol. 2013;66:847–55.
Furey SA, Hull SG, Leibowitz MT, Jayawardena S, Roth T. A randomized, double-blind, placebo-controlled, multicenter, 28-day, polysomnographic study of gabapentin in transient insomnia induced by sleep phase advance. J Clin Sleep Med. 2014;10:1101–9.
Rosenberg RP, Hull SG, Lankford DA, Mayleben DW, Seiden DJ, Furey SA, et al. A randomized, double-blind, single-dose, placebo-controlled, multicenter, polysomnographic study of gabapentin in transient insomnia induced by sleep phase advance. J Clin Sleep Med. 2014;10:1093–100.
Roth T, Arnold LM, Garcia-Borreguero D, Resnick M, Clair AG. A review of the effects of pregabalin on sleep disturbance across multiple clinical conditions. Sleep Med Rev. 2014;18:261–71.
Gomes T, Juurlink DN, Antoniou T, Mamdani MM, Paterson JM, van den Brink W. Gabapentin, opioids, and the risk of opioid-related death: A population-based nested case-control study. PLoS Med. 2017;14:e1002396.
Zaccara G, Perucca P, Gangemi PF. The adverse event profile of pregabalin across different disorders: a meta-analysis. Eur J Clin Pharmacol. 2012;68:903–12.
Savovic J, Turner RM, Mawdsley D, Jones HE, Beynon R, Higgins JPT, et al. Association between risk-of-bias assessments and results of randomized trials in Cochrane Reviews: the ROBES Meta-Epidemiologic Study. Am J Epidemiol. 2018;187:1113–22.
Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–94.
Levey DF, Gelernter J, Polimanti R, Zhou H, Cheng Z, Aslan M, et al. Reproducible genetic risk loci for anxiety: results from ∼200,000 participants in the Million Veteran Program. Am J Psychiatry. 2020;177:223–32.
Harrison PJ, Geddes JR, Tunbridge EM. The emerging neurobiology of bipolar disorder. Trends Neurosci. 2018;41:18–30.
Clark MB, Wrzesinski T, Garcia AB, Hall NAL, Kleinman JE, Hyde T, et al. Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Mol Psychiatry. 2020;25:37–47.
Hofmann F, Flockerzi V, Kahl S, Wegener JW. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev. 2014;94:303–26.
Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P. L-type Ca2+ channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal. 2014;3:15–38.
Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004;119:19–31.
Endres D, Decher N, Rohr I, Vowinkel K, Domschke K, Komlosi K, et al. New Cav1.2 Channelopathy with High-Functioning Autism, Affective Disorder, Severe Dental Enamel Defects, a Short QT Interval, and a Novel CACNA1C Loss-Of-Function Mutation. Int J Mol Sci. 2020;21:8611.
Moon AL, Haan N, Wilkinson LS, Thomas KL, Hall J. CACNA1C: Association with psychiatric disorders, behavior, and neurogenesis. Schizophr Bull. 2018;44:958–65.
Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. 2016;594:5369–90.
Tran-Van-Minh A, Dolphin AC. The α2δ ligand gabapentin inhibits the Rab11-dependent recycling of the calcium channel subunit α2δ -2. J Neurosci. 2010;30:12856–67.
Martin DJ, McClelland D, Herd MB, Sutton KG, Hall MD, Lee K, et al. Gabapentin-mediated inhibition of voltage-activated Ca2+ channel currents in cultured sensory neurones is dependent on culture conditions and channel subunit expression. Neuropharmacology 2002;42:353–66.
Dolphin AC. Voltage-gated calcium channel α2δ subunits: an assessment of proposed novel roles. F1000Res. 2018;7:F1000 Faculty Rev–830.
Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RE, et al. The α2δ -1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. 2018;22:2307–21.
Tomlinson A, Furukawa TA, Efthimiou O, Salanti G, De Crescenzo F, Singh I, et al. Personalise antidepressant treatment for unipolar depression combining individual choices, risks and big data (PETRUSHKA): rationale and protocol. Evid Based Ment Health. 2020;23:52–6.
Rees E, Owen MJ. Translating insights from neuropsychiatric genetics and genomics for precision psychiatry. Genome Med. 2020;12:43.
Spoorthy MS, Chakrabarti S, Grover S. Comorbidity of bipolar and anxiety disorders: an overview of trends in research. World J Psychiatry. 2019;9:7–29.
Lopes FL, Zhu K, Purves KL, Song C, Ahn K, Hou L, et al. Polygenic risk for anxiety influences anxiety comorbidity and suicidal behavior in bipolar disorder. Transl Psychiatry. 2020;10:298.
Kim SW, Berk L, Kulkarni J, Dodd S, de Castella A, Fitzgerald PB, et al. Impact of comorbid anxiety disorders and obsessive-compulsive disorder on 24-month clinical outcomes of bipolar I disorder. J Affect Disord. 2014;166:243–8.
Acknowledgements
This work is supported by the National Institute for Health Research (NIHR) Oxford Health Biomedical Research Centre (grant IS-BRC-1215-20005). AC is supported by the NIHR Oxford cognitive health Clinical Research Facility, by an NIHR Research Professorship (grant RP-2017-08-ST2-006), and by the NIHR Oxford and Thames Valley Applied Research Collaboration. The views expressed are those of the authors and not necessarily those of the UK National Health Service, the NIHR, or the UK Department of Health. We thank Kerensa Houghton, Alexandra Forrest, and Sarah Stockton for their contribution in conceiving the protocol of the work and/or screening abstracts in the search process.
Author information
Authors and Affiliations
Contributions
Study conception and design: AC, LZA, JRG, and PJH. Data extraction and analyses: JSWH, LZA, NA, AA, and AC. Additional input and interpretation: JRG, EMT. Paper drafted by JSWH with input from PJH, AC, EMT, JRG, and LZA. All authors reviewed and approved the submitted paper.
Corresponding authors
Ethics declarations
Competing interests
EMT and PJH have an unrestricted educational grant from Johnson & Johnson on molecular aspects of VGCC α-1 subunits unrelated to the current work. AC has received research and consultancy fees from INCiPiT (Italian Network for Paediatric Trials), CARIPLO Foundation, and Angelini Pharma outside the current work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hong, J.S.W., Atkinson, L.Z., Al-Juffali, N. et al. Gabapentin and pregabalin in bipolar disorder, anxiety states, and insomnia: Systematic review, meta-analysis, and rationale. Mol Psychiatry 27, 1339–1349 (2022). https://doi.org/10.1038/s41380-021-01386-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-021-01386-6
This article is cited by
-
Gabapentinoid consumption in 65 countries and regions from 2008 to 2018: a longitudinal trend study
Nature Communications (2023)
-
Preoperative anxiety and postoperative adverse events: a narrative overview
Anesthesiology and Perioperative Science (2023)
-
Pharmacologic Management of Cancer-Related Pain in Pregnant Patients
Drugs (2023)
