
Abstract
In acute myeloid leukemia (AML) internal tandem duplications of the FLT3 gene (FLT3-ITD) are associated with poor prognosis. Retrospectively, we investigated the prognostic and predictive impact of FLT3-ITD insertion site (IS) in 452 patients randomized within the RATIFY trial, which evaluated midostaurin additionally to intensive chemotherapy. Next-generation sequencing identified 908 ITDs, with 643 IS in the juxtamembrane domain (JMD) and 265 IS in the tyrosine kinase domain-1 (TKD1). According to IS, patients were categorized as JMDsole (n = 251, 55%), JMD and TKD1 (JMD/TKD1; n = 117, 26%), and TKD1sole (n = 84, 19%). While clinical variables did not differ among the 3 groups, NPM1 mutation was correlated with JMDsole (P = 0.028). Overall survival (OS) differed significantly, with estimated 4-year OS probabilities of 0.44, 0.50, and 0.30 for JMDsole, JMD/TKD1, and TKD1sole, respectively (P = 0.032). Multivariate (cause-specific) Cox models for OS and cumulative incidence of relapse using allogeneic hematopoietic cell transplantation (HCT) in first complete remission as a time-dependent variable identified TKD1sole as unfavorable and HCT as favorable factors. In addition, Midostaurin exerted a significant benefit only for JMDsole. Our results confirm the distinct molecular heterogeneity of FLT3-ITD and the negative prognostic impact of TKD1 IS in AML that was not overcome by midostaurin.
Similar content being viewed by others

Introduction
Internal tandem duplications of the FLT3 gene (FLT3-ITD), resulting in duplication of 3 to more than hundreds of nucleotides, are present in ~25% of younger adults with newly diagnosed acute myeloid leukemia (AML) [1,2,3,4]. The FLT3 receptor consisting of an extracellular domain of 5 immunoglobulin-like domains, a transmembrane region, a cytoplasmic juxtamembrane domain (JMD), and 2 cytoplasmic tyrosine kinase domains (TKD1 and TKD2), interrupted by a short kinase insert [5], plays an important role in self-renewal and differentiation of hematopoietic stem and progenitor cells [6,7,8]. The JMD, that can be subdivided into the JM-binding (JM-B) motif, the switch motif (JM-S), the linker/zipper peptide segment (JM-Z), and the hinge region of JMD, exerts a negative regulatory function over TKD1, consisting of beta1-sheet, nucleotide-binding loop, beta2-sheet, and 3´of beta2-sheet, as well as over TKD2 [5]. By disrupting the autoinhibitory function of the JMD or mediating constitutive phosphorylation of the TKDs, ITD mutations within JMD and TKD1, always affecting exons 14 and 15 of FLT3 and causing in-frame amino acid changes, lead to constitutive activation of the receptor tyrosine kinase and its downstream signaling pathways with consecutive dysregulated cellular proliferation [9,10,11,12]. The majority of ITD insertions (70%) occur within the JMD, whereas ITDs outside the JMD integrating in the TKD1 can be detected in ~30% of ITDs most frequently affecting the beta-1 sheet of TKD1 (~25% of all ITDs) [12, 13]. Several studies have shown that ITD mutations are associated with poor prognosis due to a high relapse rate, in particular in cases with a high mutant to wild-type allelic ratio (AR), commonly defined by a threshold of 0.50 using capillary electrophoresis [14,15,16,17,18,19], and/or insertion site (IS) located in the beta1-sheet of TKD1 [13, 18, 20].
Previous studies have demonstrated that TKD1-ITDs are associated with resistance to chemotherapy and significantly inferior outcome [13, 18, 20]. Here, in vitro studies showed that non-JMD-ITDs confer resistance to a panel of FLT3 inhibitors [21, 22]. In transfection studies performed by Arreba-Tutusaus and colleagues, 32D cells, Ba/F3 cells, and primary mouse bone marrow cells transduced with TKD1-ITD constructs displayed significantly less apoptosis compared to JMD-ITD constructs when exposed to midostaurin, and also quizartinib [22].
The natural course of FLT3 mutated AML may change with the advent of FLT3 tyrosine kinase inhibitors (TKI) that are now becoming increasingly available. In the CALGB 10603/RATIFY trial, enrolling 717 FLT3 mutated (ITD and TKD) adult AML patients, the addition of the multikinase inhibitor midostaurin to intensive chemotherapy significantly improved overall survival (OS) and event-free survival (EFS) [23]. The results from the trial led to the approval of midostaurin for first-line treatment of FLT3 mutated AML in 2017.
The aims of this large, retrospective study were to assess the molecular landscape of FLT3-ITD and to evaluate the prognostic impact of ITD IS on OS and cumulative incidence of relapse (CIR) in patients enrolled on the CALGB 10603/RATIFY trial. In addition, the predictive impact of ITD IS for response to treatment with midostaurin was evaluated.
Patients and methods
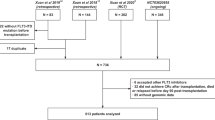
Overall, 717 patients with AML and activating FLT3 mutations (ITD and TKD mutations) were included in the CALGB 10603/RATIFY trial [23]. This exploratory post-hoc analysis focused on the prognostic and predictive impact of ITD IS in the subset of 555 FLT3-ITD positive (FLT3-ITD+) patients. Identification of FLT3-ITDs for study entry was performed by PCR-based amplification of FLT3 exon 14 and 15 using gene-specific primers followed by capillary electrophoresis (CE); stored biosamples for next-generation sequencing (NGS) analysis were available in 452/555 (81%) patients. Clinical and genetic baseline data of the patients are given in Table 1.
Molecular analyses
FLT3-ITD mutation analysis and assessment of NPM1 mutational status were performed as previously described [23, 24].
FLT3-ITD Roche 454 next-generation sequencing analysis
To investigate the relationship between ITD IS and patient outcome, Roche 454 NGS analysis was performed that allowed bi-directional sequencing with sequence read length of >500 bp and the identification of ITDs present in less than 5% of cells. A detailed methodological description is provided in the Supplementary Appendix and in Supplementary Figs. 1 and 2. Sequencing raw data were analyzed using the bioinformatics program getITD [25].
Statistical analyses
Complete remission (CR), OS, CIR, and cumulative incidence of death (CID) were defined by standard criteria [19]; responses included all CRs achieved during induction therapy. Overall survival was calculated from the date of randomization to death of any cause. The time to relapse was calculated from the date of first CR to relapse. Patients not having experienced the event of interest at the end of follow-up are censored at the date of last contact. CIR and CID were computed according to the method described by Gray [26]. The median follow-up for survival was calculated using the reverse Kaplan–Meier estimate [27]. Logistic regression and Cox proportional hazards models were used to identify prognostic variables for CR and OS [28]. CIR was analyzed using cause-specific Cox models where death in CR is considered as a competing event. Modeling the time to death in CR was not possible due to a very low number of events (22 events, 19 in the JMDsole group). Additional covariates in multivariate analysis were IS (categorized as JMDsole, JMD/TKD1, and TKD1sole due to the respective functional consequences and in line with previous studies [18, 20]), treatment with midostaurin (vs placebo), and NPM1 mutation status as dichotomous variables, as well as NGS-based calculated FLT3-ITD AR per patients (NGS-cAR) [determined as ∑variant allele frequency (VAF)/(100-∑VAF); log2 transformed], number of ITDs, white blood cell (WBC) count (log2 transformed), and age as continuous variables; allogeneic hematopoietic cell transplantation (HCT) in first CR (CR1) was included as a time-dependent variable. Kruskal-Wallis test was used for comparing quantitative variables between patient subgroups; categorical variables were compared by means of Fisher’s exact test. Associations between continuous variables were analyzed using the Spearman rank correlation coefficient. Survival distributions were estimated using the Kaplan–Meier method, and differences between groups were analyzed using two-sided log-rank tests. An effect was considered significant if its P value was less than 5%. The analyses were not adjusted for multiple testing. All statistical analyses were performed with the statistical software R 4.0.2 and/or IBM SPSS Statistics 25.
Results
Patient characteristics
Table 1 summarizes the baseline characteristics of the 452 FLT3-ITD+ patients. According to treatment arms, patients in the placebo group exhibited more frequently a normal karyotype (80.1% vs 62.2%; P < 0.001), NPM1 mutation (NPM1mut, 64.3% vs 50.0%; P = 0.008), and the favorable genotype NPM1mut/FLT3-ITDlow (23.9% vs 14.7%; P = 0.031).
Next-generation sequencing
We identified a total of 908 high confidence FLT3-ITDs in the 452 patients by NGS. The median coverage (total number of reads per patient) was 3115 (range: 482–11,616), median ITD counts were 97 (range: 2–5234), median VAF was 3.34% (range: 0.03–89.49%), and the median length was 45 base pairs (bp) (range: 6–246 bp).
Molecular characterization of FLT3-ITDs
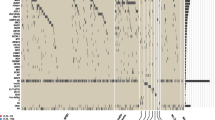
Two-hundred and ten (46.5%) of 452 FLT3-ITD + AML patients had one ITD, and 242 (53.5%) harbored more than one ITD (2 ITDs, n = 131 [29%]; 3 ITDs, n = 58 [13%]; 4 ITDs, n = 24 [5%]; 5 ITDs, n = 18 [4%]; 6 ITDs, n = 3 [1%]; 7 ITDs, n = 7 [2%]; 9 ITDs, n = 1 [0.2%]). All ITDs, except of 2 which exclusively affected exon 15, involved exon 14 of FLT3, and all ITDs were in-frame with a direct head-to-tail orientation. Eighty-eight ITDs also contained intron 14, and 15 ITDs extended to exon 15. In 619/908 (68.2%) ITDs, insertions resulted in a change of the expected amino acid sequence at the respective site. According to Griffith et al. [5] as well as previous and current releases of the e!Ensembl database (release 103, February 2021: https://www.ensembl.org/Homo_sapiens/Transcript/ProteinSummary?db=core;g=ENSG00000122025;r=13:28003274-28100592;t=ENST00000241453), molecular characterization of the ITDs revealed that in the majority (70.8%, n = 643) the IS was localized in the JMD between amino acids (aa) 573 and 609, whereas the remaining ITDs (29.2%) inserted in 3´ direction of the JMD, predominantly in the beta1-sheet of TKD1 between aa 610 and 615 (27.5%, n = 250) (Fig. 1A), which is in line with previous studies [12, 13, 18]. FLT3-ITD size was significantly correlated with IS, in that the more C-terminal the insertion of the IS, the longer the size of the inserted fragment [Rho (Spearman) = 0.530; P < 0.001] (Fig. 1B).

A correlation between ITD IS, length, as well as the relative position of the Y591-Y599 motif (depicted in black) of the 908 ITDs sorted by IS. The duplicated fragments are color-coded according to IS and the black color demonstrates the involvement of the amino acid motif Y591-Y599 which is important for intracellular signaling. The majority of the 908 ITDs included at least a part of the specific amino acid stretch Y591-599. B Correlation of ITD IS with length. C Correlation of ITD length with NGS-based calculated allelic ratio. D Circos Plot showing the interaction of concurrent IS. In 242 pts (53.5%) featuring multiple ITD clones, 698 concurrent IS were delineated; coexistent IS within JMD-Z were the most frequent interaction (41.5%) followed by the interaction between JMD-Z and beta1-sheet (20.2%), within beta1-sheet (12.2%), between JMD-Z and hinge region (11.5%), between beta1-sheet and hinge region (5.5%), and within the hinge region (4.8%).
Analysis of the duplication of specific amino acid motives
Because of the particular role of the tyrosine-rich stretch Y591 to Y599 (YVDFREYEY) for intracellular signaling [29], we determined the frequency of this motif in the ITDs. Duplication of at least one residue in this specific stretch was seen in 840/908 (92.5%) ITDs. The most frequent affected aa residues were R595 in 73.9% (n = 671), E596 (73.8%; n = 670), and Y597 (73.2%; n = 665), followed in descending order by F594 (71.0%; n = 645), D593 (68.6%; n = 623), E598 (66.1%; n = 600), V592 (60.7%; n = 551), Y599 (59.8%; n = 543), and Y591 (55.0%; n = 499). The entire Y591-Y599 motif was duplicated in 265 (29.2%) ITDs. Taking patients as denominator, in 425/452 (94.0%) patients at least one ITD showed the involvement of at least one aa of this specific motif. The entire Y591-Y599 motif was duplicated in 117 (25.9%) patients. In Fig. 1A, length of the ITDs, IS, and relative position of the Y591-Y599 motif within the ITDs are illustrated.
Mutant to wild-type FLT3-ITD allelic ratio determined by NGS (NGS-cAR)
NGS-based quantitative FLT3-ITD mutant to wild-type AR (NGS-cAR) was calculated based on VAF as VAF/(100-VAF). The NGS-cAR ranged from 0.0003 to 8.510, with a median of 0.04. The NGS-cAR correlated inversely with ITD length [Rho (Spearman) = −0.087; P = 0.009] (Fig. 1C) and aa insertion (5´ to 3´) (Rho = −0.081; P = 0.015), in that the higher the NGS-cAR, the shorter the ITD and the more 3´ the insertion site. NGS-cAR per patient [∑VAF/(100-∑VAF)] ranged from 0.0006 to 10.75, with a median of 0.27 and was correlated inversely with the number of ITDs [Rho (Spearman) = −0.142; P = 0.002], in that the higher the NGS-cAR, the lower the number of ITD mutated clones.
Characterization of co-occurring insertion sites
In 242 patients (54%) featuring multiple ITD clones, 698 co-occurring IS were detected. Figure 1D illustrates the relative interaction of concurrent IS. In detail: coexistent IS within JMD-Z were the most frequent interaction (41.5%) followed by the interaction between JMD-Z and beta1-sheet (20.2%), within beta1-sheet (12.2%), between JMD-Z and hinge region (11.5%), between beta1-sheet and hinge region (5.5%), and within the hinge region (4.8%) (Fig. 1D). Furthermore, the vast majority of patients (204/242; 84%) with more than one ITD exhibited one dominant ITD with an NGS-cAR at least twice as high as the sum of the NGS-cARs of the co-insertions (Supplementary Fig. 3).
Correlation of FLT3-ITD IS with clinical baseline characteristics, genetic features, and treatment
We correlated FLT3-ITD IS, categorized as patients having IS in (i) JMDsole (n = 251, 55.5%), (ii) JMD/TKD1 (n = 117, 25.9%), and (iii) TKD1sole (n = 84, 18.6%), with age, gender, WBC counts, bone marrow (BM) blast counts, NPM1mut, FLT3-ITD AR (based on Genescan analysis), and the four NPM1/FLT3-ITD genotypes, as defined within the 2017 European LeukemiaNet (ELN) risk categorization [19], number of ITDs, NGS-cAR, as well as treatment arm (midostaurin vs placebo) and HCT in CR1. While clinical variables did not differ among the 3 groups, JMDsole insertion was positively correlated with NPM1mut (P = 0.028), inversely with the NPM1wt/FLT3-ITDlow genotype (P < 0.001), and showed higher AR, determined by both Genescan and NGS-based (P < 0.001, each). Patients with JMD/TKD1 insertions exhibited more frequently FLT3-ITDlow (AR ≤ 0.5) (P < 0.001) and had more ITD subclones (P < 0.001). Of note, treatment with midostaurin was more frequently performed in patients with TKD1sole insertion (P = 0.012) (Table 2).
Clinical outcome
Response to induction therapy
Complete remission, including all CRs during induction cycles 1 and 2, was achieved in 274/452 (60.6%) patients. To address the impact of IS on CR, correlations of the 3 categorized IS groups [(i) JMDsole, (ii) JMD/TKD1, and (iii) TKD1sole] were performed using a logistic regression model. This model revealed NPM1mut as a favorable marker for achievement of CR (OR, 2.16; 95% confidence interval [CI], 1.36–3.44; P = 0.001), while WBC (OR for doubling of WBC, 0.83; 95% CI, 0.72–0.95; P = 0.009) and number of ITDs (OR, 0.79; 95% CI, 0.64–0.98; P = 0.035) predicted for a lower CR rate. The 3 IS groups as well as other variables had no impact on CR achievement (Fig. 2).

OR Odds ratio, CI confidence interval, cAR NGS-based calculated allelic ratio.
Survival analysis
The estimated median follow-up of the 452 FLT3-ITD + AML patients was 60.6 months (95% CI, 57.4–62.9); 248 of the 452 (54.9%) patients died. Median OS time and 4-year OS rate were 24.4 months (95% CI, 18.6–35.9 months) and 0.43 (95% CI, 0.38–0.47), respectively. HCT in CR1 was performed in 99/452 (21.9%) patients, and overall during the disease course in 257/452 (56.9%) patients.
Survival analyses according to ITD IS showed that patients with insertions in TKD1sole had a significantly inferior OS and a higher CIR compared to patients with insertion sites in JMDsole and/or JMD/TKD1 (4-year OS rates TKD1sole vs JMDsole vs JMD/TKD1: 29% vs 44% vs 50%; P = 0.032; 4-year CIR: 60% vs 45% vs 59%; P = 0.051) (Fig. 3). Supplementary Figure 4 shows CID according to ITD IS. In the JMD/TKD1 group, there was a significantly lower CID rate (4-year CID rates JMD/TKD1 vs TKD1sole vs JMDsole: 4% vs 13% vs 15%; P = 0.027), providing an explanation for the discrepancy between improved OS without decreasing the relapse rate for this subgroup.

Kaplan–Meier curves for overall survival (A) and cumulative incidence of relapse (B) for the 452 FLT3-ITD + AML patients according to ITD IS.
In Cox regression analyses on OS, FLT3-ITD IS in TKD1sole (compared to JMDsole: HR, 1.61; 95% CI, 1.10–2.34; P = 0.014, and compared to JMD/TKD1: HR, 2.17; 95% CI, 1.29–3.67; P = 0.004), higher WBC (log2) (HR, 1.12; 95% CI, 1.03–1.23; P = 0.010), and increasing age (HR, 1.17; 95% CI, 1.02–1.43; P = 0.029) revealed as unfavorable factors for OS, whereas NPM1mut (HR, 0.58; 95% CI, 0.43–0.78; P < 0.001) and HCT in CR1 (HR, 0.46; 95% CI, 0.30–0.70; P < 0.001) were both favorable. Multiclonality (higher number of ITDs) (HR, 1.13; 95% CI, 0.99–1.28; P = 0.076) and higher NGS-cAR (HR, 1.07; 95% CI, 0.99–1.15; P = 0.093) were in trend associated with inferior OS. Treatment with midostaurin had no additional impact on OS. For CIR, FLT3-ITD IS in TKD1sole (compared to JMDsole: HR, 2.20; 95% CI, 1.34–3.59; P = 0.002), higher WBC (log2) (HR, 1.18; 95% CI, 1.06–1.31; P = 0.003), and higher NGS-cAR (HR, 1.16; 95% CI, 1.04–1.29; P = 0.007) were associated with higher risk of relapse, whereas NPM1mut (HR, 0.67; 95% CI, 0.45–0.98; P = 0.037) and HCT in CR1 (HR, 0.42; 95% CI, 0.26–0.69; P < 0.001) reduced risk of relapse significantly. Treatment with midostaurin again had no impact. The results of the Cox regression analyses are summarized in Table 3.
Survival analyses for OS and CIR according to ITD IS and to treatment arms are shown in Fig. 4. The 4-year rates for OS of patients on midostaurin or placebo were 48% vs 40% (P = 0.047) for JMDsole, 53% vs 47% (P = 0.661) for JMD/TKD1, and 32% vs 26% (P = 0.256) for TKD1sole subgroups, respectively. For CIR, the 4-year rates for patients on midostaurin or placebo were 40% vs 50% (P = 0.246) for JMDsole, 56% vs 63% (P = 0.535) for JMD/TKD1, and 56% vs 63% (P = 0.997) for TKD1sole subgroups, respectively.

Kaplan–Meier curves for overall survival (A) and cumulative incidence of relapse (B) for the 452 FLT3-ITD + AML patients according to ITD IS and to treatment arms (midostaurin vs placebo).
We next performed Cox regression analyses on OS and CIR according to the 3 IS subgroups (Table 3). Within the JMDsole group, NGS-cAR (HR for OS, 1.18; 95% CI, 1.05–1.33; P = 0.006 and HR for CIR, 1.45; 95% CI, 1.21–1.73; P < 0.001) and multiclonality (HR, 1.29; 95% CI, 1.04–1.60; P = 0.022 and HR, 1.43; 95% CI, 1.02–2.02; P = 0.040) had a negative impact on both OS and CIR, older age (HR, 1.24; 95% CI, 1.03–1.51; P = 0.024) on OS only, whereas treatment with midostaurin (HR, 0.56; 95% CI, 0.37–0.84; P = 0.005 and HR, 0.57; 95% CI, 0.34–0.96; P = 0.036) and NPM1mut (HR, 0.47; 95% CI, 0.31–0.72; P < 0.001 and HR, 0.46; 95% CI, 0.27–0.78; P = 0.004) were of beneficial impact on both endpoints. A significant beneficial effect for HCT in CR1 was observed only for CIR (HR, 0.40; 95% CI, 0.20–0.80; P = 0.010). Within the TKD1sole group, HCT in CR1 (HR, 0.28; 95% CI, 0.12–0.68; P = 0.005 and HR, 0.19; 95% CI, 0.06–0.57; P = 0.003) was the only significant factor for improved OS and CIR, whereas higher WBC (log2) (HR, 1.38; 95% CI, 1.09–1.75; P = 0.008 and HR, 1.33; 95% CI, 1.04–1.70; P = 0.021) were of adverse impact for OS and CIR. The only significant factor within the JMD/TKD1 group was HCT in CR1 (HR, 0.17; 95% CI, 0.04–0.74; P = 0.018) associated with improved OS.
To further explore the prognostic impact of concurrent NPM1mut, we evaluated OS and CIR according to ITD IS with or without concurrent NPM1mut and to treatment arms. The best outcome was observed for the genotype JMDsole/NPM1mut additionally treated with midostaurin. The 4-year rates for OS and CIR for this genotype on midostaurin or placebo were 65% vs 44% (P = 0.016) and 30% vs 53% (P = 0.018), respectively. The only other genotype benefitting in trend from additional midostaurin was JMDsole/NPM1wt with 4-year rates for OS and CIR (midostaurin vs placebo) of 40% vs 28% (P = 0.075) and 53% vs 63% (P = 0.319), respectively. There was no benefit for additional midostaurin treatment within the other genotypes (Supplementary Figure 5). Of note, patient numbers in other genetic subgroups were relatively small and the study was not powered to perform subgroup analysis. Therefore, additional genotype-specific subgroup analyses (e.g., DNMT3A, RUNX1, etc.) were refrained since this will result in splitting up in small subgroups not allowing sound and meaningful statistical analyses.
Discussion
The results from this retrospective explorative analysis of the RATIFY trial confirm the distinct molecular heterogeneity of FLT3-ITD and the negative prognostic impact of TKD1 insertions. Furthermore, the data show that this negative impact might not be overcome by treatment with the multikinase inhibitor midostaurin confirming data from earlier preclinical studies. Additional midostaurin exerted a significant beneficial effect in patients with IS in JMDsole, in particular with concurrent NPM1mut, whereas no significant benefit was seen in the other patient groups. In multivariable analysis TKD1sole insertion revealed as an unfavorable prognostic factor for OS and CIR, whereas NPM1mut and HCT in CR1 were associated with improved outcome.
In this large cohort of 452 younger adult AML patients, we could confirm that ~30% of FLT3-ITDs are localized outside the JMD with a major cluster in the beta1-sheet of the TKD1 (28%), and at very low frequencies (≤1%) in the nucleotide-binding loop, the beta2-sheet of the TKD1, and 3´ of the beta2-sheet [12, 13]. As previously described, ITD size also strongly correlated with IS, in that the more C-terminal the insertion, the longer the inserted fragment [13]. Of note, NGS-cAR correlated inversely with (i) ITD length, (ii) the genomic insertion (aa, 5´to 3´), and (iii) the number of ITDs per patient. While the inverse correlation between NGS-cAR and the collinear ITD length and genomic insertion (the higher the NGS-cAR, the shorter the ITD and the more 5´ the IS) might be caused by the amplification disadvantage of larger DNA fragments, the inverse correlation between NGS-cAR and number of ITDs per patient remains elusive. In addition, in this cohort the majority of FLT3-ITD + AML patients (54%) harbored more than one ITD of different leukemic subclones which is related to the higher sensitivity of NGS compared to conventional DNA fragment analyses [25]; 84% of these patients exhibited one dominant FLT3-ITD clone with an NGS-cAR at least twice as high as the sum of the NGS-cARs of the co-occurring ITDs. In line with a previous report [13], the vast majority of patients (92%) with >1 ITD exhibited at least one insertion within the JMD.
FLT3-ITD IS were not associated with defined clinical characteristics, but with distinct genetic features: concurrent NPM1mut correlated positively with IS in JMDsole, and FLT3-ITD low AR (determined by fragment analysis) as well as number of ITDs with the JMD/TKD1 subgroup, respectively. Moreover, we observed a significant difference with respect to the 4 NPM1/FLT3-ITD genotypes: The genotype NPM1wt/FLT3-ITDlow was significantly underrepresented in the JMDsole subgroup while being highest in the JMD/TKD1 group (Table 2).
With respect to response to induction therapy the only favorable variable was NPM1mut known to predict for achievement of CR, also in AML with concurrent FLT3-ITD [13, 18]. Unfavorable factors were higher WBC counts and a higher number of ITD clones per patient (Fig. 2). This finding is at variance to previous studies that showed no impact for number of ITDs on response in multivariate analyses [13, 18]. One possible explanation might be the higher sensitivity of NGS-based ITD subclone detection as supported by the high number of patients with >1 ITD in this study compared to conventional fragment analysis used in the previous studies (54% vs 14%; P < 0.001).
The first important clinical finding of this study was the confirmation of the negative prognostic impact of TKD1 insertions [13, 18, 20]. Multivariate analysis for OS using Cox regression in the entire patient cohort identified TKD1sole IS, higher WBC counts, and older age as significant unfavorable factors. Significant favorable variables were NPM1mut and HCT in CR1 (Table 3). In the multivariate model for hazard of relapse, TKD1sole IS, higher NGS-cAR, and higher WBC counts predicted for higher risk of relapse, while HCT in CR1 reduced relapse risk (Table 3). Next, we performed multivariate analysis for OS and CIR within the 3 IS groups. The only variable that showed a consistent favorable effect across all subgroups was HCT in CR1 (with the exception of JMD/TKD1 for CIR). Of note, in the JMDsole group treatment with midostaurin – besides NPM1mut – had a strong beneficial effect on both OS and CIR (Table 3).
The second importing finding is that the beneficial effect of midostaurin was restricted to the JMDsole subgroup (Table 3 and Fig. 5) being in line with preclinical data showing sensitivity to midostaurin for JMD-ITD constructs in vitro and in vivo [22]. This benefit might be strengthened by the significant underrepresentation of the genotype NPM1wt/FLT3-ITDlow, that appears to have no benefit from midostaurin [30], in the JMDsole subgroup (Table 2) as well as by our data illustrating the impact of concurrent NPM1mut on OS and CIR in the 3 IS groups and by treatment arm (midostaurin vs placebo) (Supplementary Fig. 5). However, these results need to be interpreted with caution because the RATIFY trial was not powered to show statistically significant differences in these genetic subgroups. Nevertheless, beside the 2017 ELN risk groups our data provide evidence for additional risk stratification based on FLT3-ITD IS.

JMDsole, JMD/TKD1, TKD1sole.
The third interesting finding is that the negative prognostic impact of TKD1 IS was not overcome by additional treatment with midostaurin (Table 3, Figs. 4, 5, and Supplementary Fig. 5). The clinical observation that patients with insertion in TKD1 might not benefit from additional midostaurin does not only confirm the known resistance to chemotherapy of patients with TKD1 insertions [13, 18, 20], but also to TKI treatment, which is in line with preclinical data [21, 22]. Primary resistance of TKD1 insertions to midostaurin was first described in 2009 [21]. In a reconstitution model it could be demonstrated that TKD1-ITD627E was sufficient to confer resistance to a panel of FLT3 TKIs in vitro [21]. Subsequent transfection studies using 32D cells, Ba/F3 cells, and primary mouse bone marrow cells transduced with TKD1-ITD constructs revealed significantly reduced apoptosis for TKD1-ITD constructs compared to JMD-ITD constructs, when exposed to the multikinase inhibitor midostaurin, and also to the highly selective FLT3-inhibitor quizartinib [22]. Mechanisms mediating primary resistance of TKD1-ITDs to TKIs being discussed are upregulation of the anti-apoptotic myeloid cell leukemia 1 protein (MCL-1) and effective DNA-damage repair in TKD insertions possibly protecting from targeted and cytotoxic treatment [21, 22]. Furthermore, the significantly higher CIR rate for patients with TKD1 insertions compared to patients with JMDsole insertions might reflect different patterns of clonal evolution and acquired co-mutations mediating secondary resistance to TKIs between these groups. Complex changes in clonal architecture underlying response and resistance to TKI treatment have recently been described for quizartinib and also gilterinitib [31, 32].
In conclusion, the results from this retrospective analysis of the RATIFY trial further classify the molecular landscape of FLT3-ITD. Using a more sensitive NGS approach we identified more than one ITD clone in the majority of FLT3-ITD + AML patients. Moreover, our data confirm the negative prognostic impact of TKD1 IS that was not significantly affected by treatment with the multikinase inhibitor midostaurin. A beneficial effect of midostaurin was only found for patients with JMDsole IS. In this subset, NPM1mut also exerted a strong beneficial effect.
References
Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–86. Erratum in: J Clin Oncol. 2011;29(13):1798.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41.
Bullinger L, Döhner K, Döhner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35:934–46.
Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–78.
Matthews W, Jordan CT, Wiegand GW, Pardoll D, Lemischka IR. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell. 1991;65:1143–52.
Small D, Levenstein M, Kim E, Carow C, Amin S, Rockwell P, et al. STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc Natl Acad Sci USA. 1994;91:459–63.
Gotze KS, Ramirez M, Tabor K, Small D, Matthews W, Civin CI. Flt3high and Flt3low CD34+ progenitor cells isolated from human bone marrow are functionally distinct. Blood. 1998;91:1947–58.
Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene. 2000;19:624–31.
Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Müller C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96:3907–14.
Brandts CH, Sargin B, Rode M, Biermann C, Lindtner B, Schwäble J, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005;65:9643–50.
Breitenbuecher F, Schnittger S, Grundler R, Markova B, Carius B, Brecht A, et al. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood. 2009;113:4074–77.
Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114:2386–92.
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–59.
Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35.
Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–84.
Pratcorona M, Brunet S, Nomdedéu J, Ribera JM, Tormo M, Duarte R, et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood. 2013;121:2734–38.
Schlenk RF, Kayser S, Bullinger L, Kobbe G, Casper J, Ringhoffer M, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124:3441–49.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47.
Liu SB, Qiu QC, Bao XB, Ma X, Li HZ, Liu YJ, et al. Pattern and prognostic value of FLT3-ITD mutations in Chinese de novo adult acute myeloid leukemia. Cancer Sci. 2018;109:3981–92.
Breitenbuecher F, Markova B, Kasper S, Carius B, Stauder T, Böhmer FD, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood. 2009;113:4063–73.
Arreba-Tutusaus P, Mack TS, Bullinger L, Schnöder TM, Polanetzki A, Weinert S, et al. Impact of FLT3-ITD location on sensitivity to TKI-therapy in vitro and in vivo. Leukemia. 2016;30:1220–25.
Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017;377:454–64.
Döhner K, Schlenk RF, Habdank M, Scholl C, Rücker FG, Corbacioglu A, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–6.
Blätte TJ, Schmalbrock LK, Skambraks S, Lux S, Cocciardi S, Dolnik A, et al. getITD for FLT3-ITD-based MRD monitoring in AML. Leukemia. 2019;33:2535–39.
Gray RJ. A class of k-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–54.
Schemper M, Smith TL. A note on quantifying follow-up in studies of failure time. Control Clin Trials. 1996;17:343–6.
Cox DR. Regression models and life-tables. J R Stat Soc Ser B Stat Methodol. 1972;34:187–220.
Vempati S, Reindl C, Kaza SK, Kern R, Malamoussi T, Dugas M, et al. Arginine 595 is duplicated in patients with acute leukemias carrying internal tandem duplications of FLT3 and modulates its transforming potential. Blood. 2007;110:686–94.
Döhner K, Thiede C, Jahn N, Panina E, Gambietz A, Larson RA, et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood. 2020;135:371–80.
Smith CC, Paguirigan A, Jeschke GR, Lin KC, Massi E, Tarver T, et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood. 2017;130:48–58.
McMahon CM, Ferng T, Canaani J, Wang ES, Morrissette JJD, Eastburn DJ, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9:1050–63.
Acknowledgements
This work was supported in part by a grant from the Deutsche Forschungsgemeinschaft (SFB 1074, project B3) (KD and LB), by National Institutes of Health, National Cancer Institute grants U10CA180821 (to the Alliance for Clinical Trials in Oncology Operations Center), U10CA180882 (to the Alliance Statistics and Data Management Center), U24CA196171 (to the Alliance NCTN Biorepository and Biospecimen [CDB]), and UG1CA233338 (to ITSC for Leukemia: Novel Molecular strategies for NCTN Individualized Therapies [CDB]), and by a research grant from Novartis.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
FGR designed the study, performed research, collected, assembled, analyzed, and interpreted data, and wrote the manuscript; LD, TWP, and MH performed molecular analyses and analyzed data; TJB performed bioinformatics; AB, JK, and IG performed statistical analyses; RAL, MTV, SA, JMB, FRA, BCM, MST, MG, JS, CP, MAS, TdW, DN, TF, GE, AG, and LB collected, assembled, analyzed, and interpreted data; CDB, RMS, HD, CT, and KD designed the study, collected, assembled, analyzed and interpreted data, and wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
COMPETING INTERESTS
LD, IG, and CP are employees of Novartis. SA has acted as a consultant or advisor for Novartis and Daiichi-Sankyo. BCM has acted as a consultant or advisor for Celgene, Novartis, and Astellas and is currently employed by Roche/Genentech. MST has acted as a consultant or advisor for AbbVie, BioLineRx, Daiichi-Sankyo, Orsenix, KAHR Medical, Rigel Pharmaceuticals, Nohla, Delta Fly Pharma, Tetraphase, Oncolyze, and Jazz Pharmaceuticals; has received clinical research funding from AbbVie, Cellerant Therapeutics, Orsenix, ADC Therapeutics, and BioSight; and has received royalties from UpToDate. JS has acted as a consultant or advisor for Pfizer, Daiichi Sankyo, AbbVie, Novartis, Astellas, and Roche and is a member of the speakers bureau for Novartis, Pfizer, Daiichi Sankyo, and AbbVie. MAS has acted as a consultant or advisor for Teva Pharmaceutical Industries, Daiichi-Sankyo, Orsenix, AbbVie, Novartis, and Pfizer. DN has acted as consultants or advisors for Novartis. MH has acted as a consultant or advisor for AbbVie, Daiichi Sankyo, Novartis, Pfizer, and Bayer Pharma and has received clinical research support from Pfizer, Daiichi Sankyo, Karyopharm, BerGenBio, Bayer Pharma, Novartis, and Astellas. AG has received clinical research support from Novartis. LB has acted as an advisor for Abbvie, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Gilead, Hexal, Janssen, Jazz Pharmaceuticals, Menarini, Novartis, Pfizer, Sanofi, Seattle Genetics. RAL has acted as a consultant or advisor and has received clinical research support from Novartis. RMS has acted as a consultant or advisor for AbbVie, Actinium, and Agios; has received personal fees from Amgen, argenx, AROG, Astellas, AstraZeneca, BioLineRx, Celgene, Cornerstone, Daiichi-Sankyo, Fujifilm, Jazz Pharmaceuticals,MacroGenics, Novartis, Ono/Theradex Oncology, Orsenix, Otsuka/Astex, Pfizer, Roche, Stemline Therapeutics, Takeda, and Trovagene; and has received institutional research support from AbbVie, Agios, AROG, and Novartis. HD has acted as a consultant or advisor for AbbVie, Agios, Amgen, Astellas, Astex Pharmaceuticals, Celgene, Janssen, Jazz Pharmaceuticals, Novartis, Roche, and Seattle Genetics and has received institutional research support from Amgen, AROG Pharmaceuticals, Bristol-Myers Squibb, Celgene, Jazz Pharmaceuticals, Novartis, Pfizer, and Sunesis. CT is the Chief Executive Officer and a co-owner of Agendix, a company performing molecular diagnostics; has acted as a consult or advisor for Novartis and Astellas; and has received clinical research support from Bayer. KD has acted as a consultant or advisor for Astellas, Celgene, Daiichi Sankyo, Janssen, Novartis, and Roche and has received clinical research support from Astex, Celgene, and Novartis. The remaining authors declare no competing financial interests. Clara D. Bloomfield died on 1 March 2020.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rücker, F.G., Du, L., Luck, T.J. et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 36, 90–99 (2022). https://doi.org/10.1038/s41375-021-01323-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-021-01323-0
This article is cited by
-
Identification of a novel mutation of the FLT3 gene located on the juxtamembrane domain from acute myeloid leukemia patients
Molecular Biology Reports (2024)
-
ITDetect: a method to detect internal tandem duplication of FMS-like tyrosine kinase (FLT3) from next-generation sequencing data with high sensitivity and clinical application
BMC Bioinformatics (2023)
-
Sitravatinib as a potent FLT3 inhibitor can overcome gilteritinib resistance in acute myeloid leukemia
Biomarker Research (2023)
-
A key role of the WEE1-CDK1 axis in mediating TKI-therapy resistance in FLT3-ITD positive acute myeloid leukemia patients
Leukemia (2023)
-
Impact of FLT3-ITD location on cytarabine sensitivity in AML: a network-based approach
Leukemia (2023)
