
Abstract
The diagnosis of Waldenström’s macroglobulinemia (WM), an IgM-associated lymphoplasmacytic lymphoma, can be challenging due to the different forms of disease presentation. Furthermore, in recent years, WM has witnessed remarkable progress on the diagnostic front, as well as a deeper understanding of the disease biology, which has affected clinical practice. This, together with the increasing variety of tools and techniques available, makes it necessary to have a practical guidance for clinicians to perform the initial evaluation of patients with WM. In this paper, we present the consensus recommendations and laboratory requirements for the diagnosis of WM developed by the European Consortium of Waldenström’s Macroglobulinemia (ECWM), for both clinical practice as well as the research/academical setting. We provide the procedures for multiparametric flow cytometry, fluorescence in situ hybridization and molecular tests, and with this offer guidance for a standardized diagnostic work-up and methodological workflow of patients with IgM monoclonal gammopathy of uncertain significance, asymptomatic and symptomatic WM.
Similar content being viewed by others


Introduction
The Consensus Panel Recommendations from the Second International Workshop on Waldenström’s Macroglobulinemia (WM) [1] state that the diagnosis of WM requires the following clinical and pathological criteria: presence of infiltration of clonal lymphoplasmacytoid cells documented by bone marrow (BM) biopsy (lymphoplasmacytic lymphoma (LPL)) and presence of monoclonal IgM in the serum, irrespective of the M-protein size. The 2016 WHO classification defines lgM monoclonal gammopathy of undetermined significance (IgM-MGUS) by the presence of a serum lgM paraprotein below 30 g/l, BM lymphoplasmacytic infiltration <10%, and no evidence of end-organ damage related to the underlying lymphoproliferative disorder [2]. Recent updates of this classification have not changed this view [3, 4], although they have stressed the importance of the mutational landscape in WM. Therefore, a BM biopsy remains mandatory for the differential diagnosis between WM, IgM-MGUS and other B-cell lymphoproliferative disorders (B-LPDs) (Table 1) [5, 6]. In addition, although not yet recognized by the WHO classification, there are some patients with clinical features attributable to IgM monoclonal protein but no evidence of lymphoma (IgM-related disorders) who should also be considered for BM evaluation to rule out a WM [1].
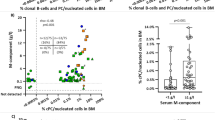
Multiparametric flow cytometry (MFC) and molecular techniques may help to confirm the diagnosis, especially to discriminate WM from other IgM-secreting disorders. MFC analysis has been shown to accurately quantify the number of clonal cells, although it may underestimate the amount of marrow infiltration compared to the BM biopsy [7], probably due to the hemodilution effect during BM aspiration. A progressive increase in the number of light-chain-isotype-positive B-cells from IgM-MGUS to smoldering and to symptomatic WM has been demonstrated [8]. However, the pattern of antigen expression and the relative fractions of individual marker expressing clonal B-cells remain stable during disease progression [9].
Main biological characteristics in WM
Important advances in understanding the biology of WM have been made in recent years, leading to an increased toolset for differential diagnosis. Using whole genome sequencing, Treon et al. [10] identified MYD88L265P as a highly recurrent (~95%) somatic mutation in patients with WM. Several studies using different techniques, such as Sanger sequencing, and allele-specific quantitative PCR (ASqPCR) [11,12,13,14], confirmed that MYD88L265P is present in >90% of WM, whereas it is absent in patients with multiple myeloma (MM) (including IgM isotype) [15], and less frequently found in marginal zone lymphoma (MZL) with plasmacytic differentiation or chronic lymphocytic leukemia (CLL) [16, 17]. Therefore, MYD88L265P assessment is considered crucial to discriminate between WM and other B-LPDs with overlapping clinical features. Mutations in the CXCR4 gene were identified as the second most common alterations in WM (30–40% patients) [18, 19], and play an important role in WM pathogenesis and disease progression [20, 21]. These mutations might also impact the clinical presentation and outcome of WM patients. Thus, MYD88L265P/CXCR4WHIM patients may present with a more aggressive clinical behavior, and inferior response to Bruton Tyrosine Kinase inhibitors (BTKi) [22]. On the other hand, higher risk of transformation to aggressive lymphoma and shorter overall survival were reported in MYD88 wild-type cases [23].
WM presents with a median of 2–3 chromosomal abnormalities per patient [24]. Deletion of 6q (−6q or del6q) is the most frequent chromosomal abnormality (40–50% of patients) [25] and it is directly related with progression from asymptomatic to symptomatic WM [26]. Deletion of 17p/TP53 is present in 8–15% of WM patients, and TP53 mutations are present in a small subset of patients with poor prognosis [27,28,29,30].
Asymptomatic patients with IgM monoclonal component below 1.5 g/dl (or 15 g/l) and normal serum free light-chain ratio have a very low-risk of progression to overt WM or other lymphoproliferative malignancies, and BM biopsy is not generally recommended at this stage, outside the context of clinical trials, and in the absence of any potential IgM-related symptom [31, 32]. However, the cut-off point of 1.5 g/dl could be misleading since in WM, there is no concordance between BM infiltration, IgM levels and patient symptoms. Thus, patients with predominant lymphocytic infiltration and poor plasmacytic differentiation may have low serum IgM levels [8, 33] and could be incorrectly classified as MGUS without a BM evaluation [34]. Consequently, although the value of BM assessment in asymptomatic individuals is not fully established, most groups currently agree that it may provide prognostic information about the risk of progression and the indication of the BM biopsy should be discussed [6].
Since WM is a rare disease and procedures may vary across different laboratories, we aim to provide consensus recommendations of the European Consortium of Waldenström’s Macroglobulinemia (ECWM) on diagnostics in this lymphoma subtype [6, 35]. We will discuss the basic and essential procedures that must be performed by local centers for the diagnosis and initial evaluation of WM patients, as well as more complex techniques that should be considered for precise pre-treatment evaluation, disease monitoring and research studies to be carried out in referral centers. In addition, detailed procedures for MFC, fluorescence in situ hybridization (FISH) and molecular tests will be provided.
ECWM-supported recommendations were made based on an international consensus reached through a Delphi survey, with two rounds of open discussion and a virtual consensus meeting; the ECWM is composed of hematologists, pathologists, and biologists/researchers in the field of WM, and all authors participated in the process. A first draft was prepared by the first, senior and corresponding authors following the usual procedures and comments made at the ECWM meetings; the initial versions were distributed to all authors with two rounds of open discussions and comments. Once the main draft was agreed, the most debated points were selected to develop 14 recommendations (10 for diagnostic purposes and 4 for helping in research), and a Delphi survey was launched among the authors. The Delphi score range from 1, completely disagree, to 9, completely agree. Nine questions were approved in a first survey round, based on a 75% agreement of 8–9, or a 90% agreement of 6–9. The five remaining recommendations were re-written considering the opinion of the dissenting authors. A second round was sufficient to reach the final consensus presented in the following paragraphs. There was an initial unanimity on 8 of the questions, and on the final 14 recommendations. The consensus recommendations represent the views of the panel and are potentially applicable to both clinical practice and biologic studies in the context of clinical trials. Future evidence might lead to updates in this guidance, which is now intended to provide a robust framework to support clinicians and avoid discrepancies in the diagnosis of WM.
Essential diagnostic requirements for WM
Although WM can present as an asymptomatic entity, most patients initially consult due to B-symptoms, such as fevers, night sweats or unintentional weight loss. Other common symptoms include fatigue, malaise, and shortness of breath, usually due to anemia, and increased bleeding or bruising that can be associated with thrombocytopenia or acquired von Willebrand disease [5]. Finally, the third group of frequent symptoms are associated with hyperviscosity, including epistaxis, headache, blurred vision, vertigo, and tinnitus. Other symptoms can be present, but a comprehensive review exceeds the intent of the present working consensus, which focuses on the laboratory steps that should follow the identification of the IgM monoclonal protein and/or the initial symptoms mentioned above.
Essential laboratory analyses in WM: general recommendations
The quality and quantity of the material required for WM diagnosis are critical. Currently, both BM and peripheral blood (PB) samples are helpful, while other tissues (lymph node, pleural effusion, cerebrospinal fluid (CSF)) may be useful to further characterize the disease. However, WM diagnostic criteria still require an histological evaluation of the BM biopsy for the final diagnosis [5]. Newer, patient-friendly molecular tools applicable to PB samples might be preferred over classical BM biopsy, but they are not yet sufficiently evaluated and standardized to provide a definitive diagnosis.
A relatively large amount of BM is needed to perform (at least) MFC and molecular studies, and during aspiration of such a volume, there is a significant risk of hemodilution. Clots may also affect the quality of samples, especially in patients with marked hyperviscosity syndrome, cold agglutinins or cryoglobulinemia. No recommendations are yet available for optimization of marrow aspiration, although normalization is an option when using MFC [36]. Clots from the BM biopsy may provide pathological material for molecular studies.
Optimally, the timing of marrow aspiration should allow rapid processing of the sample in the local laboratory or rapid shipment to a central laboratory. The central laboratory for each local site should be assigned based primarily on geographic criteria. Samples must be stored at 4 °C when long shipping times are anticipated (>48 h), while room temperature storage can be considered when samples are delivered to the central laboratory in a short time (up to 24 h). Alternatively, samples can be collected in specialized Cell-Free DNA BCT tubes (©Streck) to ensure genomic DNA stability up to 14 days.
Sample types and processing protocols: technical aspects
Bone marrow biopsy
A BM trephine biopsy with a minimal length of 20 mm containing marrow spaces is considered adequate for the histopathological diagnosis of WM/LPL. Formalin fixation and decalcification by EDTA provide the best results for morphological, immunohistochemical and molecular examination, while alternative fixatives and acid decalcification can severely compromise antigen expression and preservation of DNA and RNA. The infiltration pattern is usually divided into 3 to 4 types, including nodular, para-trabecular, (interstitial), and diffuse. According to several reports, para-trabecular invasion pattern is one of the pathological features of WM and can be useful for differentiation from MZL [1, 35, 37]. It is also important to evaluate and exonerate the presence of other BM diseases, e.g., myelodysplastic syndrome.
In addition to standard hematoxylin and eosin and/or Giemsa stains and a reticulin stain for the assessment of fibrosis, immunohistochemical stains are used for the characterization and quantification of the infiltrate [35]. The pan-B-cell marker CD20 (alternatively CD79a, which also stains the plasma cell (PC) component, or PAX5) and a minimal antibody panel containing IgM and immunoglobulin light chains should be used to demonstrate light-chain restriction [5, 6]. Markers such as CD38 (which also stains lymphocytes) and CD138 can be used to evaluate the degree of plasmacytic differentiation. Additional markers to exclude other B-cell lymphomas and MM should be included as deemed necessary depending on the availability of flow cytometric phenotyping. Although WM has a non-specific immunophenotype, CD5, CD23, CD10, cyclin D1, LEF1, and CD56 staining is helpful in excluding most differential diagnoses. However, discrimination from splenic MZL may be difficult. Presence of plasmacytoid differentiation, monoclonal PC, and increased mast cells are more suggestive of WM than MZL [37]. EDTA-decalcified BM trephines may serve as an excellent alternative source for detection of MYD88 and CXCR4 mutations by ASqPCR or sequencing [38]. In those patients with symptoms related with cardiac, renal or neurological dysfunctions, a specific search for amyloid deposits should be performed [39].
Bone marrow aspiration
During BM aspiration, representative samples should be collected for a correct initial cytomorphological evaluation of lymphoplasmacytic and PC. For standard baseline analyses, samples should be collected in at least three EDTA tubes for MFC and molecular analyses, and one sodium heparin tube for FISH purposes. The median percentage of tumor cells in BM samples of MGUS, asymptomatic and symptomatic WM is 2.2%, 8.7%, and 12.2%, respectively [8]. For these numbers, FISH studies are below the sensitivity threshold. Therefore, enrichment of BM samples for CD19+ cells by immunomagnetic approaches would be advisable [26]. This option cannot be mandatory for local centers but should be for referral centers and requires the third additional EDTA tube. Adequate cellularity of the sample is also key to perform reliable BM analyses. Therefore, the collection of 12–20 ml divided into 4 tubes (3 EDTA and 1 sodium heparin) is recommended since a standard procedure requires >3 × 107 cells.
Peripheral blood
When BM samples are difficult to obtain, in rare cases of leukemic WM, PB samples can be used as an alternative for diagnostic procedures. Whole blood cells can be used for MFC analysis and genomic studies. EDTA samples are preferred (2 tubes of 10 ml), as they allow for CD19+ cell enrichment, improving throughput and accuracy [40].
In case PB samples are used for circulating tumor DNA (ctDNA) analysis, it is important to preserve the integrity of the circulating nucleic acid; consequently, PB EDTA samples must arrive at the laboratory within 4 h after the extraction. Alternatively, ctDNA can be collected in ©Streck tubes (preferably, 2 tubes of 10 ml), specifically designed for shipment to central laboratories (Supplementary Information, Appendices A, B and D).
Cerebrospinal fluid
For cases of suspected Bing Neel Syndrome, CSF analysis should include assessment of cytology, flow cytometry, MYD88 testing, and immunoglobulin gene rearrangement analysis, along with routine biochemistry and leukocyte cell count by MFC [41,42,43]. Simultaneous MYD88 testing in PB or flow cytometric red cell quantification must be performed to identify a possible contamination of CSF with PB during lumbar puncture. This analysis should be carried out by experienced laboratories, so centralization is recommended. CXCR4 mutations have not been identified in CSF, probably due to the relatively small number of cells, which makes it difficult to obtain sufficient DNA for sequencing. Digital PCR (dPCR) for MYD88L265P detection in ctDNA from CSF is a highly sensitive method [44, 45], but requires further investigation before it can be implemented in routine clinical practice. Details on sample collection, storage and shipment are available in Appendices A and B (Supplementary Information).
Multiparametric flow cytometry protocols
Approximately 4 ml of EDTA-anticoagulated BM-aspirated sample is needed to perform an immunophenotypic analysis using an 8–12 color direct immunofluorescence stain and a lysis technique, with different combinations of monoclonal antibodies: e.g., Pacific Blue [PacB]/Pacific Orange [PacO]/fluorescein isothiocyanate [FITC]/phycoerythrin [PE]/peridinin-chlorophyll protein-cyanin 5.5 [PerCP-Cy5.5]/PE-cyanin 7 [PE-Cy7]/allophycocyanin [APC]/APCH7 (Table 2) [46].
Pre-analytical procedures are important in the evaluation of suspected WM, as the quality of the BM aspirate affects the MFC results; therefore, some authors suggest using the first aliquot of the BM sample (i.e., the “first draw”) for MFC analysis to reduce the hemodilution. Precise evaluation of the BM aspirate by MFC is necessary to determine the quality of the sample, particularly in cases with low disease burden [36]. Some approaches to normalize the sample against hemodilution can also be used [47].
The characteristic immunophenotypic features of WM clonal B-cells are intracytoplasmic and surface light-chain restriction, as well as surface expression of pan-B-cell antigens (CD19, CD20), together with CD22+dim, CD25+, CD27+ and IgM+; other antigens such as FMC7, BCL2, PAX5, CD81 and CD79b are usually positive as well, while CD10, CD11c, CD103 and CD23 are mostly absent. CD5 is expressed in 5–20% of cases [48]. CD27 and CD200 frequently show heterogeneous bimodal patterns of expression. CD305 (LAIR1) is particularly useful to detect light-chain restricted clonal B-cells due to its homogenous lack of expression in 69% of WM cases, which contrasts to the bimodal heterogeneous staining in normal B-cells [9]. WM and MZL can have an overlapping phenotypic profile, although WM usually has homogeneous expression of CD25 and weak expression of CD22 (~90% of cases), whereas MZL is usually CD22+ + CD25− (~80%) [46]. In addition, CD27 expression is usually higher in MZL. This pattern together with the histological characteristics (i.e., pattern of BM infiltration, dendritic meshwork, sinusoidal localization, mast cell presence), clinical characteristics and molecular results can help to differentiate between the two entities [37].
Among total BM nucleated cells, PC percentages are not very different among IgM-MGUS and WM patients; by contrast, a progressively higher percentage of light-chain-isotype PC is noted from IgM-MGUS to smoldering and to symptomatic WM [8]. PC are also clonally restricted, and express CD38, CD138, variable CD45, CD79A, and low levels of CD19 and CD20. They consistently lack CD56 expression, which together with CD19 expression, can be reliably used to differentiate clonal PC in WM from the clonal PC infiltrate observed in MM (usually positive for CD56 and negative for CD45 and CD19). Furthermore, the PC count can also be helpful in discriminating between WM and MZL, because the presence of clonal PC is common in WM and rare in MZL [9, 37]. According to recent findings, a correct assessment of clonal PC in WM might be important, both for the correlation with the amount of IgM paraprotein at clinical presentation, and for the potential role as predictive biomarkers of treatment response, although this should be validated in larger and prospective studies [33, 49].
For screening purposes, BM samples from patients with an IgM monoclonal gammopathy should preferably be processed following the general recommendations of the EuroFlow group (Supplementary Information, Appendix C) [36].
Most of these studies can be performed in local laboratories but quick shipment and centralization are recommended if a correct procedure cannot be warranted.
Genetic analysis
For a complete mutational analysis in WM, it is preferable to send the samples to a reference laboratory to ensure adequate sensitivity and reproducibility. However, it is also recommended that non-referral laboratories can perform MYD88 mutation screening on BM samples for diagnostic purposes. PCR methods following the operating procedures described in the Supplementary Information are recommended, and a detection limit of at least 1% is mandatory.
Non-L265P MYD88 mutations have also been identified in patients with WM, including S219C, M232T, and S243N [18]. Next generation sequencing (NGS) and Sanger sequencing of BM samples can be used to detect the MYD88 mutations outside the L265P site, but NGS is not widespread yet and Sanger sequencing usually does not have optimal sensitivity, especially for samples not enriched in CD19+ cells [50]. In addition, the application of these tools can be limited by their turn-around time, cost, quality of the sample, and BM infiltration. PCR analysis (ASqPCR [11, 12, 51], dPCR [44]) is preferred over sequencing techniques for MYD88L265P detection because of its higher sensitivity and faster turn-around time with lower costs.
Additional requirements for reference laboratories
Cytogenetics and FISH analysis
The role of conventional cytogenetics in WM is not well defined; therefore, standard karyotyping is not recommended for these patients although it may help in the differential diagnosis, especially in cases without the MYD88L265P mutation. FISH studies in CD19+ cells are well feasible. It is recommended to perform analysis of del6q and del17p at least in the central labs. Alternative methods, such as SNP arrays [52] or whole genome sequencing [18] in samples with CD19+ enrichment may also be used, but cannot yet be considered for daily laboratory practice.
Mutational studies
MYD88L265P detection
The main requirement for central laboratories is to be able to analyze MYD88L265P by molecular techniques with a detection limit of at least 1 × 10−3. The accepted techniques in terms of reproducibility and sensitivity include ASqPCR and dPCR on unselected BM samples, as well as Sanger or NGS on selected BM CD19+ cells [11,12,13,14, 44, 53]. Alternatively, BM trephines may be used for mutational screening, particularly in samples with high percentage of infiltrating tumor cells [54], being especially useful when the analysis has not been performed on fresh BM aspirate samples.
For patients that are negative for MYD88L265P by ASqPCR, complete gene sequencing should be performed looking for non-L265P mutations [18]. For these purposes, only Sanger or NGS in samples enriched for CD19+ cells can provide reliable results. When the MYD88 gene is in a full germline configuration, another diagnosis should be considered, from MZL to IgM-MM. This is also relevant when mutational analysis is used for therapeutical decision-making, as BTKi work in patients with (rare) non-L265P mutations, as well as in cold agglutinin disease, which is non-L265P mutated [55, 56].
CXCR4WHIM detection
Central laboratories are also required to provide the option to detect CXCR4 mutations. Originally, this analysis was not recommended at initial diagnosis for all WM patients, but its importance is increasing rapidly, due to the widespread use of BTKi in clinical practice. Therefore, most authors suggest performing a CXCR4 mutational screening before BTKi treatment or in case of poor response or progression, and, if possible, at initial diagnosis as well.
Although many laboratories routinely investigate only the most common variant of CXCR4, CXCR4S338X, present in nearly half of cases [20], it is important to note that there are >40 different mutations. Consequently, mutational analyses of CXCR4 should be performed by Sanger sequencing or NGS on CD19+ enriched BM samples. In addition and in contrast to MYD88L265P mutation, CXCR4 mutations are frequently sub-clonal [18, 57].
Other targets: investigational molecular tools
An attractive new tool for molecular studies in WM is the so called “liquid biopsy”, that is, the detection of ctDNA in plasma or other biological fluids. It is a less invasive, patient-friendly test that could provide a good diagnostic yield, even comparable to BM, and might allow serial mutational studies without the need for repeated BM aspirates [44, 53]. In addition, ctDNA analysis can be representative of extra-medullary disease and of the whole marrow compartment, making it a potentially cost-effective approach that avoids BM aspiration sampling bias.
Detection of MYD88 and, more recently, CXCR4 somatic mutations in ctDNA from PB of WM patients is an area under development, with initial promising results, showing a high concordance with tumor burden [53]. Recently, the introduction of dPCR has shown several practical advantages over qPCR, being particularly useful for ctDNA minimal residual disease studies [58, 59].
The newly introduced Competitive Allele-Specific TaqMan® PCR (Cast-PCR) technology is highly specific, sensitive and can detect small amounts of mutated DNA in samples with large amounts of wild-type DNA. It has already been tested to detect the MYD88L265P mutation in both tumor-derived DNA and ctDNA, showing a sensitivity of 10−3, with the possibility of using very low amounts of DNA (as low as 20 pg) [60].
However, all these techniques need to be standardized and implemented in prospective studies before they can be used in clinical practice, and the current recommendation to perform BM aspiration for MFC and molecular analyses would probably be maintained.
The analysis of TP53 mutations in CD19+ sorted cells is also being investigated, as is being done in CLL by Sanger sequencing or TP53-specific NGS approaches. The analysis of other mutations (Table 3) is also a possibility that could be considered by the physician.
In conclusion, accepted samples for mutation detection and preferred techniques can be summarized as follows:
-
Unsorted BM (either white blood cells or mononuclear cells) might be analyzed by ASqPCR, although dPCR is preferred.
-
In case of BM biopsy, EDTA-decalcified, paraffin-embedded BM trephines or paraffin-embedded BM clots (non-decalcified) can be used for ASqPCR/dPCR or NGS (except in cases of minimal BM infiltration).
-
PB is suboptimal for mutational analysis especially by ASqPCR; therefore, PB samples should be analyzed by dPCR to increase sensitivity.
-
For IgM-MGUS and follow up samples with low tumor burden, dPCR is preferable to ASqPCR.
-
CD19+ sorted BM cells can be analyzed by Sanger sequencing and NGS. Although CD19+ selection is not mandatory, it helps to increase the sensitivity of the assays and is recommended when available.
-
Plasma (ctDNA) should be analyzed by dPCR. For plasma selection, PB must be collected in EDTA tubes, if processed within 4 h from drawing, or in ©Streck tubes, if processed after 4 h from drawing (Appendix A, Supplementary Information). This method is still under research.
-
In case Bing Neel syndrome is suspected, MYD88L265P analysis on CSF should be carried out by highly sensitive methods.
Operative procedures for mutational screening can be found in the Supplementary Information.
Summary of recommendations for WM laboratory diagnosis
The work-up of suspected newly diagnosed WM patients should include pathological, MFC, and molecular studies guided by the recommendations shown in Table 4. All centers should warrant the studies shown in this table, locally or by shipment to referral centers.
References
Owen RG, Treon SP, Al-Katib A, Fonseca R, Greipp PR, McMaster ML, et al. Clinicopathological definition of Waldenström’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30:110–5.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90.
Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IB, de O, et al. The 5th edition of the World Health Organization Classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36:1720–48.
Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The International Consensus Classification of mature lymphoid neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140:1229–53.
Castillo JJ, Garcia-Sanz R, Hatjiharissi E, Kyle RA, Leleu X, McMaster M, et al. Recommendations for the diagnosis and initial evaluation of patients with Waldenström Macroglobulinaemia: a Task Force from the 8th International Workshop on Waldenström Macroglobulinaemia. Br J Haematol. 2016;175:77–86.
Pratt G, El-Sharkawi D, Kothari J, D’Sa S, Auer R, McCarthy H, et al. Guidelines on the diagnosis and management of Waldenström macroglobulinaemia—a British Society for Haematology guideline. Br J Haematol. 2022;197:171–87.
Morice WG, Chen D, Kurtin PJ, Hanson CA, McPhail ED. Novel immunophenotypic features of marrow lymphoplasmacytic lymphoma and correlation with Waldenström’s macroglobulinemia. Mod Pathol. 2009;22:807–16.
Paiva B, Montes MC, García-Sanz R, Ocio EM, Alonso J, de las Heras N, et al. Multiparameter flow cytometry for the identification of the Waldenström’s clone in IgM-MGUS and Waldenström’s Macroglobulinemia: new criteria for differential diagnosis and risk stratification. Leukemia. 2014;28:166–73.
García-Sanz R, Jimenez C, Puig N, Paiva B, Gutierrez NC, Rodríguez-Otero P, et al. Origin of Waldenstrom’s macroglobulinaemia. Best Pr Res Clin Haematol. 2016;29:136–47.
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367:826–33.
Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y, Liu X, et al. MYD88 L265P in Waldenstrom’s macroglobulinemia, IgM monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific PCR. Blood. 2013;121:2051–8.
Varettoni M, Arcaini L, Zibellini S, Boveri E, Rattotti S, Riboni R, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenström’s macroglobulinemia and related lymphoid neoplasms. Blood. 2013;88:2522–8.
Jiménez C, Sebastián E, Chillón MC, Giraldo P, Mariano Hernández J, Escalante F, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, waldenström’s macroglobulinemia. Leukemia. 2013;27:1722–8.
Poulain S, Roumier C, Decambron A, Renneville A, Herbaux C, Bertrand E, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121:4504–11.
Willenbacher W, Willenbacher E, Brunner A, Manzl C. Improved accuracy of discrimination between IgM multiple myeloma and Waldenström macroglobulinaemia by testing for MYD88 L265P mutations. Br J Haematol. 2013;161:902–4.
Martínez-Trillos A, Pinyol M, Navarro A, Aymerich M, Jares P, Juan M, et al. Mutations in TLR/MYD88 pathway identify a subset of young chronic lymphocytic leukemia patients with favorable outcome. Blood. 2014;123:3790–6.
Gachard N, Parrens M, Soubeyran I, Petit B, Marfak A, Rizzo D, et al. IGHV gene features and MYD88 L265P mutation separate the three marginal zone lymphoma entities and Waldenström macroglobulinemia/lymphoplasmacytic lymphomas. Leukemia. 2013;27:183–9.
Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–46.
Jiménez C, Prieto-Conde MI, García-Álvarez M, Alcoceba M, Escalante F, Chillón MDC, et al. Unraveling the heterogeneity of IgM monoclonal gammopathies: a gene mutational and gene expression study. Ann Hematol. 2018;97:475–84.
Roccaro AM, Sacco A, Jimenez C, Maiso P, Moschetta M, Mishima Y, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123:4120–31.
Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4 S338X somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s macroglobulinemia. Leukemia. 2015;29:169–76.
Castillo JJ, Sarosiek SR, Gustine JN, Flynn CA, Leventoff CR, White TP, et al. Response and survival predictors in a cohort of 319 patients with Waldenström macroglobulinemia treated with ibrutinib monotherapy. Blood Adv. 2022;6:1015–24.
Treon SP, Gustine J, Xu L, Manning RJ, Tsakmaklis N, Demos M, et al. MYD88 wild-type Waldenstrom macroglobulinaemia: differential diagnosis, risk of histological transformation, and overall survival. Br J Haematol. 2018;180:374–80.
Braggio E, Keats JJ, Leleu X, Van Wier S, Jimenez-Zepeda VH, Valdez R, et al. Identification of copy number abnormalities and inactivating mutations in two negative regulators of nuclear factor-kB signaling pathways in Waldenström’s macroglobulinemia. Cancer Res. 2009;69:3579–88.
Schop RFJ, Kuehl WM, Van Wier SA, Ahmann GJ, Price-Troska T, Bailey RJ, et al. Waldenström macroglobulinemia neoplastic cells lack immunoglobulin heavy chain locus translocations but have frequent 6q deletions. Blood. 2002;100:2996–3001.
García-Sanz R, Dogliotti I, Zaccaria GM, Ocio EM, Rubio A, Murillo I, et al. 6q deletion in Waldenström macroglobulinaemia negatively affects time to transformation and survival. Br J Haematol. 2021;192:843–52.
Krzisch D, Guedes N, Boccon-Gibod C, Baron M, Bravetti C, Davi F, et al. Cytogenetic and molecular abnormalities in Waldenström’s macroglobulinemia patients: correlations and prognostic impact. Am J Hematol. 2021;96:1569–79.
Gustine JN, Tsakmaklis N, Demos MG, Kofides A, Chen JG, Liu X, et al. TP53 mutations are associated with mutated MYD88 and CXCR4, and confer an adverse outcome in Waldenström macroglobulinaemia. Br J Haematol. 2019;184:242–5.
Poulain S, Roumier C, Bertrand E, Renneville A, Caillault-Venet A, Doye E, et al. TP53 mutation and its prognostic significance in Waldenstrom’s macroglobulinemia. Clin Cancer Res. 2017;23:6325–36.
Varettoni M, Zibellini S, Defrancesco I, Ferretti VV, Rizzo E, Malcovati L, et al. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017;102:2077–85.
Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378:241–9.
Khwaja J, D’Sa S, Minnema MC, Kersten MJ, Wechalekar A, Vos JM. IgM monoclonal gammopathies of clinical significance: diagnosis and management. Haematologica. 2022;107:2037–50.
Puig N, Ocio EM, Jiménez C, Paiva B, Miguel JFS, García-Sanz R. Waldenström’s macroglobulinemia immunophenotype. In: Leblond V, Treon S, Dimoploulos M, editors. Waldenström’s Macroglobulinemia. Springer, Cham; 2017. p. 21–34.
Varettoni M, Arcaini L, Rattotti S, Ferretti V, Cazzola M. Bone marrow assessment in asymptomatic immunoglobulin M monoclonal gammopathies. Br J Haematol. 2015;168:301–2.
Maqbool MG, Tam CS, Morison IM, Simpson D, Mollee P, Schneider H, et al. A practical guide to laboratory investigations at diagnosis and follow up in Waldenström macroglobulinaemia: recommendations from the Medical and Scientific Advisory Group, Myeloma Australia, the Pathology Sub-committee of the Lymphoma and Related Disease. Pathology. 2020;52:167–78.
Kalina T, Flores-Montero J, Van Der Velden VHJ, Martin-Ayuso M, Böttcher S, Ritgen M, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. 2012;26:1986–2010.
Amaador K, Vos JMI, Pals ST, Kraan W, Dobber JA, Minnema MC, et al. Discriminating between Waldenström macroglobulinemia and marginal zone lymphoma using logistic LASSO regression. Leuk Lymphoma. 2022;63:1070–9.
Schmidt J, Federmann B, Schindler N, Steinhilber J, Bonzheim I, Fend F, et al. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol. 2015;169:795–803.
Zanwar S, Abeykoon JP, Ansell SM, Gertz MA, Dispenzieri A, Muchtar E, et al. Primary systemic amyloidosis in patients with Waldenström macroglobulinemia. Leukemia. 2019;33:790–4.
Xu L, Hunter ZR, Yang G, Cao Y, Liu X, Manning R, et al. Detection of MYD88 L265P in peripheral blood of patients with Waldenström’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Leukemia. 2014;28:1698–704.
Minnema MC, Kimby E, D’Sa S, Fornecker LM, Poulain S, Snijders TJ, et al. Guideline for the diagnosis, treatment and response criteria for Bing-Neel syndrome. Haematologica. 2017;102:43–51.
Poulain S, Boyle EM, Roumier C, Demarquette H, Wemeau M, Geffroy S, et al. MYD88 L265P mutation contributes to the diagnosis of Bing Neel syndrome. Br J Haematol. 2014;167:506–13.
Muñiz C, Martín-Martín L, López A, Sánchez-González B, Salar A, Almeida J, et al. Contribution of cerebrospinal fluid sCD19 levels to the detection of CNS lymphoma and its impact on disease outcome. Blood. 2014;123:1864–9.
Drandi D, Genuardi E, Dogliotti I, Ferrante M, Jiménez C, Guerrini F, et al. Highly sensitive MYD88L265P mutation detection by droplet digital polymerase chain reaction in Waldenström macroglobulinemia. Haematologica. 2018;103:1029–37.
Hiemcke-Jiwa LS, Minnema MC, Radersma-van Loon JH, Jiwa NM, de Boer M, Leguit RJ, et al. The use of droplet digital PCR in liquid biopsies: a highly sensitive technique for MYD88 p.(L265P) detection in cerebrospinal fluid. Hematol Oncol. 2018;36:429–35.
Paiva B, Corchete LA, Vidriales M-B, Garcia-Sanz R, Perez JJ, Aires-Mejia I, et al. The cellular origin and malignant transformation of Waldenstrom macroglobulinemia. Blood. 2015;125:2370–80.
Loken MR, Chu SC, Fritschle W, Kalnoski M, Wells DA. Normalization of bone marrow aspirates for hemodilution in flow cytometric analyses. Cytom Part B Clin Cytom. 2009;76:27–36.
D’Angelo G, Hotz AM, Monti M. Lymphoplasmacytic non-Hodgkin lymphoma/Waldenström’s macroglobulinemia with CD5+, CD23+, and CD10-. Blood Res. 2013;48:300–3.
El-Ayoubi A, Wang JQ, Hein N, Talaulikar D. Role of plasma cells in Waldenström macroglobulinaemia. Pathology. 2017;49:337–45.
Wang CZ, Lin J, Qian J, Shao R, Xue D, Qian W, et al. Development of high-resolution melting analysis for the detection of the MYD88 L265P mutation. Clin Biochem. 2013;46:385–7.
Jiménez C, Chillón MC, Balanzategui A, Puig N, Sebastián E, Alcoceba M, et al. Detection of MYD88 L265P mutation by real-time allele-specific oligonucleotide polymerase chain reaction. Appl Immunohistochem Mol Morphol. 2014;22:768–73.
Poulain S, Roumier C, Galiègue-Zouitina S, Daudignon A, Herbaux C, Aiijou R, et al. Genome wide SNP array identified multiple mechanisms of genetic changes in Waldenstrom macroglobulinemia. Am J Hematol. 2013;88:948–54.
Bagratuni T, Ntanasis-Stathopoulos I, Gavriatopoulou M, Mavrianou-Koutsoukou N, Liacos C, Patseas D, et al. Detection of MYD88 and CXCR4 mutations in cell-free DNA of patients with IgM monoclonal gammopathies. Leukemia. 2018;32:2617–25.
Willenbacher E, Willenbacher W, Wolf DG, Zelger B, Peschel I, Manzl C, et al. Digital PCR in bone marrow trephine biopsies is highly sensitive for MYD88 L265P detection in lymphomas with plasmacytic/plasmacytoid differentiation. Br J Haematol. 2019;186:189–91.
Treon SP, Xu L, Hunter Z. MYD88 mutations and response to ibrutinib in Waldenström’s macroglobulinemia. N Engl J Med. 2015;373:584–6.
Malecka A, Trøen G, Tierens A, Østlie I, Malecki J, Randen U, et al. Immunoglobulin heavy and light chain gene features are correlated with primary cold agglutinin disease onset and activity. Haematologica. 2016;101:e361–4.
Poulain S, Roumier C, Venet-Caillault A, Figeac M, Herbaux C, Marot G, et al. Genomic landscape of CXCR4 mutations in Waldenström macroglobulinemia. Clin Cancer Res. 2016;22:1480–8.
Dogliotti I, Drandi D, Genuardi E, Ferrero S. New molecular technologies for minimal residual disease evaluation in B-cell lymphoid malignancies. J Clin Med. 2018;7:288.
Ferrante M, Furlan D, Zibellini S, Borriero M, Candido C, Sahnane N, et al. MYD88 L265P detection in IgM monoclonal gammopathies: methodological considerations for routine implementation. Diagnostics. 2021;11:779.
Bagratuni T, Markou A, Patseas D, Mavrianou-Koutsoukou N, Aktypi F, Liacos CI, et al. Determination of MYD88L265P mutation fraction in IgM monoclonal gammopathies. Blood Adv. 2022;6:189–99.
Acknowledgements
This study was supported by the European Consortium for Waldenström’s Macroglobulinemia.
Author information
Authors and Affiliations
Contributions
ID, CJ, SF, and RG-S were responsible for designing, writing, and editing the manuscript. RG-S designed the survey, analyzed the data, and interpreted the results. ID, CJ, MV, DT, TB, MF, JP, DD, NP, MG, MG-A, RO, WJ, AT, VL, EK, MJK, SDS, MK, WW, AMR, SP, PM, CK, FF, JV, MAD, CB, SF, and RG-S completed the survey and provided feedback on the report. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
ID, CJ, MV, TB, MF, JP, DD, NP, MG, MG-A, RO, WJ, AT, VL, SP, CK, and FF declared no conflicts of interests related to this publication. DT has received honoraria from Amgen, Novartis, Roche, BeiGene, Janssen, Antengene, CSL and EUSA; has participated in advisory boards for Roche, Janssen, Antengene, CSL, BeiGene and EUSA; received research funding from Roche and Janssen. EK has received honoraria from Amgen, Genesis Pharma, Janssen, Takeda, GSK, Pfizer; has participated in advisory boards for Janssen, GSK; received research funding from Amgen, Janssen, and received travel and accommodations reimbursement from Janssen, GSK and Sanofi. MJK has received honoraria from Celgene/BMS, Roche, Kite/Gilead, Novartis, Miltenyi Biotec and Adicet Bio (all to institution) and research support from Kite/Gilead (to institution). SDS received honoraria from BeiGene, Janssen, and Sanofi; was a consultant/advisor for Janssen, BeiGene and Sanofi; received research funding from Janssen and received travel and accommodations reimbursement from Janssen, BeiGene and Sanofi. MK has received honoraria from Eusa Pharma, Janssen, Novartis, Roche, and Takeda; has participated in advisory boards for Eusa Pharma, Novartis, and Roche; received travel and accommodations reimbursement from Takeda. WW: Steering & Safety Committees AMGEN, Celgene, DSMM, Morphosys; Employee syndena (20%); Advisory Boards AMGEN, BMS—Celgene, EUSA Pharma, Gilead, GSK, Incyte, Janssen, Kite & Consultancies Novartis, Morphosys, Merck, Pfizer, Roche, Sandoz, Sanofi, Takeda; Lectures AMGEN, Abbvie, BMS—Celgene, EUSA Pharma, Fujimoto, Gilead, GSK, Incyte, Janssen, Myelom-und Lymphomselbsthilfe Österreich, Novartis, Pfizer, Roche, Sandoz, Sanofi, Takeda; Research Funding AMGEN, BMS, Celgene, Janssen, Novartis, Roche, Sanofi, Takeda oncotyrol; European Commission (FP7—OPTATIO) Bundesland Tirol Programm: “Translational research”. AMR: Research funding from AstraZeneca, European Hematology Association, Transcan2-ERANET/FRRB, Italian Association for Cancer Research (Fondazione AIRC). Honoraria from: Amgen, Celgene, Takeda, Janssen. PM has received honoraria from BeiGene, AstraZeneca and Janssen; was consultant/advisor for BeiGene, and Janssen. JMIV: travel and accommodations reimbursement from Celgene, has participated in an advisory board and as a consultant for Sanofi, and received research support (institutional) from BeiGene. MAD: honoraria from ABBVIE and Janssen; consultant/advisor for AstraZeneca, BeiGene, Janssen; travel and accommodation reimbursement from ABBVIE, AstraZeneca, BeiGene, Janssen. CB: Consultancy: Pfizer, AbbVie, Novartis, Janssen, BeiGene, Roche, Incyte, BMS, Celgene; Morphosys; Research funding: Roche, Janssen, AbbVie, Amgen, Bayer, Celltrion, Pfizer, MSD. SF: Janssen (Consultancy, Advisory board, Speakers honoraria, Research funding); EUSA Pharma (Consultancy, Advisory board, Speakers honoraria); Gilead, Morphosys (Research funding); Incyte, Clinigen (Advisory board); Servier, Gentili (Speakers honoraria). RG-S has received honoraria from Amgen, BeiGene, Janssen, and Takeda; was a consultant/advisor for Janssen; received research funding from Gilead; holds patents, royalties, or other intellectual property from BIOMED 2 Primers, and received travel and accommodations reimbursement from Janssen and Takeda. RG-S is the current president of the Spanish Society of Hematology and Hemotherapy (http://www.sehh.es).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dogliotti, I., Jiménez, C., Varettoni, M. et al. Diagnostics in Waldenström’s macroglobulinemia: a consensus statement of the European Consortium for Waldenström’s Macroglobulinemia. Leukemia 37, 388–395 (2023). https://doi.org/10.1038/s41375-022-01762-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-022-01762-3
This article is cited by
-
There’s life in the old dog yet: immunochemotherapy in Waldenström’s macroglobulinemia
Leukemia (2024)
-
Impact of the presence and number of chromosomal abnormalities on the clinical outcome in Waldenström Macroglobulinemia: a monocentric experience
Annals of Hematology (2024)
-
Diagnostic and prognostic molecular pathology of lymphoid malignancies
Virchows Archiv (2024)
-
Ibrutinib Efficacy, Safety, and Pharmacokinetics in Chinese Patients with Relapsed or Refractory Waldenström’s Macroglobulinemia: A Multicenter, Single-Arm, Phase 4 Study
Advances in Therapy (2023)
-
Immunophenotypic assessment of clonal plasma cells and B-cells in bone marrow and blood in the diagnostic classification of early stage monoclonal gammopathies: an iSTOPMM study
Blood Cancer Journal (2023)
