
Abstract
People with psychotic disorders show abnormalities in several organ systems in addition to the central nervous system (CNS); and this contributes to excess mortality. However, it is unclear how strong the evidence is for alterations in non-CNS systems at the onset of psychosis, how the alterations in non-CNS systems compare to those in the CNS, or how they relate to symptoms. Here, we consider these questions, and suggest potential models to account for findings. We conducted a systematic meta-review to summarize effect sizes for both CNS (focusing on brain structural, neurophysiological, and neurochemical parameters) and non-CNS dysfunction (focusing on immune, cardiometabolic, and hypothalamic–pituitary–adrenal (HPA) systems) in first-episode psychosis (FEP). Relevant meta-analyses were identified in a systematic search of Pubmed and the methodological quality of these was assessed using the AMSTAR checklist (A Measurement Tool to Assess Systematic Reviews). Case–control data were extracted from studies included in these meta-analyses. Random effects meta-analyses were re-run and effect size magnitudes for individual parameters were calculated, as were summary effect sizes for each CNS and non-CNS system. We also grouped studies to obtain overall effect sizes for non-CNS and CNS alterations. Robustness of data for non-CNS and CNS parameters was assessed using Rosenthal’s fail-safe N. We next statistically compared summary effect size for overall CNSand overall non-CNS alterations, as well as for each organ system separately. We also examined how non-CNS alterations correlate CNS alterations, and with psychopathological symptoms. Case-control data were extracted for 165 studies comprising a total sample size of 13,440. For people with first episode psychosis compared with healthy controls, we observed alterations in immune parameters (summary effect size: g = 1.19), cardiometabolic parameters (g = 0.23); HPA parameters (g = 0.68); brain structure (g = 0.40); neurophysiology (g = 0.80); and neurochemistry (g = 0.43). Grouping non-CNS organ systems together provided an effect size for overall non-CNS alterations in patients compared with controls (g = 0.58), which was not significantly different from the overall CNS alterations effect size (g = 0.50). However, the summary effect size for immune alterations was significantly greater than that for brain structural (P < 0.001) and neurochemical alterations (P < 0.001), while the summary effect size for cardiometabolic alterations was significantly lower than neurochemical (P = 0.04), neurophysiological (P < 0.001), and brain structural alterations (P = 0.001). The summary effect size for HPA alterations was not significantly different from brain structural (P = 0.14), neurophysiological (P = 0.54), or neurochemical alterations (P = 0.22). These outcomes remained similar in antipsychotic naive sensitivity analyses. We found some, but limited and inconsistent, evidence that non-CNS alterations were associated with CNS changes and symptoms in first episode psychosis. Our findings indicate that there are robust alterations in non-CNS systems in psychosis, and that these are broadly similar in magnitude to a range of CNS alterations. We consider models that could account for these findings and discuss implications for future research and treatment.
Similar content being viewed by others


Introduction
Schizophrenia and related psychotic disorders have a worldwide lifetime prevalence of ~1% [1]. They are highly disabling conditions with economic costs over $60 billion per year in the United States [2, 3]. Epidemiological studies have established that people with psychotic disorders die 15–20 years earlier than the general population, and that 60% or more of this premature mortality relates to non-CNS, predominantly cardiovascular, causes [4,5,6,7,8]. Poor physical health has traditionally been blamed on the secondary effects of the illness, be that a consequence of the illness itself (e.g., negative symptoms leading to sedentary lifestyle and poor diet) [9], or a consequence of treatment (e.g., second-generation antipsychotic use) [10]. In recent years however, studies in first-episode patients have shown dysfunction in cardiometabolic [11,12,13,14,15,16], immune [17,18,19,20,21], and hypothalamic pituitary adrenal (HPA) [22,23,24,25] systems. This suggests that psychotic disorders involve multiple systems at onset. However, it is unclear how strong the evidence is for abnormalities across these systems, how alterations compare with CNS abnormalities seen in the disorder, or how they relate to symptoms. To address these questions, we perform a meta-review of the magnitude, consistency, and robustness of dysfunction across these systems as assessed using peripheral markers, and compare findings with representative CNS abnormalities in psychosis. We then review the potential models that could explain the associations, before considering both the research and clinical implications of our findings.
Methods
Systematic meta-review summarizing effect size magnitudes for central nervous system and non-central nervous system alterations in first-episode psychosis
Full details of methods employed are documented in Supplementary Information (eAppendix 1). Two systematic reviews of meta-analyses were performed according to preferred reporting items for systematic reviews and meta-analyses (PRISMA) guidelines [26] (Supplementary Information, eTable 1). Two reviewers (TP and ED) independently searched Pubmed from 1990 to May week 2 2017 for each systematic meta-review. For the meta-review focussing on non-CNS dysfunction, we focused on meta-analyses of findings in three organ systems established as showing dysregulation in schizophrenia: the immune [27], cardiometabolic [28], and HPA [29] systems. We selected meta-analyses reporting markers of immune, cardiometabolic, and HPA system differences between patient and control groups, rather than differences in rates of diagnoses of conditions based on pre-defined diagnostic criteria [30,31,32,33] (e.g., rates of diagnoses of type 2 diabetes mellitus or hypercholesterolemia). The rationale behind this methodology was threefold. First, patients with psychotic illness are less likely to seek medical attention and so there is the risk of under-reporting of diagnoses [34]. Second, certain conditions, such as glucose and lipid dysregulation, develop on a continuum and take time for serum/plasma markers to reach threshold for a diagnosis. For example, changes in glucose regulation occur 4–7 years prior to diagnosis of type 2 diabetes mellitus [35]. Third, physiological alterations that do not meet diagnostic thresholds can, nevertheless, be associated with worsened mortality/morbidity outcomes. For example, there is robust evidence that low-grade inflammation is an independent risk factor for atherosclerosis and cardiovascular disease [36]. For the meta-review focussing on CNS dysfunction, we focused on meta-analyses of parameters previously identified in an expert review as key neurobiological alterations seen in schizophrenia [37], covering alterations in brain structure, neurophysiology, and neurochemistry. The search was limited to first-episode psychosis (FEP) to limit secondary effects of illness.
Patient and control data from the studies referenced in the meta-analyses that our search terms had identified were extracted and all meta-analyses repeated. Data were only extracted for those CNS and non-CNS parameters, where there were significant differences demonstrated between FEP and controls in the original meta-analyses. A two-tailed P < 0.05 was deemed significant. A minimum of three studies was required to run a meta-analysis. For non-CNS parameters, random-effects meta-analyses were performed examining immune (interleukin-1β (IL-1β), soluble interleukin-2 receptor (sIL-2R), interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), C-reactive protein (CRP), and total lymphocyte count), cardiometabolic (fasting glucose, glucose post-oral glucose tolerance test (OGTT), fasting insulin, insulin resistance (HOMA-IR), total cholesterol, low density lipoprotein (LDL) cholesterol, and triglycerides), and HPA parameters (cortisol awakening response and prolactin concentrations). For CNS parameters, random-effects meta-analyses were performed examining brain structural (total brain volume, total grey matter volume, total CSF volume, right and left hippocampus, right and left lateral ventricle, total thalamus, right and left caudate nucleus), neurophysiological (auditory P300 amplitude and latency, duration deviant mismatch negativity), and neurochemical parameters (frontal, temporal and thalamus N-acetylaspartic acid (NAA) levels). A random-effects model was used in all analyses owing to an expectation of heterogeneity of data across studies. Standardized mean differences between patient and control cohort parameters were used as the effect size, determined using Hedges adjusted g. The 95% CI of the effect size was also calculated. A criticism of meta-analysis is that it may be based on a biased sample of studies, potentially inflating effect sizes. Thus, for each meta-analysis, Rosenthal’s “fail-safe N” [38] was used to calculate the number of additional null studies (i.e., studies where the effect size is zero) that would be required to increase the P value for a given meta-analysis to greater than 0.05. This provides an assessment of how bias could influence the results of a meta-analysis: the greater the number of estimated studies required for the finding to be no longer significant, the less likely the results are secondary to publication or small sample bias and therefore the more robust the finding. Methodological quality of each meta-analysis was assessed using the AMSTAR checklist (A Measurement Tool to Assess Systematic Reviews) (eAppendix 2). Heterogeneity scores for samples within each meta-analysis were also recorded, as assessed using the χ2 test, Q, or I2 statistic (Tables 1 and 2).
As well as individual meta-analyses being run for each parameter as described, six separate subgroup meta-analyses were performed examining data for overall immune, cardiometabolic, HPA, brain structural, neurophysiological, and neurochemical systems. Subgroup summary effect size magnitudes were calculated by running a combined analysis of all studies assigned to a subgroup (e.g., to calculate the summary effect size magnitude for immune alterations, a single analysis was performed that combined IL-1β, sIL-2R, IL-6, TNF-α, TGF-β, CRP, and total lymphocyte count data sets). If a single study provided results for more than one subgroup parameter (e.g., data for several cytokines from a single study population), then the patient and control numbers for that study were divided by the number of parameters contributed to the summary meta-analysis (e.g., if two different cytokines were reported by a single study, then the population number for that study was divided by 2). Using the same methodology, overall meta-analyses for CNS and non-CNS alterations were calculated. Sensitivity analyses for antipsychotic naive FEP were performed.
Statistical comparison of effect sizes for central nervous system and non-central nervous system alterations in first-episode psychosis
After obtaining effect size estimates for each CNS (brain structural, neurophysiological, and neurochemical) and non-CNS system (immune, cardiometabolic, and HPA), we next performed bivariate comparisons of each of these six effect sizes against one another using a Wald-type test. We determined statistical significance by entering each pair of effect size estimates into a fixed effects model (given that the residual heterogeneity had previously been accounted for in the initial random effects meta-analyses). P < 0.05 was deemed significant. This method was also used to compare overall summary CNS and non-CNS effect sizes. Sensitivity analyses were performed restricting analyses to antipsychotic naive cohorts. All statistical tests were conducted using the metafor package [39] in the R statistical programming language.
Results
For non-CNS dysfunction, of 365 citations retrieved, 15 meta-analyses met inclusion criteria [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25] (Table 1). For CNS dysfunction, of 446 citations retrieved, 13 meta-analyses met inclusion criteria [40,41,42,43,44,45,46,47,48,49,50,51,52] (Table 2). Data were extracted from a total of 165 case–control studies (eAppendix 3). After excluding overlapping samples, the total sample size was 13,440 (6806 patients and 6634 controls), with 6817 in the non-CNS sample (3300 patients and 3517 controls), and 6623 in the CNS sample (3506 patients and 3117 controls). The quality of studies was medium to high (AMSTAR scores 6–10, Tables 1 and 2).
Meta-analytic outcomes for central nervous system and non-central nervous system alterations in first-episode psychosis
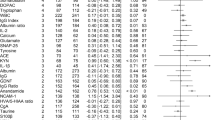
Figure 1a depicts a forest plot for magnitude of immune, cardiometabolic, HPA, brain structural, neurophysiological, and neurochemical alterations in first-episode psychosis compared with healthy controls, as well as overall summary effect sizes for CNS and non-CNS alterations. As per Cohen’s guidelines [53], a medium overall summary effect size for non-CNS alterations in FEP was observed, (g = 0.58 (95% CI: 0.44–0.72). A medium overall summary effect size for CNS alterations in FEP was also observed (g = 0.50 (95% CI: 0.44–0.56). Similar results were observed in antipsychotic naive sensitivity analyses (effect size for non-CNS alterations: g = 0.51 (95% CI: 0.34–0.67); effect size for CNS alterations: g = 0.48 (95% CI: 0.39–0.58), eFigure 3).
The immune system
Five meta-analyses examining immune disturbances in FEP were identified [17,18,19,20,21]. Four meta-analyses examined inflammatory mediators [17,18,19,20], and one lymphocyte counts [21]. After allowing for overlapping studies, data were extracted for a total sample size of 1343 patients and 1643 controls. FEP is associated with elevated blood cytokine levels, specifically IL-1β, sIL2R, IL-6, TNFα, TGFβ, CRP, and elevated total lymphocyte counts (effect size range: 0.61–1.62) (Fig. 1a). The summary effect size of immune alterations in FEP is 1.19 (95% CI: 0.82–1.56). Fail-safe N calculations demonstrated that between 17 and 1639 additional negative studies would be required for these findings to lose significance. Heterogeneity of studies was low to high (I2: 0–98%), and study quality medium to high (AMSTAR: 6–9). Antipsychotic naive FEP is associated with elevated blood cytokine levels, specifically IL-1β, sIL2R, IL-6, and TNFα (effect size: 1.00–1.86). The overall effect size for magnitude of immune alterations in antipsychotic naive FEP is 1.46 (95% CI: 0.74–2.18) (eFigure 3).
The cardiometabolic system
Six meta-analyses examining cardiometabolic dysfunction in FEP were identified [11,12,13,14,15,16]. Three meta-analyses focused on glucose and insulin disturbance [11,12,13], two on lipid disturbance [14, 16], and one on oxidative stress [15]. After allowing for overlapping studies, data were extracted for a total sample size of 1556 patients and 1480 controls. All data extracted were for antipsychotic naive individuals. Antipsychotic naive FEP is associated with elevated fasting glucose, glucose following the oral glucose tolerance test, fasting insulin, and insulin resistance (effect size range 0.20–0.61), raised triglycerides (effect size: 0.14), and reduced total cholesterol and LDL cholesterol (effect size range: −0.22 to −0.19) (Fig. 1a). We were unable to extract sufficient data to allow for a meta-analysis of oxidative stress parameters in FEP. The summary effect size of cardiometabolic alterations in antipsychotic naive FEP is 0.23 (95% CI: 0.15–0.31). Fail-safe N calculations demonstrate that between 26 and 97 additional negative studies would be required for findings to lose significance. Heterogeneity was low to high (I2: 0–97%), and study quality medium to high (AMSTAR: 7–10).
The hypothalamic–pituitary–adrenal system
Four meta-analyses examining HPA dysregulation in FEP were identified [22,23,24,25]. Two meta-analyses reported on morning cortisol [23, 24], one on cortisol awakening response [22], and one on prolactin levels [25]. Prolactin levels were included as a marker of HPA axis activation owing to previous evidence that its levels increase in response to various stressors, with a direct correlation observed between prolactin levels and both adrenocorticotrophic hormone and cortisol levels in healthy controls [54]. Data were extracted for a total sample size of 401 patients and 394 controls. The cortisol awakening response is blunted in FEP (effect size: 0.62), and prolactin levels are elevated in antipsychotic naive FEP [25] (effect size: 0.74) (Fig. 1a). The summary effect size of HPA alterations in FEP is 0.68 (95% CI: 0.32–1.04). Fail-safe N calculations demonstrate that between 75 and 125 negative studies would be required for these findings to lose significance. Heterogeneity was low to high (I2: 24–83%), and study quality high (AMSTAR: 9–10). There were insufficient data to allow for a meta-analysis of cortisol awakening response in antipsychotic naive FEP.
Central nervous system: brain structural changes
Eight meta-analyses examining brain structure in FEP were identified [40, 42,43,44,45,46,47, 55]. Data were extracted for a total sample size of 1937 patients and 1656 controls. FEP is associated with reductions in both total and regional brain volumes (effect size: 0.26–0.58), and an increase in CSF volume (effect size: 0.34) (Fig. 1a). The summary effect size of brain structural alteration in FEP is 0.40 (95% CI: 0.33–0.47). Fail-safe N calculations demonstrate that between 27 and 663 additional negative studies would be required for these findings to lose significance. Heterogeneity was low to medium (I2: 9–56%), and study quality medium (AMSTAR: 4–7). Antipsychotic naive FEP is also associated with total and regional brain volume reduction (effect size: 0.23–0.87), and an in increase in total CSF volume (effect size: 0.32). The summary effect size of brain structural alterations in antipsychotic naive FEP is 0.44 (95% CI: 0.34–0.54) (eFigure 3).

An overview and comparison of CNS and non-CNS alterations in first-episode psychosis. Figure 1a: Forest plot for magnitude of immune, cardiometabolic, HPA, brain structural, neurophysiological, and neurochemical alterations in first-episode psychosis compared with healthy controls. Each line represents a summary effect size for a meta-analysis in one parameter: squares represent the summary effect size for that parameter, with the horizontal line running through each square illustrating the width of the overall 95% CI. Blue diamonds represent summary effect sizes for immune, cardiometabolic, HPA, structural, neurophysiological, and neurochemical systems: the middle of each diamond represents the summary effect size, and the width of the diamond depicts the width of the overall 95% CI. Red diamonds represent summary effect sizes and accompanying 95% CI for non-CNS and CNS effect sizes. ES effect size, CNS central nervous system, FEP first-episode psychosis, HPA hypothalamic pituitary adrenal axis, IL1β interleukin-1β, sIL2-R soluble interleukin-2 receptor, IL6 interleukin-6, TGFβ transforming growth factor-β, CRP C-reactive protein, NAA N-acetylaspartic acid, N number. Figure 1b: Heat map comparing relative magnitude of effect sizes (ES) for immune, hypothalamic pituitary adrenal (HPA) axis, cardiometabolic, brain structural, neurophysiological, and neurochemical alterations in first-episode psychosis (FEP). The map is read from left to right, comparing parameters on the y axis with parameters on the x axis. A negative Wald score (blue squares) demonstrates that the parameter ES on the y axis is numerically lower compared with the intersecting parameter ES on the x axis. A positive Wald score (red squares) demonstrates that the parameter ES on the y axis is numerically higher than the intersecting parameter ES on the x axis. Numbers within the squares are the P values that accompany the Wald score, e.g., structural abnormalities show significantly smaller patient-control differences compared to immune abnormalities, and significantly greater differences compared to cardiometabolic abnormalities
Central nervous system: neurophysiological changes
Four meta-analyses examining neurophysiological changes in FEP were identified [48,49,50,51]. Data were extracted for a total sample size of 1051 patients and 980 controls. FEP is associated with decreased auditory P300 amplitude (effect size: 0.83), and reduced duration-deviant mismatch negativity (effect size: 0.77) (Fig. 1a). The summary effect size for magnitude of neurophysiological alteration in FEP is 0.80 (95% CI: 0.64–0.96). Fail-safe N calculations demonstrate that between 450 and 1100 additional negative studies would be required for these findings to lose significance. Sample heterogeneity was medium to high (I2: 55–86%), and study quality medium to high (AMSTAR: 4–8). Antipsychotic naive FEP is associated with a reduction in auditory P300 amplitude (effect size: 0.86) and latency (effect size: 0.63), as well as a reduction in duration-deviant mismatch negativity (effect size: 0.67). The overall effect size for magnitude of neurophysiological alterations in antipsychotic naive FEP is 0.70 (95% CI: 0.47–0.94) (eFigure 3).
Central nervous system: neurochemical changes
One meta-analysis examining brain chemistry in FEP was identified [52]. Data were extracted for a total sample size of 518 patients and 481 controls. FEP is associated with decreased levels of N-acetyl aspartate concentrations across multiple brain regions (effect size: 0.35–0.50) (Fig. 1a). The overall effect size for magnitude of neurochemical alteration in FEP is 0.43 (95% CI: 0.26–0.60). Fail-safe N calculations demonstrate that between 19 and 359 additional negative studies would be required for these findings to lose significance. Sample heterogeneity was low to medium (I2: 23–63%), and study quality medium (AMSTAR: 5). Antipsychotic naive FEP is associated with a reduction in frontal cortical NAA (ES: 0.34) (eFigure 3).
Statistical comparison of effect sizes for central nervous system and non-central nervous system alterations in first-episode psychosis
Overall, CNS and non-CNS effect sizes were compared using a Wald-type test. There was no significant difference between the overall effect size of alterations in the CNS compared with the overall effect size of alterations in the non-CNS systems examined (P = 0.283), and this remained the case when analyses were restricted to studies of antipsychotic naive patients (P = 0.825).
Individual system comparisons
A heat map of respective Wald scores and associated significance values between each of the systems examined (immune, cardiometabolic, HPA, brain structural, neurochemical, and neurophysiological) was constructed to allow graphical representation of the relative effect size magnitude difference between systems (Fig. 1b). Wald test comparisons of overall immune effect size with CNS effect sizes in FEP demonstrated significantly higher effect size magnitudes for immune alterations compared with brain structural (P < 0.001) and neurochemical (P < 0.001) alterations, and no significant difference when compared with neurophysiological alterations (P = 0.053). For antipsychotic naive FEP, summary effect size for immune alterations was significantly higher in antipsychotic naive FEP compared with brain structural (P = 0.006) and neurochemical alterations (P = 0.005), and no significant difference when compared with neurophysiological alterations (P = 0.05) (eFigure 4). Comparisons of the summary effect size for cardiometabolic alterations with those for CNS alterations in antipsychotic naive FEP demonstrated significantly reduced effect size magnitudes for cardiometabolic alterations compared with brain structural (P = 0.001), neurochemical (P = 0.04), and neurophysiological alterations (P < 0.001). Comparisons of summary effect size of HPA with those for CNS alterations in FEP demonstrated no significant difference when compared with brain structural (P = 0.14), neurochemical (P = 0.217), and neurophysiological (P = 0.541) alterations.
Correlations between non-central nervous system parameters and central nervous system parameters
In view of evidence of both CNS and non-CNS alterations in FEP, we examined evidence for associations between these parameters. Only a limited number of observational studies have investigated relationships between CNS and non-CNS measures in FEP. Studies in broader psychotic illness, including schizophrenia, present data that is conflicting, which may reflect the heterogeneity of the population studied, but also the multi-faceted roles of the metabolic, immune, and endocrine parameters we have examined. For example, IL-6 has both neurodegenerative [56] and neuroprotective properties [57]. Thus, conflicting outcomes with regard to correlations between pro-inflammatory cytokines and brain structural alterations might be expected. Indeed, in schizophrenia, hippocampal volumes correlate directly with the pro-inflammatory cytokine interleukin-18 [58], while pro-inflammatory IL-1β titres correlate indirectly with Broca’s area volume in a “pro-inflammatory” subgroup of patients [59]. Thus, we have evidence of elevated pro-inflammatory cytokines being differentially associated with regional brain volume alterations: further studies are required to clarify regional brain structural alterations in the context of systemic inflammation in FEP. In terms of HPA axis alterations and influence on regional brain structure, although one study has failed to demonstrate a relationship between cortisol levels and hippocampal volume in FEP [60], Mondelli and colleagues have reported an inverse correlation between blood cortisol levels and hippocampal volumes [61], and the degree of left-sided hippocampal volume reduction has been associated with a blunted cortisol awakening response in FEP [62]. Beyond brain structural alterations, in schizophrenia, diffusion tensor imaging has demonstrated that peripheral IL-6 levels inversely correlate with measures of fractional anisotropy in the forceps major, inferior longitudinal fasciculus (ILF), and inferior fronto-occipital fasciculus (IFOF), a relationship not demonstrated in healthy controls. In both the ILF and IFOF, IL-6 levels correlate directly with radial diffusivity measures. CRP levels in schizophrenia also show an inverse correlation with fractional anisotropy within the forceps major [63]. Thus, in psychosis, selected neural pathways may be differentially susceptible to systemic immune alterations, although replication of these findings is required. We were unable to find any studies in FEP exploring correlations between CNS parameters and cardiometabolic disturbances. In individuals with type 2 diabetes who do not have a psychotic disorder, insulin resistance is associated with hippocampal atrophy and memory impairment [64]. Since cognitive impairment is a key feature of psychotic illness, and we have demonstrated that antipsychotic naive FEP is associated with both insulin resistance and reduced hippocampal volumes, future structural imaging studies combined with metabolic assays are required to clarify if a correlation between impaired glucose homeostasis and hippocampal structural alterations also exists in FEP.
Correlations between non-CNS parameters and symptom severity
Although the evidence presented indicates that cardiometabolic, immune, and HPA alterations are present in early psychosis, this does not indicate whether these abnormalities are linked to the clinical expression of the disorder. As such, we also examined evidence of a “biological gradient” between these markers and symptom measures. In antipsychotic naive FEP, positive symptom severity, as assessed using the positive and negative syndrome scale (PANSS), correlates indirectly with fasting glucose levels [65] and insulin resistance [66], indicating more severe symptoms are associated with less marked glucoregulatory disturbance. As part of a meta-analysis examining CRP titres in schizophrenia performed by Fernandes and colleagues [67], meta-regression of effect size for CRP changes on PANSS-positive scores demonstrated that the greater the severity of positive symptoms, the greater the increase in CRP (r = 0.12; 95% CI: 0.03–0.23; P = 0.013). Broader evidence for a biological gradient between symptom severity and inflammatory cytokines in psychosis is however inconsistent and contradictory. For example, in antipsychotic naive FEP, levels of the anti-inflammatory cytokine IL-10 inversely correlate with PANSS-negative scores [68], indicating this anti-inflammatory marker is associated with less severe symptoms, however PANSS-positive scores in FEP have been observed to inversely correlate with pro-inflammatory IL-6 [69]. Studies in broader psychotic illness have demonstrated that IL-6 levels correlate directly with total psychopathology [70, 71], however others have failed to demonstrate an association [72,73,74]. No significant correlations have been reported between TNF-α, IL-1β, IL-12, and TGF-β levels with symptomatology in FEP [75,76,77,78]. Correlations between HPA axis alterations and symptom severity in FEP are similarly inconsistent and contradictory. For example, although cortisol levels have been observed to directly correlate with symptom severity [79,80,81], there have been negative studies [82,83,84], and inverse correlations with illness severity reported [85]. Varied outcomes may be a consequence of heterogeneity of patient populations and parameter measurement techniques between studies. Future projects may benefit from recruitment of a more homogenous group of participants, either by applying more stringent diagnostic inclusion criteria (e.g., focussing on individuals with predominant negative or positive symptoms, as demonstrated by Kirkpatrick and colleagues who defined differences in glucose tolerance between deficit and non-deficit schizophrenia) [86] or by stratifying patients based on a pre-defined physiological parameter (e.g., a “pro-inflammatory” group based on cytokine titres) [59].
Discussion
The evidence analyzed in this review, compiled from 165 case–control studies and an overall sample size of 13,440 subjects, indicates that there are a range of significant non-CNS as well as CNS alterations in patients with first-episode psychosis (summarized in Fig. 2a), and that the overall magnitudes of alteration for CNS and non-CNS alterations are not significantly different. Fail-safe N calculations, which provide a surrogate marker of how robust these findings are, demonstrate that large numbers of null studies would need to be added to these meta-analyses for both CNS and non-CNS outcomes to lose statistical significance (fail-safe N ranges: immune 17–1639; HPA 75–125; cardiometabolic 26–97; brain structural 27–663; neurophysiological 450–1100; neurochemical 19–359). Although our review of observational studies in psychosis suggests that there may be a link between certain non-CNS and CNS alterations as well as some non-CNS parameters and symptom severity, the number of these correlative studies is small and the results are inconsistent, making definitive inferences difficult.

A summary of non-CNS alterations in first-episode psychosis, and a consideration of potential pathoetiology. Figure 2a: First-episode psychosis shows alterations in multiple systems in addition to the central nervous system. OGTT oral glucose tolerance test, HDL high-density lipoprotein, LDL low-density lipoprotein. Figure 2b–d: Models of the relationship between psychosis and non-CNS dysfunction. Figure 2b: Model 1: A risk factor induces non-CNS dysfunction, which may consequently impact CNS function to increase the risk of psychosis. Figure 2c: Model 2: A risk factor induces CNS dysfunction and thence psychotic symptoms, which may consequently trigger non-CNS dysfunction. Figure 2d: Model 3: A shared risk factor may result in the development of psychosis and non-CNS dysfunction through independent mechanisms. CNS central nervous system
Strengths and limitations
The major strength of this meta-review is its focus on FEP and antipsychotic naive data. Thus, where possible the confounding effects of illness chronicity and treatment were limited. There are of course limitations to studies examining physical dysfunction in FEP (see limitations box). It is important to recognize that the FEP population is inevitably diagnostically heterogenous. To investigate the degree of heterogeneity, we extracted the diagnoses of patients from each study where reported (see eAppendix4). This shows that the overall proportion of FEP patients included in our analyses who had a diagnosis of schizophrenia was 74% in the non-CNS group and 84% in the CNS group. While some of the patients without a diagnosis of schizophrenia will go on to develop schizophrenia, it still important to recognize that some will never develop schizophrenia and, consequently, our findings should not be taken as being specific to schizophrenia. While the difference in the proportion with schizophrenia between groups is modest, we cannot exclude that this has moderated the effect sizes. Thus, we recommend future FEP studies include follow-up to determine diagnoses, and report findings separately by diagnosis to enable this to be tested. The specificity of findings is also low, with other serious mental illnesses showing similar physiological dysfunction [87, 88]. However, it should be recognized that many of the same factors apply to CNS alterations, and reverse causality is possible, with lifestyle factors potentially impacting both CNS and non-CNS measures. For example, smoking [89], stress [90], drug [91], and alcohol [92] abuse are associated with gray matter volume loss. Despite our focus on early psychosis, studies often fail to report or examine the impact of duration of untreated psychosis. As such, it remains possible that non-CNS changes are secondary to emerging psychotic symptoms, e.g., due to resultant poor lifestyle habits, social isolation, and stress [93, 94]. Moreover, inconsistencies in studies investigating a “biological gradient” between CNS and non-CNS alterations, and non-CNS alterations with symptom severity, limit arguments regarding non-CNS alterations contributing to pathological causality in psychosis as per Bradford Hill’s guidelines [95]. Although meta-analysis as a methodology is a powerful tool to summarize research knowledge in a field, it has limitations [96]. These limitations relate to the reliability of a meta-analysis’ outcome being dependent on the quality of the data meta-analyzed, the size of the samples included, the potential for type 1 error owing to publication bias toward “positive” results, and the heterogeneity of study populations included. However, our quality assessment of meta-analyses selected for this review was reassuring, with only medium-high quality analyses included. Although heterogeneity of studies documented by these meta-analyses varied, our meta-analyses used a random-effects model which is robust to heterogeneity. The fail-safe N approach has limitations [97]. For example, the formula assumes that the mean effect of unpublished null studies is zero, whereas studies may also have negative effect sizes, thereby requiring fewer studies to nullify the mean effect. However, the fail-safe N was applied across organ systems, thus, assuming there is similar publication bias across CNS and non-CNS research fields in psychosis, we can be confident that the fail-safe N is a reasonable quantitative measure by which the relative strength of the evidence supporting alterations in CNS and non-CNS parameters can be assessed. Indeed, fail-safe N calculations suggest outcomes for certain CNS and non-CNS parameters are more robust than others (Fig. 1a). Of the 15 comparative Wald analyses between CNS and non-CNS alterations in FEP, only four contrasts changed significance (based on α < 0.05) when moving from medicated to antipsychotic naive sensitivity analyses. There was no difference overall between CNS and non-CNS effect size magnitudes in both medicated and antipsychotic naive cohorts. This suggests that antipsychotic treatment does not significantly moderate effect sizes documented, providing further evidence that CNS and non-CNS alterations co-occur in FEP.
The putative nature of the relationship between central nervous system and non-central nervous system abnormalities in first-episode psychosis
Figure 2b–d illustrates three putative models that could explain our findings of both non-CNS and CNS abnormalities in FEP. Model 1 (Fig. 2b) shows how a risk factor inducing non-CNS dysfunction may lead to development of non-CNS disorders as well as impacting CNS function leading to psychosis. An example is that of paraneoplastic-induced anti-N-methyl-D-aspartate (NMDA)-receptor encephalitis, which is associated with psychosis. Resection of the causative tumor is associated with resolution of psychotic experiences [98]. More broadly, immune dysregulation could be responsible for co-development of psychosis and cardiometabolic disease. Pre-clinical studies demonstrate that peripheral inflammation can induce neuro-inflammation [99], which could potentially contribute to the pathogenesis of psychosis [100]. There is also evidence of inflammatory cytokines modulating dopaminergic and glutamatergic neurotransmission [101, 102]. As dysfunction in these neurotransmitters is implicated in the development of psychosis [103], this could link cytokine alterations to development of psychosis. As chronic inflammation is associated with accelerated atherosclerotic plaque formation, insulin resistance, and increased cardiovascular risk [36], elevated cytokines in psychosis could also be playing a role in the increased risk of cardiovascular disease in this population. Altered lipid turnover both peripherally and centrally may also be a consequence of an inflammatory process in FEP. Total and LDL cholesterol are reduced in antipsychotic naive FEP compared with healthy controls, a finding that is maintained in body mass index (BMI) sensitivity analyses [14, 16]. It has been hypothesized that the pro-inflammatory state of FEP is responsible for a “paradoxical” reduction in cholesterol via a similar mechanism seen in inflammatory arthritides [16]. Whether a similar mechanism modulates CNS lipid metabolism in schizophrenia and thus plays a role in disease pathogenesis through resultant synaptic dysfunction remains unclear. Such a mechanism would fit with model 1. In contrast, model 2 (Fig. 2c) shows how a risk factor can induce CNS dysfunction resulting in psychotic symptoms, which then trigger non-CNS dysfunction. An example is that the stress of psychosis could lead to HPA axis activation. Supporting this, psychological stress increases cortisol levels [104]. Cortisol excess is associated with hypertension, obesity, insulin resistance, dyslipidemia, and cardiovascular disease [105]. Thus, hypercortisolemia associated with psychosis may be contributing to cardiometabolic disease. It should however be recognized that, in addition to causing non-CNS dysfunction, stress may also contribute to the development of psychosis [106], suggesting a model whereby an exposure contributes jointly to CNS and non-CNS dysfunction. Finally, model 3 (Fig. 2d) proposes that a common risk factor has independent and parallel effects that result in the separate development of psychosis and non-CNS dysfunction. For example, population-based cohort studies have demonstrated that cardiometabolic disease [107] and psychosis share risk factors, including low-birth weight [108], pre-term birth [109, 110], and maternal malnutrition [111, 112]. Nutritional deficiencies in utero may result in neurodevelopmental changes increasing vulnerability to psychosis [113]. Nutritional deficiencies may also result in epigenetic changes relating to metabolic function, leading to metabolic alterations and ultimately diabetes [114]. Similarly, there may be shared genetic risk between psychosis and non-CNS disturbances. Genome-wide association studies have demonstrated pleiotropic enrichment between genes conferring risk for schizophrenia and non-CNS alterations, including immune and metabolic processes [115, 116].
Critique and comparison of the models
The models discussed above are intended to summarize the main potential relationships that exist between CNS and non-CNS alterations in psychosis, and it should be recognized that there is evidence for and against each model. The relative strengths and weakness of the three models proposed are summarized in Supplementary Information (eBox 1). Model 1 is supported by cases where a non-CNS disorder clearly predates psychosis, which resolves when the non-CNS disorder is treated, such as the example of NMDA auto-antibodies leading to psychosis discussed above, but these cases are rare [98]. Moreover, population-based cohort studies have observed that elevated levels of serum CRP [117] and IL-6 [118] in childhood are associated with increased risk of psychotic experience and schizophrenia in adulthood. Furthermore, the North American Prodrome Longitudinal Study (NAPLS) [119] has provided evidence that hypercortisolemia, inflammation, and elevated oxidative stress in individuals already deemed to be at risk for developing psychosis are risk factors for transition to FEP. However, many associations between non-CNS dysfunction and psychosis have not shown evidence of causation to date. To demonstrate causality, non-CNS dysfunction needs to be addressed prior to the onset of psychosis, for example in the prodrome, and show that the development of psychosis is prevented. In addition, although model 1 explains rare cases of psychosis, it is unlikely to account for typical cases of schizophrenia where CNS alterations are thought to occur early in neurodevelopment [106], unless non-CNS alterations also occur very early in development. Model 2, where non-CNS dysfunction emerges as a consequence of psychosis, is supported by meta-analytic evidence that resolution of acute psychosis is associated with normalization of previously elevated cytokines (IL-1β, IL-6, and TGF-β) [19]. Also, there is only limited evidence for alterations of certain non-CNS parameters in the prodrome (e.g., glucose and lipid disturbances), suggesting their later development may be a consequence of psychosis or its treatment. However, as described in support of model 1, several non-CNS alterations have been demonstrated in the prodrome. In addition, the observed reduction in levels of cytokines in association with the resolution of an acute psychotic episode could be part of the therapeutic action of treatment (supportive of model 1 rather than 2). Model 3, where a shared risk factor plays a role in development of psychosis and non-CNS alterations through divergent mechanisms, is supported by the lack of consistent relationships between a number of CNS and non-CNS alterations. Also, the heterogeneity in both non-CNS and CNS findings between patients could suggest divergent mechanisms underlie them [120]. However, we have identified certain correlations, e.g., between glucose dysregulation [65], insulin resistance [66], and PANSS-positive scores in FEP, that point toward these non-CNS alterations being linked with the clinical expression of psychosis, consistent with, although not proving, a common pathoetiological mechanism. Moreover, there is a paucity of studies testing relationships between non-CNS and CNS alterations in psychosis, limiting any conclusions regarding common causality. Overall, there is both supportive and contradictory evidence for all three models, and aspects of each model that remain to be fully tested. The contradictory evidence suggests that one model is unlikely to account for all cases of psychosis. Moreover, while the models provide an overarching framework, the specific mechanisms that might link risk factors, CNS and non-CNS alterations need to be investigated. Further work is clearly needed, particularly to investigate the causal nature of relationships, and how common they are to patients in general.
Does the involvement of multiple systems in first-episode psychosis indicate that psychosis is a multisystem disorder?
Conceptually, multisystem disorders can be categorized into two groups: [121] (1) conditions where multiple organ systems are pervasively affected with no single predominant organ involved (e.g., inborn errors of metabolism); (2) conditions where one organ system is predominantly affected, but where other organs may concurrently be involved (e.g., rheumatoid arthritis). Since psychosis is by definition a description of psychological phenomena, it would be hard to argue that psychosis meets the first definition. However, consistent with the second definition, the evidence reviewed above suggests that early in psychosis dysfunction is present across multiple organ systems. Given the large effect sizes and number of negative studies required for many of these to become no longer significant, these findings appear robust, at least as robust as for many of the brain structural/functional alterations seen in psychosis. Moreover, we found that there is some, albeit limited, evidence that non-CNS measures are linked to symptoms and CNS changes in FEP. This could be taken as suggesting that psychosis, and by extension schizophrenia, should be considered a multisystem disorder. However, the International Classification of Disease-11 2010 Steering Group discussion paper on multisystem diseases [121], defined these as “diseases that regularly manifest without involvement of a common single system and with concomitant major involvement of several systems”, and, on this basis, a disorder such as rheumatoid arthritis would not be considered multisystem because, while it affects multiple systems, its predominant manifestation is musculoskeletal. By the same token, given that psychosis by definition is a disorder of thought and behavior, it is clear that CNS dysfunction bears the most direct relationship with the clinical expression of the disorder. This argues against psychosis being a multisystem disorder. However, this argument is inherently circular because psychotic disorders are diagnosed solely on the basis of mental symptoms. Here, while the evidence reviewed above goes some way to showing the magnitude of non-CNS effects is similar, in some cases larger, than CNS effects, what is currently lacking is robust evidence that the changes in non-CNS systems have commensurate clinical impacts, for example on functioning, prognosis, or mortality. Evidence is also needed on the nature of the relationship between CNS and non-CNS changes: if non-CNS changes are found to be due to a common pathoetiology or risk factor (model 3) or lead to CNS changes (model 1), this could support psychosis being a multisystem disorder, while finding that non-CNS alterations are secondary to mental symptoms (model 2) would not. A consequence of this would potentially be that diagnostic and prognostic assessment might incorporate assessment of non-CNS organ dysfunction in addition to assessment of thought and behavior. For example, if the pathophysiology of FEP includes a hypercortisolemic, pro-inflammatory state, then a biomarker “fingerprint” of antipsychotic naive FEP could conceivably include evidence of HPA axis dysregulation (e.g., blunted cortisol awakening response) with raised peripheral cytokines (e.g., IL-6) as well as relevant measures of CNS function.
Implications and future directions
Further research is required to elucidate whether non-CNS dysfunction is a cause or a consequence of psychosis. Longitudinal studies of non-CNS parameters starting in people at clinical high risk for psychosis and continuing through development of psychosis is a potential approach, and has the advantage of including a control group exposed to the same risk factors (those individuals who do not develop psychosis), as well as the research advantage that a number will develop other mental disorders. This group could help address another key question, which is the degree to which non-CNS alterations are specific to psychosis or are a common feature of a number of mental disorders, potentially consistent with a model whereby the stress of mental illness leads to changes in other systems [122] (model 2). Other key areas that our review has highlighted as requiring further work are the degree to which alterations in non-CNS systems are linked to psychotic symptoms, and other clinical outcomes, and whether common pathoetiological mechanisms underlie both CNS and non-CNS alterations.
In terms of clinical practice, the majority of excess mortality seen in schizophrenia is due to non-CNS causes, predominantly cardiovascular disease [8], and life expectancy in schizophrenia has failed to improve relative to the general population over recent decades [1]. Our findings of cardiometabolic, inflammatory, and HPA axis alterations in FEP suggest that processes underlying excess mortality are present early in schizophrenia and other psychotic disorders. One implication for clinicians is to routinely consider these systems in the assessment of psychotic disorders. Studies are needed to determine if addressing non-CNS alterations early reduces development of physical co-morbidity, and ultimately reduces mortality in psychotic disorders (see summary box).
Conclusions
Abnormalities in multiple organ systems in addition to the CNS are seen at onset of psychotic disorders with similar magnitudes to those seen in the CNS. While the causal relationship between non-CNS and CNS alterations remains to be determined, this evidence indicates that psychosis involves multiple systems from illness onset, although is not sufficient to define it as a multisystem disorder.
Competing interests
Professor Howes has received investigator-initiated research funding from and/or participated in advisory/speaker meetings organized by Astra-Zeneca, Autifony, BMS, Eli Lilly, Heptares, Janssen, Lundbeck, Lyden-Delta, Otsuka, Servier, Sunovion, Rand, and Roche. Neither Professor Howes nor his family have been employed by or have holdings/a financial stake in any biomedical company. Drs Pillinger, D’Ambrosio, and McCutcheon report no financial relationships with commercial interests.
Change history
18 October 2018
Updated online [insert date]: This article was originally published under standard licence, but has now been made available under a [CC BY 4.0] license. The PDF and HTML versions of the paper have been modified accordingly.
References
Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64:1123–31.
Whiteford HA, Baxter AJ. The Global Burden of Disease 2010 Study: what does it tell us about mental disorders in Latin America? Rev Bras Psiquiatr. 2013;35:111–2.
Knapp M, Mangalore R, Simon J. The global costs of schizophrenia. Schizophr Bull. 2004;30:279–93.
Crump C, Winkleby MA, Sundquist K, Sundquist J. Comorbidities and mortality in persons with schizophrenia: a Swedish national cohort study. Am J Psychiatry. 2013;170:324–33.
Hoang U, Goldacre MJ, Stewart R. Avoidable mortality in people with schizophrenia or bipolar disorder in England. Acta Psychiatr Scand. 2013;127:195–201.
Laursen TM, Nordentoft M, Mortensen PB. Excess early mortality in schizophrenia. Annu Rev Clin Psychol. 2014;10:425–48.
Brown S, Kim M, Mitchell C, Inskip H. Twenty-five year mortality of a community cohort with schizophrenia. Br J Psychiatry. 2010;196:116–21.
Correll CU, Solmi M, Veronese N, et al. Prevalence, incidence and mortality from cardiovascular disease in patients with pooled and specific severe mental illness: a large-scale meta-analysis of 3,211,768 patients and 113,383,368 controls. World Psychiatry. 2017;16:163–80.
McCreadie RG. Scottish Schizophrenia Lifestyle G. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534–9.
Correll CU, Frederickson AM, Kane JM, Manu P. Metabolic syndrome and the risk of coronary heart disease in 367 patients treated with second-generation antipsychotic drugs. J Clin Psychiatry. 2006;67:575–83.
Pillinger T, Beck K, Gobjila C, Donocik JG, Jauhar S, Howes OD. Impaired glucose homeostasis in first-episode schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74:261–9.
Perry BI, McIntosh G, Weich S, Singh S, Rees K. The association between first-episode psychosis and abnormal glycaemic control: systematic review and meta-analysis. Lancet Psychiatry 2016;3:1049–1058.
Greenhalgh AM, Gonzalez-Blanco L, Garcia-Rizo C, Fernandez-Egea E, Miller B, Arroyo MB et al. Meta-analysis of glucose tolerance, insulin, and insulin resistance in antipsychotic-naive patients with nonaffective psychosis. Schizophr Res 2017;179:57–63.
Misiak B, Stanczykiewicz B, Laczmanski L, Frydecka D. Lipid profile disturbances in antipsychotic-naive patients with first-episode non-affective psychosis: A systematic review and meta-analysis. Schizophr Res 2017;190:18–27.
Flatow J, Buckley P, Miller BJ. Meta-analysis of oxidative stress in schizophrenia. Biol Psychiat. 2013;74:400–9.
Pillinger T, Beck K, Stubbs B, Howes OD. Cholesterol and triglyceride levels in first-episode psychosis: systematic review and meta-analysis. Br J Psychiatry 2017;211:339–349.
Upthegrove R, Manzanares-Teson N, Barnes NM. Cytokine function in medication-naive first episode psychosis: a systematic review and meta-analysis. Schizophr Res. 2014;155:101–8.
Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry 2016;21:1696–1709.
Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B. Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70:663–71.
Fernandes BS, Steiner J, Bernstein HG, et al. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol Psychiatry. 2016;21:554–64.
Miller BJ, Gassama B, Sebastian D, Buckley P, Mellor A. Meta-analysis of lymphocytes in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2013;73:993–9.
Berger M, Kraeuter AK, Romanik D, Malouf P, Amminger GP, Sarnyai Z. Cortisol awakening response in patients with psychosis: systematic review and meta-analysis. Neurosci Biobehav Rev. 2016;68:157–66.
Chaumette B, Kebir O, Mam-Lam-Fook C, et al. Salivary cortisol in early psychosis: New findings and meta-analysis. Psychoneuroendocrinology. 2016;63:262–70.
Girshkin L, Matheson SL, Shepherd AM, Green MJ. Morning cortisol levels in schizophrenia and bipolar disorder: a meta-analysis. Psychoneuroendocrinology.. 2014;49:187–206.
Gonzalez-Blanco L, Greenhalgh AM, Garcia-Rizo C, Fernandez-Egea E, Miller BJ, Kirkpatrick B. Prolactin concentrations in antipsychotic-naive patients with schizophrenia and related disorders: a meta-analysis. Schizophr Res. 2016;174:156–60.
Moher D, Liberati A, Tetzlaff J, Altman DG, Grp P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol. 2009;62:1006–12.
Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry. 2015;2:258–70.
Mitchell AJ, Vancampfort D, Sweers K, van Winkel R, Yu W, De Hert M. Prevalence of metabolic syndrome and metabolic abnormalities in schizophrenia and related disorders--a systematic review and meta-analysis. Schizophr Bull. 2013;39:306–18.
Phillips LJ, McGorry PD, Garner B, et al. Stress, the hippocampus and the hypothalamic-pituitary-adrenal axis: implications for the development of psychotic disorders. Aust N Z J Psychiatry. 2006;40:725–41.
Expert Panel on Detection E, Treatment of High Blood Cholesterol in A. Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). JAMA. 2001;285:2486–97.
Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–52.
Alberti KG, Zimmet P, Shaw J. Group IDFETFC. The metabolic syndrome--a new worldwide definition. Lancet. 2005;366:1059–62.
World Health Organization. Definition, diagnosis and classification of diabetes mellitus and its complications: report of a WHO consultation. Part 1: diagnosis and classification of diabetes mellitus. Geneva: World Health Org.; 1999.
M DEH, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry. 2011;10:52–77.
Harris MI, Klein R, Welborn TA, Knuiman MW. Onset of NIDDM occurs at least 4-7 yr before clinical diagnosis. Diabetes Care. 1992;15:815–9.
Danesh J, Whincup P, Walker M, et al. Low grade inflammation and coronary heart disease: prospective study and updated meta-analyses. BMJ. 2000;321:199–204.
Keshavan MS, Tandon R, Boutros NN, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 Part 3: neurobiology. Schizophr Res. 2008;106:89–107.
Rosenthal R. The file drawer problem and tolerance for null results. Psychol Bull. 1979;86:638–41.
Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. 2010;36:1–48.
Adriano F, Caltagirone C, Spalletta G. Hippocampal volume reduction in first-episode and chronic schizophrenia: a review and meta-analysis. Neuroscientist. 2012;18:180–200.
Walter A, Suenderhauf C, Harrisberger F, et al. Hippocampal volume in subjects at clinical high-risk for psychosis: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2016;71:680–90.
Adriano F, Spoletini I, Caltagirone C, Spalletta G. Updated meta-analyses reveal thalamus volume reduction in patients with first-episode and chronic schizophrenia. Schizophr Res. 2010;123:1–14.
Haijma SV, Van Haren N, Cahn W, Koolschijn PC, Hulshoff Pol HE, Kahn RS. Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull. 2013;39:1129–38.
Vita A, de Peri L. Hippocampal and amygdala volume reductions in first-episode schizophrenia. Br J Psychiatry. 2007;190:271.
de Peri L, Crescini A, Deste G, Fusar-Poli P, Sacchetti E, Vita A. Brain structural abnormalities at the onset of schizophrenia and bipolar disorder: a meta-analysis of controlled magnetic resonance imaging studies. Curr Pharmaceutical Des. 2012;18:486–94.
Vita A, De Peri L, Silenzi C, Dieci M. Brain morphology in first-episode schizophrenia: A meta-analysis of quantitative magnetic resonance imaging studies. Schizophr Res. 2006;82:75–88.
Fusar-Poli P, Radua J, McGuire P, Borgwardt S. Neuroanatomical maps of psychosis onset: Voxel-wise meta-analysis of antipsychotic-naive vbm studies. Schizophr Bull. 2012;38:1297–307.
Erickson MA, Ruffle A, Gold JM. A meta-analysis of mismatch negativity in schizophrenia: from clinical risk to disease specificity and progression. Biol Psychiatry. 2016;79:980–7.
Qiu YQ, Tang YX, Chan RC, Sun XY, He J. P300 aberration in first-episode schizophrenia patients: a meta-analysis. PLoS ONE. 2014;9:e97794.
Chen KC, Lee IH, Yang YK, et al. P300 waveform and dopamine transporter availability: a controlled EEG and SPECT study in medication-naive patients with schizophrenia and a meta-analysis. Psychol Med. 2014;44:2151–62.
Haigh SM, Coffman BA, Salisbury DF. Mismatch negativity in first-episode schizophrenia: a meta-analysis. Clin EEG Neurosci. 2017;48:3–10.
Brugger S, Davis JM, Leucht S, Stone JM. Proton magnetic resonance spectroscopy and illness stage in schizophrenia--a systematic review and meta-analysis. Biol Psychiatry. 2011;69:495–503.
Cohen J. Statistical power analysis for the behavioral sciences. 2nd ed. Hillsdale, N.J.: L. Erlbaum Associates; 1988.
Lennartsson AK, Jonsdottir IH. Prolactin in response to acute psychosocial stress in healthy men and women. Psychoneuroendocrinology. 2011;36:1530–9.
Bora E, Fornito A, Radua J, et al. Neuroanatomical abnormalities in schizophrenia: a multimodal voxelwise meta-analysis and meta-regression analysi7s. Schizophr Res. 2011;127:46–57.
Morales I, Farias G, Maccioni RB. Neuroimmunomodulation in the pathogenesis of Alzheimer’s disease. Neuroimmunomodulation. 2010;17:202–4.
Peng YP, Qiu YH, Lu JH, Wang JJ. Interleukin-6 protects cultured cerebellar granule neurons against glutamate-induced neurotoxicity. Neurosci Lett. 2005;374:192–6.
Bossu P, Piras F, Palladino I, et al. Hippocampal volume and depressive symptoms are linked to serum IL-18 in schizophrenia. Neurol Neuroimmunol Neuroinflamm. 2015;2:e111.
Fillman SG, Weickert TW, Lenroot RK, et al. Elevated peripheral cytokines characterize a subgroup of people with schizophrenia displaying poor verbal fluency and reduced Broca’s area volume. Mol Psychiatry. 2016;21:1090–8.
Gunduz-Bruce H, Szeszko PR, Gueorguieva R, et al. Cortisol levels in relation to hippocampal sub-regions in subjects with first episode schizophrenia. Schizophr Res. 2007;94:281–7.
Mondelli V, Pariante CM, Navari S, et al. Higher cortisol levels are associated with smaller left hippocampal volume in first-episode psychosis. Schizophr Res. 2010;119:75–78.
Pruessner M, Lepage M, Collins DL, Pruessner JC, Joober R, Malla AK. Reduced hippocampal volume and hypothalamus-pituitary-adrenal axis function in first episode psychosis: evidence for sex differences. Neuroimage Clin. 2015;7:195–202.
Prasad KM, Upton CH, Nimgaonkar VL, Keshavan MS. Differential susceptibility of white matter tracts to inflammatory mediators in schizophrenia: an integrated DTI study. Schizophr Res. 2015;161:119–25.
Willette AA, Xu G, Johnson SC, et al. Insulin resistance, brain atrophy, and cognitive performance in late middle-aged adults. Diabetes Care. 2013;36:443–9.
Zhang XY, Chen DC, Tan YL, et al. Glucose disturbances in first-episode drug-naive schizophrenia: Relationship to psychopathology. Psychoneuroendocrinology. 2015;62:376–80.
Chen S, Broqueres-You D, Yang G, et al. Relationship between insulin resistance, dyslipidaemia and positive symptom in Chinese antipsychotic-naive first-episode patients with schizophrenia. Psychiatry Res. 2013;210:825–9.
Fernandes B, Steiner J, Bernstein H, et al. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol Psychiatry. 2016;21:554–64.
Xiu MH, Yang GG, Tan YL, et al. Decreased interleukin-10 serum levels in first-episode drug-naive schizophrenia: relationship to psychopathology. Schizophr Res. 2014;156:9–14.
Stojanovic A, Martorell L, Montalvo I, et al. Increased serum interleukin-6 levels in early stages of psychosis: associations with at-risk mental states and the severity of psychotic symptoms. Psychoneuroendocrinology. 2014;41:23–32.
Frommberger UH, Bauer J, Haselbauer P, Fraulin A, Riemann D, Berger M. Interleukin-6-(IL-6) plasma levels in depression and schizophrenia: comparison between the acute state and after remission. Eur Arch Psychiatry Clin Neurosci. 1997;247:228–33.
Pae CU, Yoon CH, Kim TS, et al. Antipsychotic treatment may alter T-helper (TH) 2 arm cytokines. Int Immunopharmacol. 2006;6:666–71.
Akiyama K. Serum levels of soluble IL-2 receptor alpha, IL-6 and IL-1 receptor antagonist in schizophrenia before and during neuroleptic administration. Schizophr Res. 1999;37:97–106.
Maes M, Bocchio Chiavetto L, Bignotti S, et al. Effects of atypical antipsychotics on the inflammatory response system in schizophrenic patients resistant to treatment with typical neuroleptics. Eur Neuropsychopharmacol. 2000;10:119–24.
Dimitrov DH, Lee S, Yantis J, et al. Differential correlations between inflammatory cytokines and psychopathology in veterans with schizophrenia: potential role for IL-17 pathway. Schizophr Res. 2013;151:29–35.
Song XQ, Lv LX, Li WQ, Hao YH, Zhao JP. The interaction of nuclear factor-kappa B and cytokines is associated with schizophrenia. Biol Psychiatry. 2009;65:481–8.
Kubistova A, Horacek J, Novak T. Increased interleukin-6 and tumor necrosis factor alpha in first episode schizophrenia patients versus healthy controls. Psychiatr Danub. 2012;24:S153–156.
Crespo-Facorro B, Carrasco-Marin E, Perez-Iglesias R, et al. Interleukin-12 plasma levels in drug-naive patients with a first episode of psychosis: effects of antipsychotic drugs. Psychiatry Res. 2008;158:206–16.
Borovcanin M, Jovanovic I, Radosavljevic G, et al. Elevated serum level of type-2 cytokine and low IL-17 in first episode psychosis and schizophrenia in relapse. J Psychiatr Res. 2012;46:1421–6.
Strous RD, Maayan R, Lapidus R, et al. Increased circulatory dehydroepiandrosterone and dehydroepiandrosterone-sulphate in first-episode schizophrenia: relationship to gender, aggression and symptomatology. Schizophr Res. 2004;71:427–34.
Garner B, Phassouliotis C, Phillips LJ, et al. Cortisol and dehydroepiandrosterone-sulphate levels correlate with symptom severity in first-episode psychosis. J Psychiatr Res. 2011;45:249–55.
Belvederi Murri M, Pariante CM, Dazzan P, et al. Hypothalamic-pituitary-adrenal axis and clinical symptoms in first-episode psychosis. Psychoneuroendocrinology. 2012;37:629–44.
Pruessner M, Boekestyn L, Bechard-Evans L, et al. Sex differences in the cortisol response to awakening in recent onset psychosis. Psychoneuroendocrinology. 2008;33:1151–4.
Venkatasubramanian G, Chittiprol S, Neelakantachar N, et al. Insulin and insulin-like growth factor-1 abnormalities in antipsychotic-naive schizophrenia. Am J Psychiatry. 2007;164:1557–60.
Ryan MC, Sharifi N, Condren R, Thakore JH. Evidence of basal pituitary-adrenal overactivity in first episode, drug naive patients with schizophrenia. Psychoneuroendocrinology. 2004;29:1065–70.
Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175:47–53.
Kirkpatrick B, Fernandez-Egea E, Garcia-Rizo C, Bernardo M. Differences in glucose tolerance between deficit and nondeficit schizophrenia. Schizophr Res. 2009;107:122–7.
Lopresti AL, Maker GL, Hood SD, Drummond PD. A review of peripheral biomarkers in major depression: the potential of inflammatory and oxidative stress biomarkers. Prog Neuropsychopharmacol Biol Psychiatry. 2014;48:102–11.
Goldstein BI, Young LT. Toward clinically applicable biomarkers in bipolar disorder: focus on BDNF, inflammatory markers, and endothelial function. Curr Psychiatry Rep. 2013;15:425.
Pan P, Shi H, Zhong J, et al. Chronic smoking and brain gray matter changes: evidence from meta-analysis of voxel-based morphometry studies. Neurol Sci. 2013;34:813–7.
Ansell EB, Rando K, Tuit K, Guarnaccia J, Sinha R. Cumulative adversity and smaller gray matter volume in medial prefrontal, anterior cingulate, and insula regions. Biol Psychiatry. 2012;72:57–64.
Connolly CG, Bell RP, Foxe JJ, Garavan H. Dissociated grey matter changes with prolonged addiction and extended abstinence in cocaine users. PLoS ONE. 2013;8:e59645.
Xiao PR, Dai ZY, Zhong JG, Zhu YL, Shi HC, Pan PL. Regional gray matter deficits in alcohol dependence: A meta-analysis of voxel-based morphometry studies. Drug Alcohol Depend. 2015;153:22–28.
Juutinen J, Hakko H, Meyer-Rochow VB, Rasanen P, Timonen M. Study-70 research G. body mass index (BMI) of drug-naive psychotic adolescents based on a population of adolescent psychiatric inpatients. Eur Psychiatry. 2008;23:521–6.
Koivukangas J, Tammelin T, Kaakinen M, et al. Physical activity and fitness in adolescents at risk for psychosis within the Northern Finland 1986 Birth Cohort. Schizophr Res. 2010;116:152–8.
Howick J, Glasziou P, Aronson JK. The evolution of evidence hierarchies: what can Bradford Hill’s ‘guidelines for causation’ contribute? J Roy Soc Med. 2009;102:186–94.
Ioannidis JP, Lau J. Pooling research results: benefits and limitations of meta-analysis. Jt Comm J Qual Improv. 1999;25:462–9.
Becker BJ. Failsafe n or file-drawer number, in publication bias in meta-analysis: prevention, assessment and adjustments. Chichester: John Wiley & Sons; 2005.
Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36.
Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur J Pharmacol. 2004;500:399–411.
Bloomfield PS, Selvaraj S, Veronese M, et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [(11)C]PBR28 PET brain imaging study. Am J Psychiatry. 2016;173:44–52.
Song C, Merali Z, Anisman H. Variations of nucleus accumbens dopamine and serotonin following systemic interleukin-1, interleukin-2 or interleukin-6 treatment. Neuroscience. 1999;88:823–36.
Barry S, Clarke G, Scully P, Dinan TG. Kynurenine pathway in psychosis: evidence of increased tryptophan degradation. J Psychopharmacol. 2009;23:287–94.
Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol. 2015;29:97–115.
Skoluda N, Strahler J, Schlotz W, et al. Intra-individual psychological and physiological responses to acute laboratory stressors of different intensity. Psychoneuroendocrinology. 2015;51:227–36.
Whitworth JA, Williamson PM, Mangos G, Kelly JJ. Cardiovascular consequences of cortisol excess. Vasc Health Risk Manag. 2005;1:291–9.
Howes OD, Murray RM. Schizophrenia: an integrated sociodevelopmental-cognitive model. Lancet. 2014;383:1677–87.
Lawlor DA, Davey Smith G, Clark H, Leon DA. The associations of birthweight, gestational age and childhood BMI with type 2 diabetes: findings from the Aberdeen Children of the 1950s cohort. Diabetologia. 2006;49:2614–7.
Sorensen HJ, Gamborg M, Sorensen TI, Baker JL, Mortensen EL. Childhood body mass index and risk of schizophrenia in relation to childhood age, sex and age of first contact with schizophrenia. Eur Psychiatry. 2016;34:64–69.
Morrison KM, Ramsingh L, Gunn E, Streiner D, Van Lieshout R, Boyle M et al. Cardiometabolic Health in Adults Born Premature With Extremely Low Birth Weight. Pediatrics 2016;138.
Nosarti C, Reichenberg A, Murray RM, et al. Preterm birth and psychiatric disorders in young adult life. Arch Gen Psychiatry. 2012;69:E1–8.
St Clair D, Xu M, Wang P, et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959-61. JAMA. 2005;294:557–62.
Ravelli AC, van der Meulen JH, Michels RP, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351:173–7.
Brown AS, Susser ES. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr Bull. 2008;34:1054–63.
Lee HS. Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients. 2015;7:9492–507.
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Malan-Muller S, Kilian S, van den Heuvel LL, et al. A systematic review of genetic variants associated with metabolic syndrome in patients with schizophrenia. Schizophr Res. 2016;170:1–17.
Metcalf SA, Jones PB, Nordstrom T, et al. Serum C-reactive protein in adolescence and risk of schizophrenia in adulthood: a prospective birth cohort study. Brain Behav Immun. 2017;59:253–9.
Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry. 2014;71:1121–8.
Addington J, Cadenhead KS, Cornblatt BA, et al. North American Prodrome Longitudinal Study (NAPLS 2): overview and recruitment. Schizophr Res. 2012;142:77–82.
Brugger SP, Howes OD. Heterogeneity and homogeneity of regional brain structure in schizophrenia: a meta-analysis. JAMA Psychiatry. 2017;74:1104–11.
Aymé C, Chute, Jakob. ICD Revision: discussion paper: multisystem disorders. http://bit.ly/2nnilKW. 2010.
Calcia MA, Bonsall DR, Bloomfield PS, Selvaraj S, Barichello T, Howes OD. Stress and neuroinflammation: a systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology. 2016;233:1637–50.
Acknowledgements
We thank Dr Stefan Brugger of the MRC London Institute of Medical Sciences for providing additional data.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pillinger, T., D’Ambrosio, E., McCutcheon, R. et al. Is psychosis a multisystem disorder? A meta-review of central nervous system, immune, cardiometabolic, and endocrine alterations in first-episode psychosis and perspective on potential models. Mol Psychiatry 24, 776–794 (2019). https://doi.org/10.1038/s41380-018-0058-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-018-0058-9
This article is cited by
-
Immunophenotypes in psychosis: is it a premature inflamm-aging disorder?
Molecular Psychiatry (2024)
-
Dance/movement therapy for improving metabolic parameters in long-term veterans with schizophrenia
Schizophrenia (2024)
-
How can we improve the care of patients with schizophrenia in the real-world? A population-based cohort study of 456,003 patients
Molecular Psychiatry (2023)
-
Molecular mapping of a core transcriptional signature of microglia-specific genes in schizophrenia
Translational Psychiatry (2023)
-
Molecular-Genetic and Immunological Aspects of the Formation of Psychopathological Symptoms in Schizophrenia
Neuroscience and Behavioral Physiology (2023)
