
Abstract
In this Perspective, we discuss criteria for defining a new disease entity or variant of a recognized disease or disorder. We do so in the context of the current topography of the BCR::ABL-negative myeloproliferative neoplasms (MPNs) where two new variants are reported: clonal megakaryocyte dysplasia with normal blood values (CMD-NBV) and clonal megakaryocyte dysplasia with isolated thrombocytosis (CMD-IT). The cardinal feature of these variants is bone marrow megakaryocyte hyperplasia and atypia corresponding the WHO histological criteria for primary myelofibrosis (myelofibrosis-type megakaryocyte dysplasia-MTMD). Persons with these new variants have a different disease course and features from others in the MPN domain. In a broader context we suggest myelofibrosis-type megakaryocyte dysplasia defines a spectrum of related MPN variants including CMD-NBV, CMD-IT, pre-fibrotic myelofibrosis and overt myelofibrosis, which differ from polycythemia vera and essential thrombocythemia. Our proposal needs external validation and we stress the need for a consensus definition of the megakaryocyte dysplasia which is the hallmark of these disorders.
Similar content being viewed by others


We recently described two new disease variants we claimed belonging to the BCR::ABL-negative myeloproliferative neoplasms (MPNs) domain [1, 2]. The first is a re-formatting of our 1991 article reporting 18 cases of an atypical myeloproliferative disorder with high risk of thrombosis and slow disease progression [3]. The second is defined by a platelet concentration ≥450 × 10E + 9/L without other blood or laboratory signs of myeloproliferation or disease activity. The cardinal feature of both variants is bone marrow megakaryocyte hyperplasia and atypia corresponding the WHO description of megakaryocytes alterations included in the diagnostic criteria for primary myelofibrosis (PMF): dense or loose clustering and frequent endosteal translocation of megakaryocytes with hyper-chromatic, hypo-lobulated, bulbous or irregularly folded nuclei and an aberrant nuclear/cytoplasmic ratio [4]. (Fig. 1). We termed the first variant clonal megakaryocyte dysplasia with normal blood values (CMD-NBV) and the second clonal megakaryocyte dysplasia with isolated thrombocytosis (CMD-IT; Table 1).

The picture show megakaryocte hyperplasia and cellular atypia.
Our belief is that these reports represent an advance in the field of MPNs by reinforcing the concept of MPNs as an array of phenotypes and by highlighting that the number of these phenotypes is higher than previously recognized. This speculation is contrasted by the unavoidable limitations of our papers. These included retrospective analyses of a single center observational database without external validation, and bone marrow histology classified by one pathologist rather than by consensus amongst pathologists. However, the strongest criticism to our articles was these variants had insufficiently consistent phenotypic uniqueness to be considered new entities.
In the last few decades genetic studies of the MPNs indicate 3 underlying driver mutations, JAK2V617F, CALR and MPL. Data from next generation sequencing (NGS) has uncovered additional molecular complexity altering how we classify the MPNs. The revised 2017 and 2022 World Health Organization (WHO) criteria reflected these advances [4, 5]. For example, pre-fibrotic myelofibrosis (pre-fibrotic MF) has been added as has MPN, unclassifiable.
Despite this progress, classification of MPNs is not biology-driven. Patho-physiological mechanisms distinguishing polycythemia vera (PV) from essential thrombocythemia (ET) and pre-fibrotic MF from ET are incompletely defined nor understood. Consequently, as in many areas of medicine, classification of the MPNs follows the statute of the nominal law which prescriptively assigns names to entities with empirical repeatability. A disease becomes an entity if its existence is plausible without needing to know its causality or biology.
We think criticism of the new variants we described is more philosophical than biological. The underlying question is: when can we claim the existence of a new disease or disease variant? Our position is that the only relevant question for the acceptance is utility. The utility of our proposed new variants is that people with the features we describe have an unique disease course and features, and can be removed from the intellectually unsatisfying MPN, unclassifiable grab bag.
None of the 15 subjects with CMD-NBV had signs of disease progression after a median follow-up of more than 8 years, Subjects with CMD-IT lived longer than those with pre-fibrotic MF and those with overt- MF (HR = 0.42 [95% Confidence interval (CI) 0.23, 0.75], P = 0.003; and HR = 0.13 [0.075, 0.23], P < 0.001). Subjects with CMD-NBV had a significantly higher incidence of post-diagnosis thrombosis compared with persons with ET or PV (3.9 events per 100 subject-years versus 1.7 and 2.7). In contrast, subjects with CMD-IT had a significantly lower risk of thrombosis compared with persons with ET or pre-fibrotic MF [1.03 (0.53, 1.79) events per 100 subject-years versus 3.09 and 2.09].
Distinguishing features of the new variants we propose are of considerable clinical import. Moreover, they reinforce the concept of the MPNs as a spectrum of disorders promoted by specific constitutive and genetic features. For example, subjects with CMD-IT are more often female, have a higher frequency of type-2/type-2-like CALR mutations and a lower JAK2V617F allele burden compared with persons with PMF. They also have a lower frequency of genetic variants correlated with risk of developing PMF such as the 46/1 haplotype and VEGFA rs3025039 polymorphism [3].
CMD-NBV is a special disease variant characterized by lack of hematological abnormalities or marginally elevated blood values. Subjects are mostly diagnosed in the context of thromboses. 13 of our 15 subjects with CMD-NBV had a synchronous symptomatic thrombotic event including portal vein thrombosis, Budd-Chiari syndrome, peripheral arterial thrombosis, myocardial infarction, spleen infarction or an incidentally detected portal cavernoma at diagnosis.
CMD-NBV forces us to rethink the generalizability of diagnostic reasoning in MPNs. The WHO criteria for the classical MPNs starts with considering an abnormal hematological value. In contrast, in CMD-NBV the diagnosis is triggered by clinical features. Positive predictive value of MPN diagnosis depends on patient- and practice setting-related co-variates. This results in a high level of uncertainty in CMD-NBV diagnostic reasoning requiring ad hoc rules to guide physicians to move from a circumstantial diagnostic metric to a metric based on clinical features, histology and genetics, and suggests the real frequency of CMD-NBV may be much greater than thought.
A further advancement in the MPN field of the introduction of the new disease variants is that they share the histological megakaryocyte characteristics typical of PMF, and that megakaryocyte morphology deviation was a major criterion to diagnose them. To define these megakaryocyte characteristics we now propose to introduce the term myelofibrosis-type megakaryocyte dysplasia (MTMD) and to highlight its relevance we conceptualize that this morphology identifies a disease category among MPNs.
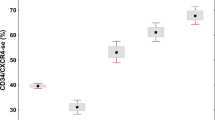
The theory of MTMD has been accepted in the 2022 WHO [5] and International Consensus Classification (ICC) [6] to distinguish pre-fibrotic MF from ET. With our proposal, now MTDT defines a group of related MPN variants including CMD-NBV, CMD-IT, pre-fibrotic MF and overt MF (Fig. 2). In our dataset these variants constitute 2, 13, 37 and 48 percent of this category. If representative, our data suggest the indolent non-fibrotic variants are more common than overt MF.

PV polycythemia vera, ET essential thrombocythemia, CMD-NBV clonal megakaryocyte dysplasia with normal blood values, CMD-IT clonal megakaryocyte dysplasia with isolated thrombocytosis, pre-MF pre-fibrotic myelofibrosis, overt MF overt myelofibrosis.
This MTMD paradigm, born from an acute morphological vision, is now supported by biological evidence. In fact, hypo-morphic GATA1 mutations selectively decrease GATA1 in megakaryocytes and induce myelofibrosis in mice and a bone marrow histology like primary myelofibrosis in humans [7]. Also, megakaryocytes from humans with PMF have low levels of GATA1 probably maintained by ribosome abnormalities induced by driver mutations [8]. Recently, aurora kinase A was reported to be over-expressed in PMF and a selective inhibitor promotes polyploidization and differentiation of megakaryocytes with PMF-associated mutations in mice [9]. Finally, evidence that megakaryocyte morphology in PMF is unique among MPNs is suggested by the observation that in vitro PMF-derived megakaryocytes display nuclei with a bulbous appearance, and are smaller than those ET- or PV-derived [10].
The MTMD concept contrasts with megakaryocyte dysplasia in other myeloid disorders such as myelodysplastic syndromes (MDS). Histological criteria distinguishing megakaryocyte dysplasia in MDS from that in PMF is proposed but unvalidated [11]. A useful classification tool could be the megakaryocyte dysplasia score proposed to predict response in people with PMF receiving a haematopoietic cell transplant but is unvalidated in non-transplanted persons [12].
In conclusion, the recognition of 2 new variants of MPN including subjects previously considered under the rubric of MPN, unclassifiable addressed us to hypothesize a broad category of MPNs bearing different phenotypes may be included in an unique category according to the megakaryocyte morphology. Agreeing on MTMD as a category needs standardization of relevant bone marrow features. Since the histological assessment of bone marrow in MPNs remains constrained by a reliance on subjective and qualitative criteria, we emphasize the use of more precise methods. Computational methods designed to systematically capture the key morphological characteristics of megakaryocytes were proved able to associate with particular MPN subtypes [13, 14]. This strategy seems to have significant potential for translation into a better definition of our proposed category of megakaryocyte morphology. Biomarkers of this histological diagnostic criteria would also help and should be investigated.
References
Barosi G, Rosti V, Massa M, Campanelli R, Villani L, Catarsi P, et al. Clonal Megakaryocyte Dysplasia with Normal Blood Values Is a Distinct Myeloproliferative Neoplasm. Acta Haematol. 2022;145:30–7.
Barosi G, Campanelli R, Massa M, Catarsi P, Carolei A, Abbà C, et al. Clonal Megakaryocyte Dysplasia with Isolated Thrombocytosis Is a Distinct Myeloproliferative Neoplasm Phenotype. Acta Haematol. 2023;146:14–25.
Barosi G, Buratti A, Costa A, Liberato LN, Balduini C, Cazzola M, et al. An atypical myeloproliferative disorder with high thrombotic risk and slow disease progression. Cancer. 1991;68:2310–8.
Swerdow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2017. Revised.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–19.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
Vannucchi AM, Bianchi L, Cellai C, Paoletti F, Rana RA, Lorenzini R, et al. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1(low) mice). Blood. 2002;100:1123–32.
Vannucchi AM, Pancrazzi A, Guglielmelli P, Di Lollo S, Bogani C, Baroni G, et al. Abnormalities of GATA-1 in megakaryocytes from patients with idiopathic myelofibrosis. Am J Patho.l 2005;167:849–58.
Wen QJ, Yang Q, Goldenson B, Malinge S, Lasho T, Schneider RK, et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by AURKA inhibition. Nat Med. 2015;21:1473–80.
Balduini A, Badalucco S, Pugliano MT, Baev D, De Silvestri A, Cattaneo M, et al. In vitro megakaryocyte differentiation and proplatelet formation in Ph-negative classical myeloproliferative neoplasms: distinct patterns in the different clinical phenotypes. PLoS ONE. 2011;6:e21015.
Della Porta MG, Travaglino E, Boveri E, Ponzoni M, Malcovati L, Papaemmanuil E, et al. Clinical Network. Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes. Leukemia. 2015;29:66–75.
Khanlari M, Wang X, Loghavi S, Wang SA, Li S, Thakral B, et al. Value and pitfalls of assessing bone marrow morphologic findings to predict response in patients with myelofibrosis who undergo hematopoietic stem cell transplantation. Ann Diagn Pathol. 2022;56:151860.
Sirinukunwattana K, Aberdeen A, Theissen H, Sousos N, Psaila B, Mead AJ, et al. Artificial intelligence-based morphological fingerprinting of megakaryocytes: a new tool for assessing disease in MPN patients. Blood Adv. 2020;4:3284–94.
Ryou H, Sirinukunwattana K, Aberdeen A, Grindstaff G, Stolz BJ, Byrne H, et al. Continuous Indexing of Fibrosis (CIF): improving the assessment and classification of MPN patients. Leukemia. 2023;37:348–58.
Funding
Supported by AIRC 5 × 1000 call “Metastatic disease: the key unmet need in oncology” to MYNERVA project, #21267 (MYeloid Research Venture AIRC); by Ricerca Corrente IRCCS Policlinico San Matteo Foundation, Pavia, Italy, project number 874, code number 08054517, received by Vittorio Rosti (www.sanmatteo.org).
Author information
Authors and Affiliations
Contributions
GB wrote the first version of the typescript. VR and RPG revised and discussed the typescript. All authors agreed to the published version of the paper.
Corresponding author
Ethics declarations
Competing interests
RPG is a consultant to BeiGene Ltd., Fusion Pharma LLC, LaJolla NanoMedical Inc., Mingsight Parmaceuticals Inc., Kite Pharma and CStone Pharmaceuticals; Advisor to Antegene Biotech LLC, Medical Director and FFF Enterprises Inc.; Partner in AZACA Inc.; Board of Directors, RakFond Foundation for Cancer Research Support; Scientific Advisory Board, StemRad Ltd.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barosi, G., Rosti, V. & Gale, R.P. Myelofibrosis-type megakaryocyte dysplasia (MTMD) as a distinct category of BCR::ABL-negative myeloproliferative neoplasms. Challenges and perspectives. Leukemia 37, 725–727 (2023). https://doi.org/10.1038/s41375-023-01861-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-023-01861-9
