
Abstract
The accumulation of somatic mutations in hematopoietic stem cells during aging, leading to clonal expansion, is linked to a higher risk of cardiovascular mortality and hematologic malignancies. Clinically, clonal hematopoiesis is associated with a pro-inflammatory phenotype of hematopoietic cells and their progeny, inflammatory conditions and a poor outcome for patients with hematologic neoplasms and solid tumors. Here, we review the relevance and complications of clonal hematopoiesis for the treatment of hematologic malignancies with cell therapeutic approaches. In autologous and allogeneic hematopoietic stem cell transplantation native hematopoietic and immune effector cells of clonal origin are transferred, which may affect outcome of the procedure. In chimeric antigen receptor modified T-cell therapy, the effectiveness may be altered by preexisting somatic mutations in genetically modified effector cells or by unmodified bystander cells harboring clonal hematopoiesis. Registry studies and carefully designed prospective trials will be required to assess the relative roles of donor- and recipient-derived individual clonal events for autologous and allogeneic cell therapies and to incorporate novel insights into therapeutic strategies.
Similar content being viewed by others

Introduction
Somatic mutations accumulate in aged tissues and may contribute to malignant transformation [1,2,3,4,5,6,7,8]. Most of the work so far has focused on genomic alterations in exons, however mutations are also observed in non-coding regions [9]. Common alterations are single base exchanges and short insertions and deletion (indels) resulting from error-prone repair mechanisms. In addition to genetic changes, malignant transformation is fueled by epigenetic modifications, such as altered expression patterns of genes involved in tumorigenesis like oncogenes and tumor-suppressor genes [10,11]. Signs of aging are further the increased frequency of other mutational events including larger chromosome breaks, that accumulate throughout the life span of a tissue, like hematopoiesis [12,13,14,15,16,17].
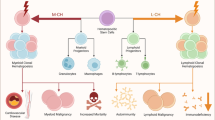
In hematopoiesis, the clonal dominance of a subset of hematopoietic stem and progenitor cells (HSPC) and their progeny, as detectable by patterns of somatic mutations, is termed clonal hematopoiesis (CH, Fig. 1). Besides aging, also smoking and germline genetic variants are associated with emerging CH and CH could even be observed in newborns [9,18,19,20]. Therefore, varying factors influence the frequency of CH in a given cohort, which is additionally influenced by the sensitivity of the applied detection method. Whereas targeted deep sequencing of specific gene regions may detect 1/1000 mutated alleles in a given sample, whole genome sequencing typically detects VAF of 2–5% [9,21,22].

CH, which describes the accumulation of somatic mutations through aging and stress over an individual’s lifetime, may result in clonal hematopoiesis of indeterminate potential, CHIP. Various pathological conditions have been associated with CH dominance. Pictograms: BioRender.com.
CH is not restricted to malignant conditions like acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) but can be also observed in ostensibly healthy persons. Indeed, the terms age-related clonal hematopoiesis and clonal hematopoiesis of indeterminate potential (CHIP) have been introduced to describe the occurrence of somatic mutations in healthy persons without hematologic abnormalities [7,23]. CHIP is defined as the manifestation of cancer-associated somatic driver-mutations with a variant allele frequency (VAF) of a least 0.02 (corresponding to roughly 4% of cells for heterozygous mutations) [23]. The prevalence of CHIP is also dependent on age, while about 20% of people aged 70 and older harbor CHIP, this affects <1% of the population below 40.
Exon alterations in CH are restricted to a well-defined set of genes. Two-thirds of clonal events associated with CHIP are dominant-negative or loss-of-function mutations in DNMT3A, TET2, and ASXL1, all of them being epigenetic regulators. Other recurring mutations observed in CHIP most frequently relate to DNA damage response (DDR) (e.g., TP53, PPM1D), growth factor signaling (e.g., JAK2, CBL) and components of the spliceosome machinery (e.g., SF3B1, U2AF1). Some of these mutations have been detected in HSPC and provide increased self-renewal, enhanced repopulating activity and reduced differentiation capacity [24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41]. Extrinsic factors, however, further influence the fate of mutated HSPC. For example, mutations in DDR genes like PPM1D confer survival advantage under repetitive cytotoxic conditions [42,43]. Other mechanisms that propagate clonal progression are mostly unknown, but first functional studies in preclinical models suggest an increased activity of inflammatory circuits being involved in CHIP progression [44].
The transition of somatic mutations from HSPC to all mature lineages may be expected. In experimental models and in human samples with high allele frequency (typically ≥ 0.02 VAF), mutational burden was instead highest in cells of the myeloid lineage, indicative for a preferential myeloid differentiation, termed myeloid skewing/bias [29,36,38,45,46,47,48]. By using more sensitive assays it was observed that in samples with low allele frequency (<0.02 VAF) the mutational burden was equally distributed between different lineages [21]. As the mutational spectrum differs between high and low mutational burden samples, myeloid skewing probably does not represent a phenomenon of CH in general, instead it could reflect a feature of clones acquiring dominance at later time points, potentially due to pro-inflammatory cytokine loops [44,49]. Despite myeloid skewing and risk of transformation to myeloid neoplasms (MDS/AML; low in absolute numbers) being common, CHIP should not be regarded as a disorder restricted to the myeloid lineage but has pleiotropic consequences. First, in individuals with CHIP the incidence of lymphatic neoplasms is also elevated but the pathophysiological link to CH is less obvious. In contrast to myeloid neoplasms, these lymphomas do not necessarily arise out of mutated HSPC by stepwise acquisition of additional mutations [50,51,52,53,54]. Second, CH enhances the risk of inflammatory diseases like cardiovascular disease (CVD) [7,18,55,56] and adult onset autoinflammatory disease [57]. CH patients are described to be enriched in illnesses like hemophagocytic lymphohistiocytosis [58], severe COVID-19 [59], anti-neutrophil antibody-associated vasculitis [60], to name a few examples of the expanding list. Third, cytostatic therapy, but not immune checkpoint blockade, is a major risk factor for development of CH and shapes the mutational spectrum [15,61,62,63]. Conversely, the presence of CH negatively impacts the prognosis of patients with solid cancers [62] and aggressive lymphomas [64] undergoing cytostatic therapy. Although the mechanistic link between CH and this large number of various disorders remains to be elucidated, CH-associated inflammation appears as the overarching principle. Diverse mediators of innate and adaptive immunity have been described to be modulated by the occurrence of specific CH-associated somatic mutations [65,66]. Not only cellular components of the myeloid lineage like monocytes/macrophages [56,67,68,69,70], mast cells [71], neutrophils [72], but also lymphatic cells [73,74,75,76,77] can exert altered function.
In view of the plethora of consequences of CH, all aspects of cellular therapies, from cell harvest to cell processing and product/graft function (Fig. 2 showing exemplary CAR T-cell therapy) might be affected. The purpose of this review is to (1) summarize the available information on the clinical relevance of CH in the context of both hematopoietic cell transplantation (HCT) as well as the application of genetically engineered T cells, and to (2) provide some insights into published preclinical information, which may stimulate ongoing and future translational research studies.

CH might theoretically impact CAR T-cell processing and treatment (gray text) at all stages. Already observed effects attributed to CH (black text). Pictograms: BioRender.com.
Clonal hematopoiesis and autologous hematopoietic cell transplantation
Somatic mutations associated with myeloid malignancies (≥2% VAF) can be detected in 5–30% of specimen from unselected, heavily pretreated patients undergoing autologous transplantation [77,78,79,80]. Again, the frequency of CH also depends on the sensitivity of the applied method. Even in less intensively treated patients undergoing autologous HCT, the prevalence of CH is 11–22% when lowering the VAF threshold to 0.01 and 0.02, respectively [81]. In summary, the prevalence of CH in patients undergoing autologous HCT is elevated due to previous exposure to conditions that promote CH (e.g., chemotherapy) when compared to epidemiological studies in hematological healthy persons.
Autologous grafts are predominantly obtained by chemotherapy combined with G-CSF, or G-CSF alone (steady state mobilization). In poor mobilizers, G-CSF is combined with plerixafor to increase HSC yield. There is no systematic data available in humans how these various mobilization strategies differentially influence graft composition with regard to the proportion of contained clonal cells. For example, apheresis following chemotherapy might theoretically enrich for HSC bearing mutations in DDR genes. More important, the presence of CH has been suggested to affect process-related results and to influence short- and long-term clinical outcome parameters. To this end, the presence of CH has been associated with inferior mobilization capacity [76,80,81,82,83] and delayed engraftment [84], however, inferior mobilization is not a consistent finding [79,84].
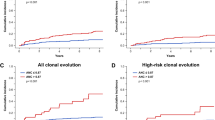
The presence of CH in the autologous graft has in a few retrospective studies been linked to an increased risk for therapy-related myeloid neoplasms (tMN) [78,80,83] and reduced overall survival (OS) [80,81], partly due to an increase in CVD [80]. Assuming that the detection of somatic mutations in the autologous graft simply reflects the presence of CH, the increase in myeloid neoplasms and CVD-associated mortality, when compared to non-CH patients, merely confirms the known risk profile. It remains unclear whether the mobilization process itself (e.g., by enrichment of CH clones) can accelerate the progression of CH-associated diseases and the increase in non-relapse mortality (NRM). Future research should investigate whether purging of CH from the graft might have positive effects on the overall outcome after autologous HCT. Longitudinal tracking of somatic mutations in patients receiving autologous HCT revealed that impressive changes (increase in VAF and a gain in new mutations) could only be observed in temporal proximity to autologous HCT. In subsequent peripheral blood (pB) samples (without continued chemotherapy), mutational burden remained mostly stable [43,84]. Therefore, hematopoietic stress might help CH to dominate wild-type hematopoiesis and might accelerate CH-associated non-hematologic diseases. However, in other studies no abrupt and only modest changes in mutational burden directly after autologous HCT was described [76,77]. Remarkably, presence of CH in the context of autologous HCT did not necessarily translate into decreased OS or excess in NRM [43,79]. In patients with MRD negative MCL after first-line therapy, four patients developed tMN. Only one of them was CH positive at the time point of transplantation [43]. Also in MM, presence of CHIP at autologous transplantation was not predictive for tMN [81]. In lymphoma patients, an increase in tMN was not associated with bulk CH, but with mutation in DDR genes [78] (Fig. 3).

The presence of CH in the graft has been associated with various findings in allogeneic, as well as autologous hematopoietic stem cell transplantation. The potential influence of different mobilization procedures on CH recovery within the graft is not depicted. Image showing blood transfusion: scraffitto at the University Hospital Dresden by Alfred Hesse, photo: T. Albrecht.
The cumulative incidence of relapse usually did not differ between CH and non-CH patients, suggesting that presence of CH did not influence the therapeutic efficacy of autologous transplantation [43,80].
In summary, the prevalence of CH at the time point of autologous transplantation mainly depends on age and pretreatment intensity. Autologous HCT, like other hematopoietic stressors, represents a risk factor for CH. The contribution of various procedures (e.g., graft, mobilization, conditioning regimen) however remains vague and the impact of CH on outcome may vary between different cohorts, likely depending on continued treatment, affected genes, and mutational burden. To date, the effectivity of autologous HCT for the underlying disease seems unaffected by the presence of CH. Therefore, the presence of CH should currently not impact on clinical decision-making in the context of autologous HCT.
Clonal hematopoiesis and allogeneic hematopoietic cell transplantation
Donor cell-derived myeloid malignancies, summarized as donor cell leukemia (DCL), include AML and MDS and occur after allogeneic HCT. It is conceivable that DCL may be promoted by transplantation of grafts that are burdened by CH (Fig. 3). In a European survey, the frequency of DCL was 0.8%. In 28% of evaluable patients, transplanted CH could be retrospectively identified [85]. Rare cases of simultaneous development of myeloid neoplasms in donor and recipient, originating from the same clone, further underscore transplantability of preleukemic clones [86].
In contrast to autologous HCT, where grafts are harvested from the aged patient, the unrelated donors in allogeneic HCT are usually younger [87]. These younger donors were less exposed to hematopoietic, CH-boosting stressors, the expected CHIP (≥0.02 VAF) prevalence is therefore <1% [6,7,8]. However transplantation and GvHD have been suggested to accelerate aging of transplanted donor-derived HSPC [88], which in turn could foster emergence of CH.
Sibling donors are often older than unrelated donors, consequently in grafts from sibling donors ≥55 years CHIP prevalence was 16% [89]. After allogeneic HCT, unexplained cytopenia has been associated with the presence of donor-engrafted CHIP and indeed the donors with CHIP were older and predominantly siblings [90]. In the most comprehensive study so far, presence of CHIP in the graft of donors ≥55 years was a risk factor for emergence of DCL. It has been shown that almost all donor-derived CHIP successfully engrafted the recipients. CHIP-transplanted patients showed an increase in the incidence of chronic graft versus host disease (cGvHD), but not in cGvHD severity and also not in the incidence of acute GvHD. Remarkably, increase in cGvHD was attributable to a significant excess of ocular GvHD, while skin and visceral cGvHD were equally distributed between both cohorts. This organ tropism is so far unexplained and leaves room for different hypotheses. In the CHIP positive cohort, a numerical, but not statistically significant rise in NRM could also be observed. For the entire population, donor CHIP was associated with a significantly reduced cumulative incidence of relapse/progression (CIR/P), but this observation was mainly attributable to patients being not in complete remission (non-CR) before allogeneic HCT. Finally, OS of the entire population was not influenced by the presence of CHIP in the donor. Again only in the subpopulation of non-CR patients, presence of CHIP in the donor was associated with preferable OS [89]. In another study with donors ≥40 years, the presence of DNMT3A mutations (but not other mutations) in the graft with ≥0.01 VAF was associated with improved progression-free survival in a large heterogenous cohort of patients. The association even remained statistically significant in a multivariable model. The effect was driven by patients who did not receive posttransplant cyclophosphamide. These patients showed a reduced relapse risk and an increase in cGvHD [91]. Further results are expected, as this study is not fully published yet.
To sum up, donor CH, specifically donor CHIP, is a risk factor for DCL (Fig. 3). Because of the low incidence of DCL, a systemic screening of the entire donor cohort will probably not substantially influence OS of the entire recipient population. In selected cases (older donors and several alternative donors available) CH-screening might help in the donor selection process. This might be of special interest in related donors, as the incidence of CHIP has been described to be elevated in relatives of patients with myeloid neoplasms [89]. First retrospective analyses suggest a potential impact of donor CHIP on relapse incidence and chronic GvHD [89], however further confirmatory analyses in independent cohorts are required.
Clonal hematopoiesis and genetically engineered autologous T cells
The vast majority of immune effector cells (IEC) for genetic modification (e.g., chimeric antigen receptor CAR T cells) are extracted from pB samples after unstimulated leukapheresis. Clinical grade IEC are furthermore subject to a variety of procedures including viral transduction and expansion of the requested population in vitro (Fig. 2). Somatic mutation associated with CHIP might directly (mutation in transduced T cells) and indirectly (mutation in bystander cells) affect IEC activity.
To date, clinical data about the interference of CH with CAR T-cell therapy (CART) is still scarce. Circumstantial evidences however suggest that CH might influence CART toxicity as well as effectivity. Treatment with CAR T cells is frequently complicated by the cytokine release syndrome (CRS). Recipient myeloid cells play a critical role in CRS development. Monocyte/macrophage-derived IL-1 and IL-6 were demonstrated to orchestrate this inflammatory process [92,93]. Presence of CH in men has been associated with various inflammatory conditions and in TET2-mutant mice monocytes/macrophages showed pro-inflammatory characteristics and substantially contributed to progression of arteriosclerotic disease by secretion of IL-1 and IL-6 [56,67]. Thus, it appears possible that patients with CH have an increased risk for inflammatory side effects of CART. Larger cohort studies investigating such associations are pending.
Patients undergoing CART have been heavily pretreated and therefore have an elevated risk for the presence of CH. Although CH frequency in CART patients has not been systematically reported, we expect a CHIP prevalence at least as high as in lymphoma patients undergoing autologous HCT. Sterile inflammation (clinically recognized as CRS) is a prototypic side-effect of CART. Under clinical aspects, CRS is very limited in time, but it cannot be excluded that this inflammatory episode is sufficient to push clonal progression and to increase the risk of tMN. Furthermore, prolonged cytopenia is a frequent observation after CART. Etiology of delayed hematopoietic recovery remains vague but it has been associated with occurrence of severe CRS [94]. In autologous HCT, presence of CH was linked to a delay in engraftment [84]. Thus, it is conceivable that presence of CH negatively affects hematological regeneration. Again, clinical data that underpin these hypotheses are missing.
Some genes frequently mutated in CH also play a pivotal role in regulation of lymphocyte activity and might influence clinical outcome parameters (toxicity, effectivity) of CAR T cells. TET-family of genes are critical for the development and function of various lymphocyte subset. Homozygous loss of function of TET-genes has been shown to promote autoinflammation [95,96] and to alter antigen-specific responses [74]. Accordingly, homozygous loss-of-function mutations of TET2 can lead to changes in activity of human T-cell subsets genetically modified to express CAR. One patient with chronic lymphocytic leukemia, 2 months after a second infusion of anti-CD19 CAR T cells with CD28 co-stimulation domain, developed an atypical, delayed expansion of monoclonal CD8+ CAR T cells with central memory phenotype, which was associated with emergence of CRS. The CAR T-cell population showed long-term persistence and the patient experienced sustained clinical response. These monoclonal CD8+ CAR T cells were characterized by compound heterozygous loss-of-function of TET2. One TET2 allele was disrupted by CAR lentiviral integration, the second allele by a E1879Q mutation, which was present also in non-CAR hematopoietic cells therefore representing CH. After CAR-specific stimulation, CAR T cells with compound heterozygous TET2-mutation showed an increased expression of various cytokines including INF-γ compared to CAR T cells with lentiviral integration sites outside TET2. Compound heterozygous TET2-mutated CAR T cells were less prone to senescence and showed prolonged activation capacity after repetitive CAR-specific stimulation [97]. This example suggests that both CAR integration site [98] and somatic mutations in the context of CH might directly and indirectly influence CAR T-cell activity and clinical outcome.
Summary and conclusion
Several observations indicate that CH might impact most cellular therapies currently established for patients with hematologic malignancies. In patients undergoing autologous HCT, it remains to be elucidated precisely how the presence of CH impacts on clinical outcomes. Manipulation of CH in the graft could represent a therapeutic approach, for example by targeted inhibition of pathways driven by somatic mutations [99]. In the context of allogeneic HCT, donor testing for CH has not been included in routine diagnostic procedures. Despite emerging evidences toward an impact of donor CH on transplant outcome, the long-term clinical consequence of the transfer of grafts with CH is so far less well-defined. Future studies will have to address whether CHIP should be integrated in donor selection algorithms. Patients with detectable CHIP after autologous or allogeneic HCT may be at an increased risk for tMN and monitoring strategies are warranted. In CART, the impact of CH on clinical outcome (toxicity and efficacy) has so far not been investigated, but first evidences for interaction are emerging. Purging of autologous CAR T-cell products and usage of allogeneic CAR T-cell products would offer therapeutic options. More importantly, harnessing the epigenetic effects of certain mutations involved in CH to improve the efficacy of CAR T cells is an exciting area of ongoing research. In summary, while detailed studies are necessary and critical, it is apparent that considering CH status of patients may further pave the way toward effective and patient-centered, personalized treatment regimes.
References
Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, Mori Y, et al. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Rep. 2018;24:1777–89.
Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–6.
Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911–7.
Blokzijl F, De Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–4.
Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, Grimsby J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364:eaaw0726.
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8.
Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–52.
Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28.
Cypris O, Božić T, Wagner W. Chicken or egg: is clonal hematopoiesis primarily caused by genetic or epigenetic aberrations? Front Genet. 2019;10:785.
Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47:1402–7.
Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44:642–50.
Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–8.
Gao T, Ptashkin R, Bolton KL, Sirenko M, Fong C, Spitzer B, et al. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nat Commun. 2021;12:338.
Terao C, Suzuki A, Momozawa Y, Akiyama M, Ishigaki K, Yamamoto K, et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature. 2020;584:130–5.
Zekavat SM, Lin SH, Bick AG, Liu A, Paruchuri K, Uddin MM, et al. Hematopoietic mosaic chromosomal alterations and risk for infection among 767,891 individuals without blood cancer. medRxiv. 2020. https://doi.org/10.1101/2020.11.12.20230821.
Wang L, Swierczek SI, Drummond J, Hickman K, Kim S, Walker K, et al. Whole-exome sequencing of polycythemia vera revealed novel driver genes and somatic mutation shared by T cells and granulocytes. Leukemia. 2014;28:935–8.
Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586:763–8.
Lin SH, Wang Y, Hartley SW, Karyadi DM, Lee OW, Zhu B, et al. In utero exposure to zidovudine-containing antiretroviral therapy and clonal hematopoiesis in HIV-exposed uninfected newborns. AIDS. 2021. https://doi.org/10.1097/QAD.0000000000002894.
Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484.
Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559:400–4.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16.
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301.
Arends CM, Galan-Sousa J, Hoyer K, Chan W, Jäger M, Yoshida K, et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia. 2018;32:1908–19.
Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 2017;170:1079–95.
Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210:2641–59.
Shide K, Kameda T, Shimoda H, Yamaji T, Abe H, Kamiunten A, et al. TET2 is essential for survival and hematopoietic stem cell homeostasis. Leukemia. 2012;26:2216–23.
Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA. 2011;108:14566–71.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24.
Koya J, Kataoka K, Sato T, Bando M, Kato Y, Tsuruta-Kishino T, et al. DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat Commun. 2016;7:10924.
Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell. 2014;15:350–64.
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31.
Izzo F, Lee SC, Poran A, Chaligne R, Gaiti F, Gross B, et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat Genet. 2020;52:378–87.
Ostrander EL, Kramer AC, Mallaney C, Celik H, Koh WK, Fairchild J, et al. Divergent effects of Dnmt3a and Tet2 mutations on hematopoietic progenitor cell fitness. Stem Cell Rep. 2020;14:551–60.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33.
Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111:2548–53.
Cole CB, Russler-Germain DA, Ketkar S, Verdoni AM, Smith AM, Bangert CV, et al. Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J Clin Invest. 2017;127:3657–74.
Jeong M, Park HJ, Celik H, Ostrander EL, Reyes JM, Guzman A, et al. Loss of Dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep. 2018;23:1–10.
Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17:584–96.
Mullally A, Poveromo L, Schneider RK, Al-Shahrour F, Lane SW, Ebert BL. Distinct roles for long-term hematopoietic stem cells and erythroid precursor cells in a murine model of Jak2V617F-mediated polycythemia vera. Blood. 2012;120:166–72.
Hsu JI, Dayaram T, Tovy A, De Braekeleer E, Jeong M, Wang F, et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell. 2018;23:700–13.
Eskelund CW, Husby S, Favero F, Klausen TW, Rodriguez-Gonzalez FG, Kolstad A, et al. Clonal hematopoiesis evolves from pretreatment clones and stabilizes after end of chemotherapy in patients with MCL. Blood. 2020;135:2000–4.
Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. 2018;23:833–49.
Buscarlet M, Provost S, Zada YF, Bourgoin V, Mollica L, Dubé MP, et al. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood. 2018;132:277–80.
Dai YJ, Wang YY, Huang JY, Xia L, Shi XD, Xu J, et al. Conditional knockin of Dnmt3a R878H initiates acute myeloid leukemia with mTOR pathway involvement. Proc Natl Acad Sci USA. 2017;114:5237–42.
Ishii T, Bruno E, Hoffman R, Xu M. Involvement of various hematopoietic-cell lineages by the JAK2 V617F mutation in polycythemia vera. Blood. 2006;108:3128–34.
Wong TN, Miller CA, Klco JM, Petti A, Demeter R, Helton NM, et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood. 2016;127:893–7.
Cai Z, Lu X, Zhang C, Nelanuthala S, Aguilera F, Hadley A, et al. Hyperglycemia cooperates with Tet2 heterozygosity to induce leukemia driven by proinflammatory cytokine-induced lncRNA Morrbid. J Clin Invest. 2021;131:e140707.
Lewis NE, Petrova-Drus K, Huet S, Epstein-Peterson ZD, Gao Q, Sigler AE, et al. Clonal hematopoiesis in angioimmunoblastic T-cell lymphoma with divergent evolution to myeloid neoplasms. Blood Adv. 2020;4:2261–71.
Tiacci E, Venanzi A, Ascani S, Marra A, Cardinali V, Martino G, et al. High-risk clonal hematopoiesis as the origin of AITL and NPM1 -mutated AML. N Engl J Med. 2018;379:981–4.
Mouly E, Ghamlouch H, Della-Valle V, Scourzic L, Quivoron C, Roos-Weil D, et al. B-cell tumor development in Tet2-deficient mice. Blood Adv. 2018;2:703–14.
Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38.
Klimkowska M, Nannya Y, Gran CE, Mansson R, Douagi I, Ogawa S, et al. Absence of a common founder mutation in patients with co-occurring myelodysplastic syndrome and plasma cell disorder. Blood. 2020;137:1260–3.
Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4:25–33.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–21.
Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020;383:2628–38.
Miller PG, Sperling AS, Gibson CJ, Viswanathan K, Castellano C, McConkey M, et al. Contribution of clonal hematopoiesis to adult-onset hemophagocytic lymphohistiocytosis. Blood. 2020;136:3051–5.
Bolton KL, Koh Y, Foote MB, Im H, Jee J, Sun CH, et al. Clonal hematopoiesis is associated with risk of severe Covid-19. medRxiv. 2020. https://doi.org/10.1101/2020.11.25.20233163.
Arends CM, Weiss M, Christen F, Eulenberg-Gustavus C, Rousselle A, Kettritz R, et al. Clonal hematopoiesis in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Haematologica. 2020;105:e264–7.
Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52:1219–26.
Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21:374–82.
Miller PG, Gibson CJ, Mehta A, Sperling AS, Frederick DT, Manos MP, et al. Fitness landscape of clonal hematopoiesis under selective pressure of immune checkpoint blockade. JCO Precis Oncol. 2020;4:1027–33.
Amini RM, Ljungström V, Abdulla M, Cavelier L, Pandzic T, Hollander P, et al. Clonal hematopoiesis in patients with high-grade B-cell lymphoma is associated with inferior outcome. Am J Hematol. 2020;95:e287–9.
Cook EK, Luo M, Rauh MJ. Clonal hematopoiesis and inflammation: partners in leukemogenesis and comorbidity. Exp Hematol. 2020;83:85–94.
Jaiswal S. Clonal hematopoiesis and nonhematologic disorders. Blood. 2020;136:1606–14.
Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–7.
Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. 2017;55:56–70.
Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–93.
Abplanalp WT, Cremer S, John D, Hoffmann J, Schuhmacher B, Merten M, et al. Clonal hematopoiesis-driver DNMT3A mutations alter immune cells in heart failure. Circ Res. 2020;128:216–28.
Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C, et al. Dnmt3a restrains mast cell inflammatory responses. Proc Natl Acad Sci USA. 2017;114:E1490–9.
Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10:eaan8292.
Kramer AC, Kothari A, Wilson WC, Celik H, Nikitas J, Mallaney C, et al. Dnmt3a regulates T-cell development and suppresses T-ALL transformation. Leukemia. 2017;31:2479–90.
Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E, et al. The loss of TET2 promotes CD8 + T cell memory differentiation. J Immunol. 2018;200:82–91.
Spegarova JS, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 loss of function causes childhood immunodeficiency and lymphoma. Blood. 2020;136:1055–66.
Berger G, Kroeze LI, Koorenhof-Scheele TN, De Graaf AO, Yoshida K, Ueno H, et al. Early detection and evolution of preleukemic clones in therapy-related myeloid neoplasms following autologous SCT. Blood. 2018;131:1846–57.
Wong TN, Miller CA, Jotte MRM, Bagegni N, Baty JD, Schmidt AP, et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun. 2018;9:455.
Husby S, Favero F, Nielsen C, Sørensen BS, Bæch J, Grell K, et al. Clinical impact of clonal hematopoiesis in patients with lymphoma undergoing ASCT: a national population-based cohort study. Leukemia. 2020;34:3256–68.
Chitre S, Stölzel F, Cuthill K, Streetly M, Graham C, Dill C, et al. Clonal hematopoiesis in patients with multiple myeloma undergoing autologous stem cell transplantation. Leukemia. 2018;32:2020–4.
Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35:1598–605.
Mouhieddine TH, Sperling AS, Redd R, Park J, Leventhal M, Gibson CJ, et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat Commun. 2020;11:2996.
Gifford G, Hesson L, Wong JWH, Carroll A, Gabrielli S, Bai L, et al. Poor mobilization of autologous CD34+ peripheral blood stem cells in haematology patients undergoing autologous stem cell transplantation is associated with the presence of variants in genes implicated in clonal haematopoiesis of indeterminant potential. Br J Haematol. 2021:193:841–4.
Soerensen JF, Aggerholm A, Kerndrup GB, Hansen MC, Ewald IKL, Bill M, et al. Clonal hematopoiesis predicts development of therapy-related myeloid neoplasms post-autologous stem cell transplantation. Blood Adv. 2020;4:885–92.
Ortmann CA, Dorsheimer L, Abou-El-Ardat K, Hoffrichter J, Assmus B, Bonig H, et al. Functional dominance of CHIP-mutated hematopoietic stem cells in patients undergoing autologous transplantation. Cell Rep. 2019;27:2022–8.
Engel N, Rovo A, Badoglio M, Labopin M, Basak GW, Beguin Y, et al. European experience and risk factor analysis of donor cell-derived leukaemias/MDS following haematopoietic cell transplantation. Leukemia. 2019;33:508–17.
Herold S, Kuhn M, von Bonin M, Stange T, Platzbecker U, Radke J, et al. Donor cell leukemia: evidence for multiple preleukemic clones and parallel long term clonal evolution in donor and recipient. Leukemia. 2017;31:1637–40.
Dehn J, Spellman S, Hurley CK, Shaw BE, Barker JN, Burns LJ, et al. Selection of unrelated donors and cord blood units for hematopoietic cell transplantation: guidelines from the NMDP/CIBMTR. Blood. 2019;134:924–34.
Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019;25:1408–14.
Frick M, Chan W, Arends CM, Hablesreiter R, Halik A, Heuser M, et al. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. 2019;37:375–85.
Gibson CJ, Kennedy JA, Nikiforow S, Kuo FC, Alyea EP, Ho V, et al. Donor-engrafted CHIP is common among stem cell transplant recipients with unexplained cytopenias. Blood. 2017;130:91–4.
Gibson CJ, Kim HT, Murdock HM, Hambley B, Zhao L, Green L, et al. DNMT3A clonal hematopoiesis in older donors is associated with improved survival in recipients after allogeneic hematopoietic cell transplant. Blood. 2020;136:26.
Giavridis T, Van Der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade letter. Nat Med. 2018;24:731–8.
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24:739–48.
Jain T, Knezevic A, Pennisi M, Chen Y, Ruiz JD, Purdon TJ, et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood Adv. 2020;4:3776–87.
Yue X, Lio CWJ, Samaniego-Castruita D, Li X, Rao A. Loss of TET2 and TET3 in regulatory T cells unleashes effector function. Nat Commun. 2019;10:2011.
Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med. 2016;213:377–97.
Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–12.
Nobles CL, Sherrill-Mix S, Everett JK, Reddy S, Fraietta JA, Porter DL, et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J Clin Invest. 2020;130:673–85.
Kahn JD, Miller PG, Silver AJ, Sellar RS, Bhatt S, Gibson C, et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood. 2018;132:1095–105.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
CT is managing director of AgenDix. All other authors declare no competing financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
von Bonin, M., Jambor, H.K., Teipel, R. et al. Clonal hematopoiesis and its emerging effects on cellular therapies. Leukemia 35, 2752–2758 (2021). https://doi.org/10.1038/s41375-021-01337-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-021-01337-8
This article is cited by
-
Atherosclerotic cardiovascular disease in aging and the role of advanced cardiovascular imaging
npj Cardiovascular Health (2024)
-
Klonale Hämatopoese – Bedeutung für die Zelltherapie
Die Innere Medizin (2022)
