
Abstract
It has been nearly half a century since angioimmunoblastic T-cell lymphoma (AITL) was characterized in the early 1970’s. Our understanding of the disease has dramatically changed due to multiple discoveries and insights. One of the key features of AITL is aberrant immune activity. Although AITL is now understood to be a neoplastic disease, pathologists appreciated that it was an inflammatory condition. The more we understand AITL at cellular and genetic levels, the more we view it as both a neoplastic and an inflammatory disease. Here, we review recent progress in our understanding of AITL, focusing on as yet unsolved questions.
Similar content being viewed by others


Introduction
The cancer microenvironment is closely associated with inflammation and the immune response. In lymphoid malignancies, a tumor arises in tissue that was once part of the immune system, and thus, inflammatory/immune cells constitute a more dominant component of the microenvironment than in other malignancies.
Angioimmunoblastic T-cell lymphoma (AITL) exemplifies a neoplasm characterized by intense inflammatory and immune reactions, as evidenced by its clinical, pathologic, cellular, and biologic properties. Because tumor cells phenotypically resemble T follicular helper (Tfh) cells [1,2,3], they are considered to function similarly to some extent to nonneoplastic Tfh cells seen in reactive follicular hyperplasia. However, follicles are not hyperplastic but are rather depleted or destroyed in vast majority of AITL cases.
Stepwise understanding of AITL—historic overview
In the early-mid 1970’s, different groups proposed what turned out to be a fundamentally identical disease based on observations of independent patient cohorts, namely, a condition variously described as immunodysblastic disease [4], angioimmunoblastic lymphadenopathy with dysproteinemia (AILD) [5], or immunoblastic lymphadenopathy (IBL) [6]. All groups initially proposed that the disease was a nonneoplastic, hyperimmune reaction. Early on, however, investigators recognized that the condition was marked by a broad histologic spectrum and speculated that some patients’ disease might be neoplastic, although they could not make the distinction. Later, a proportion of AILD/IBL cases were diagnosed as malignant lymphoma of either B-cell [7] or T-cell [8] origin. By the late 1980s, the consensus was that most cases described as AILD/IBL were T-cell lymphoma [9, 10]. At the same time, accurate diagnosis of AILD/IBL was facilitated by staining of follicular dendritic cells (FDCs) with anti-CD21 and -CD23 antibodies [11].
In 1994 the term “angioimmunoblastic T-cell lymphoma” was introduced in the Revised European and American Classification of Lymphoid Neoplasms [12]. WHO classifications [13,14,15] followed this designation, although debate continued until the 2000s over whether true nonneoplastic AILD/IBL exists and whether it represents a premalignant state [13].
Such confusion was in part attributable to uncertainty over tumor cell identity. In the 1990s a consensus was gradually reached that “clear cells” rather than “immunoblasts” represented neoplastic cells [16]. By the early 2000s, important insight had been gained by the discovery of expression of CD10 [17] and C-X-C motif chemokine ligand 13 (CXCL13) [1, 2] in clear cells. Global gene expression patterns seen in whole AITL tissues and isolated tumor cells were then shown to resemble those seen in Tfh cells [3]. These findings confirmed the idea that Tfh cells are the normal cellular counterparts of AITL tumor cells [14]. It is noteworthy that Tfh cell physiology was defined and characterized in parallel [18, 19].
A 2011 report of frequent somatic mutations in the tet methylcytosine dioxygenase 2 (TET2) gene provided insight into AITL genetics [20], as did discovery soon after of recurrent somatic mutations in DNA methyltransferase 3A (DNMT3A) [21] and isocitrate dehydrogenase 2 (IDH2) [22]. Interestingly, these three genes encode enzymes functioning in epigenetic regulation [23,24,25] and were originally identified as recurrent mutations in acute myeloid leukemia (AML) and other myeloid malignancies [26,27,28]. The AITL disease-defining G17A mutation in ras-homology family member A (RHOA), RHOAG17V, was identified in 2014 [29, 30]. In AITL, TET2, DNMT3A, IDH2, and RHOAG17V mutations are seen at around ~80% [29, 31, 32], 20–40% [29,30,31,32], 20–30% [22, 29, 31,32,33], and 50–70% [29,30,31,32, 34, 35] of the cases, respectively, followed by mutations in several T-cell receptor (TCR)-related genes, such as CD28 and phospholipase C gamma 1 (PLCG1) [31, 34, 36]. Of the former four, all but IDH2 are mutated at similar frequencies in AITL, follicular T-cell lymphoma (FTCL), and in a subset of lymphomas classified by the 2008 version of the WHO classification as peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS) [14]. That subset corresponded to PTCL-NOS with Tfh phenotype [29, 32, 34, 37]. Based on these discoveries, a new umbrella category, namely, “AITL and other nodal T-cell lymphomas of Tfh origin” (hereafter, designated as Tfh lymphomas), was proposed in the 2016 WHO classification to include three diseases, namely, AITL, FTCL, and newly-defined nodal PTCL with Tfh phenotype (nPTCL-Tfh) [15]. In that classification, PTCL-NOS was defined as excluding nPTCL-Tfh. However, diagnosis was not based on this new classification in most of the literature cited here. Here, when we refer to “PTCL-NOS”, we include nPTCL-Tfh, which should largely overlap with PTCL-NOS with Tfh gene expression profiles (GEP) (PTCL-NOS-Tfh) [33, 38].
AITL incidence—regional differences
The International T-Cell Lymphoma Project (ITCLP) analyzed 1153 PTCL cases (excluding leukemic and cutaneous types and inappropriately diagnosed cases from an original total of 1314) collected from Europe [n = 450, 34.2%], North America [n = 332, 25.3%], and Asia [n = 532, 40.5%]. In that analysis, AITL accounted for 21.1% of PTCL [39], making it the second most frequent PTCL after PTCL-NOS (29.5%). This report confirmed anticipated variations by geographic region for several subtypes such as adult T-cell leukemia/lymphoma (ATLL), extranodal NK/T-cell lymphoma, nasal type (ENKTL), and enteropathy-associated T-cell lymphoma. Unexpectedly, apparent regional variations were also observed in AITL and PTCL-NOS, which accounted for 28.7% and 34.3% of PTCL in Europe, 16.0% and 34.4% in North America, and 17.9% and 22.4% in Asia, respectively. In Asia, ATLL caused by human T-cell leukemia virus type 1 infection (particularly in Japan) and ENKTL closely associated with Epstein–Barr virus (EBV) (particularly in continental east Asia) were markedly overrepresented, which lowered frequencies of other subtypes. However, there remained differences that could not be explained solely by ATLL and ENKTL incidence, as described in the next paragraph.
AITL was recently reported to account for 36.1% of PTCL (excluding cutaneous types) in a combined cohort of 2046 PTCL cases in France, making it more frequent than PTCL-NOS (26.9%) [40]. These incidences are apparently discordant from those reported in the European cohort by the ITCLP. Such discordance could be explained by selection bias, choice of diagnostic tools used at different times of analysis, or differences in 2001 and 2008 WHO classifications on which the ITCLP and French studies, respectively, were based. True geographic variations within Europe have additionally been discussed, particularly with relevance to the Swedish national registry data (2000–2009), in which AITL was diagnosed only in 14% while PTCL-NOS was diagnosed in 29% of non-cutaneous and non-leukemic PTCL (n = 755) [40].
In Japan, a national registry for hematologic malignancies based on the 2008 WHO classification was begun in 2012. To compare the incidences of AITL and PTCL-NOS in Japan with those in others, we looked into this registry data with the permission by the Japanese Society of Hematology (Fig. 1). In the 7 years between 2012 and 2018, 11 403 (10 920 non-cutaneous) PTCL cases were registered (Supplementary Table 1). The most frequent diagnosis was ATLL (n = 4143; 37.9% in non-cutaneous types), followed by PTCL-NOS (n = 2424; 22.2%), AITL (n = 1855; 17.0%), ENKTL (n = 1063; 9.7%), and ALK(−) ALCL (n = 567; 5.2%). As noted, extremely high incidence of ATLL in Japan hampers comparison with other cohorts. If ATLL were excluded, the incidence of AITL and PTCL-NOS would be 27.4% and 35.8%, respectively, figures comparable to the European cohort by the ITCLP (Fig. 1). This factor should be considered in discussions of regional differences in AITL incidence.

IPTCLP International PTCL Project, ALK anaplastic lymphoma kinase, ALCL anaplastic large cell lymphoma, EATL enteropathy-associated T-cell lymphoma. *1, calculated as 87.8% (frequency of PTCL and ENKTL after excluding non-PTCL cases in IPTCLP) of 450 (number of cases registered in Europe in IPTCLP). *2, calculated excluding ATLL from *1. a Frequencies including ATLL. b Frequencies excluding ATLL.
Prognostic indicators
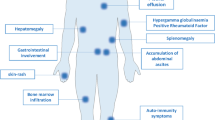
Clinical features of AITL include generalized lymphadenopathy with systemic inflammation/immune-related symptoms/signs, developing most often in the elderly. In five large cohort studies (n = 246, 77, 157, 207, and 243, respectively), major clinical parameters of patients were similar: median age (62–67 years), Ann Arbor stage III/IV (81–98%), B symptoms (60–77%), and anemia (51–65% after exclusion of one study with undefined hemoglobin levels) [40].
Five-year overall survival (OS) and event/progression/failure-free survival were 32–41% [40] and 18–38% [41,42,43], respectively. In the French study (n = 157) in which patients were chosen from two prospective clinical trials, a plateau in both OS and EFS was observed around 6 years (7-year OS and EFS, 29% and 23%, respectively) [41]. Plateaus were unclear in other studies, although 7-year OS and PFS were relatively high at 35% and 26%, respectively, in the Japanese retrospective study [42]. In short, the first third dies within a year, the second third between 1 and 5 years, and the last third survives longer. AITL has been described as fatal, but at the same time, long-term survivors have been reported since the first papers were published in the 1970s.
Biomarkers indicative of prognosis have been long sought. Among clinical parameters, high international prognostic index and prognostic index for PTCL-U scores, plus a low platelet count are reproducibly predictive of poor prognosis to some degree [41,42,43]. No pathologic parameters have been shown to be prognostic indicators, including the presence or absence of clear cells, which are neoplastic but undetectable in ~30% of samples diagnosed as AITL [40]. EBV status in AITL cases is also controversial. In a study evaluating the efficacy of combining rituximab with CHOP, the presence of EBV DNA in the circulation was correlated with the number of EBV+ cells in tissues, based on in situ hybridization (ISH) for the EBV-encoded small RNA (EBER; EBER-ISH) in lymph nodes, and with shorter PFS [44]. Others, however, report that the presence of EBV+ cells does not alter prognosis [41, 42]. A prognostic model has also been proposed based on gene expression signatures [45]. Focusing on 34 genes whose expression are significantly correlated with clinical outcomes, three functional scores with B-cell, monocytic, and p53-induced signatures have been validated. Among these, a B-cell-associated signature predicted a favorable outcome, while the other two were associated with poorer outcomes. It will be of interest to determine whether the B-cell-associated signature predicts long-term (>5 years) survivors, who might not need aggressive treatments such as autologous transplantation at upfront settings [46].
Pathology and the Tfh reaction
Typical AITL lymph nodes (pattern III, roughly 80% of AITL) [17, 47] show complete structural effacement. Infiltrating cellular components include clear cells, blastic cells, and arborizing vessels composed of high endothelial venules (HEVs), which have been identified morphologically in 69%, 94%, and 98% of AITL, respectively, based on hematoxylin–eosin staining [40]. Other inflammatory cells are also present, such as small lymphocytes, usually with a normal CD4+ and CD8+ cell ratio [48], eosinophils, macrophages, and plasma cells. Clear cells correspond to neoplastic cells, but morphologically identifiable clear cells are absent in ~30% of AITL. Biopsied tissues can also exhibit EBV+ cells, which are found in 66–91% [37, 40,41,42, 49] of AITL. Immunostaining reveals the presence of increased FDCs in 93% [40] of cases. Quantity and distribution of all of these cells can differ, resulting in highly significant variation [47, 50]. In addition, there are two nonclassical types of AITL: AITL with hyperplastic follicles (pattern I) and AITL with depleted follicles (pattern II) [17, 47, 50]. Immunohistochemically, CD3, CD4, and CD5 are positive in most cases (100%, 90–95%, and 85–95%, respectively) [41, 42]. CD7 is aberrantly downregulated in many PTCLs [51] and not detectable in 50–70% of AITL cases [41, 42]. AITL neoplastic cells frequently lack cell surface CD3 [51, 52].
Among Tfh cell markers, AITL specimens stain positively for CXCL13, programmed cell death-1 (PD1/PDCD1/CD279; hereafter PD1), CD10, B-cell lymphoma 6 (BCL6), and inducible T-cell co-stimulator (ICOS) in 76–100%, 62–100%, 30–89%, 62-91%, and 98% of cases, respectively [37, 40,41,42, 49]. It is noteworthy that BCL6 expression in clear cells in AITL had been documented even before Tfh cells were characterized [16, 53]. It is also of interest that a fraction of functional Tfh cells expresses CD10 in normal secondary lymphoid organs [54], although CD10 was initially thought to be aberrantly expressed in AITL neoplastic cells [17]. Based on these and other observations, the 2016 WHO classification requires that Tfh lymphomas be diagnosed by positive immunostaining for at least 2 (ideally 3) of the following 7 antigens: CD10, BCL6, PD1, CXCL13, C-X-C motif chemokine receptor 5 (CXCR5), ICOS, and signaling lymphocytic activation molecule-associated protein (SAP) [15].
Understanding of Tfh cell function in germinal center (GC) formation and reactions has greatly expanded over the past two decades; in particular, progress in the last decade has been swift due to identification of BCL6 as a master transcription factor for Tfh cells [55,56,57]. Priming of naïve CD4+ T cells to Tfh cells is the first step of GC formation and thus of all humoral immunity. Complex activities involving multiple factors then proceed, including interactions between Tfh and B cells and between B cells and FDCs. (See a recent review [19] for a comprehensive summary of these processes.) In brief (Fig. 2a), interaction of naïve CD4+ T cells with dendritic cells (DCs) at the T-cell zone governs the choice toward either a Tfh cell fate based on BCL6 expression or a non-Tfh cell fate marked by expression of B lymphocyte-induced maturation protein-1 [55,56,57,58]. Expression of the co-stimulatory molecule ICOS by DC-activated naïve CD4+ T cells is an early event in Tfh priming [58]. Tfh-primed cells migrate to the T/B border and start interacting with activated antigen-specific B cells [19]. It is suggested that ligation between PD1 and PDL1 controls Tfh cell positioning [59].

(a) Germinal center reaction in physiologic condition. Both Tfh-primed cells and B cells together form the GC, where GC B cells begin proliferating at the dark zone and undergo somatic hypermutation. In the light zone, FDCs participate in the network and contribute to selection of affinity-matured GC B cells. In the light zone, GC B cells make one of the three choices: re-entry to the dark zone, differentiation into plasmablast/plasma cells, or differentiation into memory B cells. For all of these processes, CD40 ligand (CD40L) expressed on the Tfh cell membrane and interleukin-21 (IL-21), IL-4, and CXCL13 secreted by Tfh cells play important roles in GC B-cell activation. Engagements of MHC class II, CD40, and ICOS ligand on GC B cells with the TCR, CD40L, and ICOS on Tfh cells, respectively, are of particular importance in terms of direct cell-to-cell contact. Also see the text for additional explanations. (b) Germinal center reaction in the presence of Tet2 disruption in B cells shown in mice. Follicular hyperplasia is caused by impaired exit of GC B cells from the GC light zone. Tfh follicular helper T cell, GCB germinal center B cell, activated B activated B cell, Tfh-primed CD4+ Tfh-primed CD4+ T cell, naive CD4+ naive CD4+ T cell, memory B memory B cell, mDC myeloid dendritic cell, FDC follicular dendritic cell, HSC hematopoietic stem cell, Th1 T helper 1 cell, eosino eosinophil. ICOSL ICOS ligand, MHC/Ag antigen presented on major histocompatibility complex, TCR T-cell receptor, CD40L CD40 ligand, BCR B-cell receptor, VEGF vascular endothelial growth factor. GC germinal center, LZ light zone, DZ dark zone, BM bone marrow, LN lymph nodes. SHM somatic hypermutation, mut mutation. Red closed circles indicate antigen localized on FDC.
Tfh activity is closely associated with numerous pathologies, including infectious, allergic, autoimmune, atherosclerotic, and neoplastic disease [19]. AITL (other than pattern II), however, is unique because the physiologic GC reaction described above is completely abrogated. Neoplastic Tfh cells are hypothesized to function in disease initiation and development. Understanding AITL pathology requires defining stage(s) of Tfh development and activity that differ between physiologic and neoplastic Tfh cells (see Figs. 2, 3).

In the bone marrow (BM), somatic mutations in TET2 (or TET2 plus DNMT3A; marked as TET2 and DNMT3A) result in clonal hematopoiesis. TET2 alone or TET2 plus DNMT3A mutated hematopoietic stem cells (HSC) can give rise to thymocytes. TET2 plus DNMT3A mutated HSC generate more CD4+ T cells than CD8+ T cells. TET2* indicates TET2 mutation alone or TET2 plus DNMT3A mutations. TET2 alone or TET2 plus DNMT3A mutated naive CD4+ T cells are primed to Tfh cells by the contact with myeloid DC cells, and migrate to the T-B border. The TET2 mutation (or TET2 plus DNMT3A mutations)-carrying Tfh-primed cells contact with the TET2-mutated activated B cells, then acquire the RHOAG17V mutation (RHOA) before or after differentiation into Tfh cells (Tfh). These Tfh cells should further interact with B cells (“B”) derived from activated B cells at the follicle-destroyed lymph nodes. The Tfh cells carrying TET2 (or TET2 plus DNMT3A) plus RHOA mutations may further acquire the IDH2 mutation (IDH2). RHOA mutation alone or RHOA plus IDH2 mutations are designated as RHOA*. Ultimately, mutations in TCR-related genes (TCRr) are acquired. TET2-mutated B cells are infected with EBV and/or acquire mutations in genes such as NOTCH1. These Tfh cells and B cells with individually accumulated mutations still contact and activate each other through ICOS-L and ICOS ligation and still unknown mechanisms (designated as “?”). Some of the B cells take a morphology as immunoblasts or HRS cells, and B-cell lymphoma arises. See the text for the further scenario. LN lymph nodes, Thy thymus. HSC hematopoietic stem cell, naive CD4+ naive CD4+ T cell, Tfh-primed Tfh-primed CD4+ T cell, Tfh follicular helper T cell, activated B activated B cell, Th17 T helper 17 cell, Th1 T helper 1 cell, Th2 T helper 2 cell, eosino eosinophil, plasma plasma cell, FDC follicular dendritic cell, HEV high endothelial venule. ICOSL ICOS ligand, MHC/Ag antigen presented on major histocompatibility complex, TCR T-cell receptor, BCR B-cell receptor, VEGF vascular endothelial growth factor.
AITL genetics, Tfh cell specification and differentiation, and AITL modeling
AITL is characterized by several recurrent mutations, many analyzed molecularly in mice. Surprisingly, functionally distinct mutations respectively promote Tfh cell specification or differentiation. Thus, further understanding is required of how these changes lead to AITL/Tfh lymphoma development. What follows is a list of some of the most predominant mutations.
TET2 mutations
TET2 is the most frequently mutated gene in AITL: TET2 somatic variations are seen in ~80% of patient specimens, with many exhibiting 2 or more TET2 mutations [29, 31, 32]. TET2 encodes a dioxygenase that transfers oxygen to 5-methylcytosine (5mC) in DNA and converts it into 5-hydroxymethylcytosine (5hmC) and further sequentially to 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC) [23, 60]. TET2 mutations promote loss-of-function of the TET2 enzyme [61] and this is true for AITL [20].
To model TET2 mutations in animals, several groups have analyzed mice with Tet2-disruption or -downregulation in either germline or whole blood cells. In general, mutant mice develop both chronic myelomonocytic leukemia-like disease [20, 62] and mature T-cell lymphoma with Tfh phenotype [63]. Skewed Tfh cell differentiation in splenic CD4+ T cells prior to lymphoma development has also been reported [63]. In this report, mice died at a median survival period of 67 weeks, developing T-cell lymphoma with Tfh-like features. It has also been demonstrated that Bcl6 is upregulated by Tet2 downregulation via hypermethylation at a Bcl6 intronic silencer region [63].
TET2 loss-of-function could affect any stage of T-cell development [20, 63]. One critical stage may be that of naive CD4+ cells, in which TET2 downregulation would enhance BCL6 expression and initiate skewed differentiation toward Tfh cells (Fig. 3).
RHOA G17V mutations
RHOA mutations, most of which are missense and result in Gly to Val substitution at amino acid 17, are found in 50–70% of AITL [29,30,31,32, 34, 35]. RHOA mutations are seen at relatively high frequencies in other lymphomas such as ATLL [64] and pediatric Burkitt’s lymphoma [65], and diffuse-type gastric cancer [66]. In these neoplasms, however, mutations occur at hotspots different from Gly17. RHOAG17V mutations are reportedly confined to a microdissected PD1+ cell fraction enriched for cells with Tfh features, which are absent in the CD20+ cell fraction enriched for B cells [67], strongly indicating that RHOAG17V mutations are acquired after divergence of the T-cell fate from the B-cell fate. Wild-type RHOA functions as a small GTPase, a function lost in RHOAG17V due to loss of nucleotide binding capacity [29, 30, 35]. Instead, RHOAG17V acquires a neo-function, namely, binding capacity to VAV1 protein [68], which normally functions as a guanine nucleotide exchange factor (GEF) and GEF-independent adaptor in the TCR signaling pathway [69, 70]. Both GEF-dependent and -independent functions are regulated by tyrosine phosphorylation [68, 71, 72]. TCR-associated FYN and LCK are among candidate tyrosine kinases responsible for VAV1 phosphorylation [71, 73, 74]. Studies in Jurkat cells show that RHOAG17V increases TCR signaling through enhanced VAV1 phosphorylation at Tyr174 [68]. In vivo, this likely results in stronger TCR signaling input to RHOAG17V-expressing naive CD4+ T cells, which can contribute to preferential commitment to Tfh rather than non-Tfh lineages upon engagement with DCs [75].
Multiple mouse models established by retroviral transfer [76], knock in [77], or transgenic [78] techniques have been used to assess the effects of expression of RHOAG17V or its murine version RhoaG17V. Analysis of RhoaG17V knock in and RHOAG17V transgenic models in which either is driven by the CD4 promoter show that both RhoaG17V and RHOAG17V per se induce Tfh specification and differentiation in spleen [77, 78]. In the presence of RhoaG17V, Icos expression is induced in vitro by stimulation of CD4+ cells with irradiated antigen-presenting cells and anti-CD3 antibody [77]. ICOS functions as a co-stimulatory molecule and a migration receptor for Tfh cells, and its signaling is essential at both the initial step of Tfh specification [58, 79] and in maintenance of Tfh phenotypes [80]. RHOAG17V transgenic mice produce antibodies to double-stranded DNA and exhibit autoimmune-like phenotypes, such as skin rash and renal immune complex deposition [78]. RHOAG17V/RhoaG17V expression likely perturbs many other regulatory programs in CD4+ T cells; for example, it enhances CD4+ T-cell proliferation and reduces differentiation into T helper type 1 (Th1) cells [77]. The effect of RHOAG17V/RhoaG17V expression in CD4+ T cells on differentiation to regulatory T cells remains controversial [77, 78].
Taken together, the presence of RHOAG17V enhances both TCR and ICOS signaling possibly through independent mechanisms, specifies Tfh lineage, maintains Tfh phenotypes, and provokes autoimmune reactions, all reminiscent of outcomes associated with AITL and other Tfh lymphomas (Fig. 3). These outcomes may explain why RHOAG17V mutations are genetically selected specifically in Tfh lymphomas.
Combined Tet2 loss-of-function and RHOA G17V or Rhoa G17V expression—an AITL model
Tet2 disruption or RHOAG17V/RhoaG17V expression independently induces Tfh differentiation, as discussed in the two previous subsections. Although T-cell lymphoma with Tfh phenotypes is seen following Tet2 downregulation in mice, histology of lymphoma tissues does not resemble AITL [63]. It is thus of great interest to assess phenotypes in mice with combined Tet2 disruption and RHOAG17V or RhoaG17V expression. So far, using independent approaches to engineer such mouse models, three groups have reported development of lymphoma with Tfh features, and lymphoma tissue histology more resembling classical AITL, including increased numbers of blood vessels [77, 78, 81].
Comparison of these models provides insight into mechanisms underlying AITL development. In the first model [77], Tet2 disruption and knocked-in RhoaG17V expression were both driven by the mouse CD4 promoter using the Cre-lox system (CD4-Tet2-Rhoa), confining both mutations to the T-cell compartment. In the second model [78], Tet2 disruption was driven by the VAV1 promoter-Cre and transgenic RHOAG17V expression by the mouse CD4 promoter (VAV1-Tet2/CD4-RHOA); thus, Tet2 is disrupted in all hematopoietic cells, while RHOAG17V expression is confined to T cells. In the third model [81], Tet2 disruption was driven by the Mx promoter-Cre following pIpC injection and transgenic RHOAG17V expression was driven by the human CD2 promoter (Mx-Tet2/CD2-RHOA); thus, Tet2 disruption and RHOAG17V expression are controlled in largely the same manner as in the VAV1-Tet2/CD4-RHOA model.
In the first model, AITL-like lymphoma did not naturally develop: bone marrow transplantation from Tet2-Rhoa-CD4 mice to syngenic mice followed by immunization with sheep red blood cells (SRBCs) was required for development of AITL-like lymphoma in recipients [77]. By contrast, mice in the third model showed development of AITL-like lymphoma [81]. These outcomes suggest that, in patients, TET2 loss-of-function mutations in non-T-cell microenvironmental cells may be an important inducer of AITL in the context that RHOAG17V mutations are present in T cells. TET2 loss-of-function mutations in T cells might also contribute to AITL establishment in the presence of RHOAG17V mutations in T cells, if the host is exposed to strong immunization. SRBC immunization induces GC hyperplasia and, as described below, TET2 mutations in B cells may also contribute to GC hyperplasia [82]. Therefore, induction of GC hyperplasia could be a critical event in promoting AITL development.
In summary, both TET2 and RHOAG17V mutations can enhance Tfh differentiation from naïve CD4+ T cells. Signaling inducing GC hyperplasia is necessary and sufficient for AITL establishment in the context of enhanced Tfh differentiation. TET2 mutations alter transcription programs in both T and B cells, which may activate signaling that induces GC hyperplasia. Nevertheless, gaps remain in our understanding of GC hyperplasia (Fig. 2b) versus AITL (Fig. 3) pathology.
IDH2 R172 mutations
In AITL, IDH2R172 mutations occur at a frequency of 20–30% [22, 29, 31,32,33], while IDH1R132 and IDH2R140 mutations, which recur in AML [25, 83], are very rare. IDH enzymes normally convert isocitrate to α-ketoglutarate (αKG); AML/AITL-associated mutant IDH proteins acquire a neo-function leading to production of the oncometabolite 2-hydroxyglutarate (2HG) [25, 83, 84]. 2HG inhibits iron- and αKG-dependent dioxygenase activity [25]. TET2 and IDH1/2 mutations are found in a mutually exclusive manner in AML, and IDH1/2 mutations in AML promote a DNA hypermethylation signature similar that is associated with TET2 mutations. Therefore, TET2 is considered an important target of 2HG in AML [25].
In AITL, in contrast, IDH2R172 mutations are identified almost exclusively in samples that also harbor TET2 mutations [22, 29, 31,32,33], and cells in those samples generally show markedly increased H3K27me3 levels plus hypermethylation of promoter regions genome wide. Thus, histone demethylases are likely important targets of 2HG generated by IDH2R172S/M/G/K in AITL [32], as has been shown in glioma marked by IDH1R132H and IDH2R172K mutations [85].
In a mouse model, expression of IDH2R172K but not I IDH1R132H mutants increases 2HG levels in T cells, corresponding to the specificity of IDH2R172 mutations with T-cell lymphpoma [86]. Microdissection of clinical AITL samples has identified that IDH2R172 mutations are unique to PD1+ cells [67] and correspondingly, IDH2R172-specific antibody reacts specifically with ICOS+ cells [86]. It is thus thought that acquisition of IDH2R172 mutations occurs shortly before or after Tfh commitment.
Importantly, among Tfh lymphomas, IDH2R172 mutations are mostly confined to AITL and are rare in FTCL [37] and PTCL-NOS with Tfh phenotype or GEP [31, 33, 37, 38, 40], unlike recurrent mutations in TET2, DNMT3A, and RHOA, which are common to all types of Tfh lymphomas. IDH2R172 mutation-positive AITL cases define a discrete subpopulation in both GEP [32, 33] and genetic profiles [33]. IDH2R172 mutation-positive AITL cases generally show downregulation of Th1 differentiation-associated genes and upregulation of Tfh-associated genes [33]. Furthermore, IDH2R172 mutation-positive samples exhibit increased expression of vascular endothelial cell growth factor (VEGF) [33], which may be associated with increased vascularization seen in AITL and account for why this mutation is unique to AITL-specific histology. Genetically, a gain of chromosome 5 (Chr5) uniquely characterizes AITL and is seen in 43% (15/35) of samples [33]. IDH2R172 mutations are also associated with Chr5 gain (10/14 chr5 gain-positive cases vs. 4/18 negative cases). Interleukin-4 (IL-4) is localized on Chr5 and its expression increases in cases marked by Chr5 gain [33], an outcome corresponding to accelerated Tfh differentiation observed in IDH2R172 mutation-positive cases.
IDH2R172 mutations may be associated with enhanced Tfh features and histopathologic characteristics of AITL such as increased blood vessels, through changing epigenetic regulation and gene expression signatures (Fig. 3).
TCR-related gene variations
Variations in TCR-associated genes, such as PLCG1, CD28, VAV1, and FYN, are seen in approximately half of AITL cases [30, 33, 34]. Moreover, structural variations such as gene fusions have been identified for CD28, VAV1, and ITK in AITL samples [87, 88]. Given that strong TCR signaling in naïve CD4+ T cells dictates the choice toward a Tfh fate [75], these mutations may contribute to AITL development. However, most of them (with the exception of particular mutations and fusions of CD28 [33, 88]) are not specific to AITL or Tfh lymphomas but occur equally or even with greater frequency in PTCL-NOS [38] or ATLL [89]. Thus, their consequences likely differ significantly from mutations in TET2, RHOAG17V, and IDH2R172 with respect to AITL. Variations in TCR signaling pathway genes may broadly influence T-cell activities, such as proliferation (Fig. 3), resistance to apoptosis, migration, or cytokine production, and generally contribute to development of multiple PTCLs. Abnormal RHOAG17V-VAV1 signaling is seen in AITL and nPTCL-Tfh [68], but VAV1 mutations/fusions may play different roles in PTCLs other than Tfh lymphomas.
DNMT3A mutations
DNMT3A mutations in AITL occur at a frequency of 20–38.5% [29,30,31,32], a frequency similar to that seen in PTCL-NOS with Tfh phenotype/GEP [29, 32, 33, 38]. However, two recent studies report frequencies of DNMT3A mutations in PTCL-NOS categorized as PTCL-NOS-GATA3 or PTCL-NOS-TBX21 [45] as <15% [33, 38], indicating aberrant accumulation of these mutations in Tfh lymphomas. Currently, there is no evidence that DNMT3A mutations directly perturb Tfh cell specification or differentiation. Interestingly, most DNMT3A mutations in Tfh lymphomas occur together with TET2 mutations [29,30,31,32]. Tet2/Dnmt3a double-knockout mice show synergistic effects on expression of lineage-specifying transcription factor genes via epigenetic mechanisms [90]. Cooperation between Tet2 knockout and DNMT3AR882H expression is also shown by the transplantation experiment with the Tet2-null hematopoietic stem/progenitor cells retrovirally expressing DNMT3AR882H [91]. In this report, serial transplantation results in development of AITL-like disease. It will be of interest to determine whether DNMT3A mutations actually enhance the effect of TET2 mutations on promoting Tfh cell differentiation. DNMT3A and TET2 mutations are often co-identified in clonal hematopoietic cells in Tfh lymphoma patients [21, 29, 67]. Similar to the profile of DNMT3A mutations found in clonal hematopoiesis in healthy individuals, mutations observed in Tfh lymphoma patients do not accumulate in the DNMT3A methyltransferase domain, and hotspot R882(H) mutations are less frequent in AITL samples [29, 33, 38]. Overall, DNMT3A mutations should be involved in clonal hematopoiesis, but it still remains unclear whether they are directly involved in the Tfh specification or differentiation in Tfh lymphomas (Fig. 3).
RC3H1/Roquin and GAPDH—AITL mouse models not based on patient genetics
Clinical AITL samples do not show variations in the ring finger and CCCH-type domain 1 (RC3H1/ROQUIN) [92]; nonetheless, mice heterozygous for the Roquin missense allele Roquinsan develop AITL-like lymphoma with an approximately 50% penetrance [93]. Roquin is an RNA-binding ring-type ubiquitin ligase discovered in a mutagenesis screen for autoimmune phenotypes in mice [94]. Homozygous missense mutations (M199R) in Rc3h1/Roquin (san/san) cause systemic lupus erythematosus-like disease with marked generalized lymphadenopathy. Heterozygous san/+ mice, which develop asymmetric lymphadenopathy, have been analyzed as a lymphoma model [93]. Wild-type Roquin binds to Icos mRNA to promote its degradation, and mutant Roquin encoded by Roquinsan binds to Icos mRNA with a higher affinity than does the wild-type protein, leading to slower mRNA decay [95], aberrant expression of Icos on CD4+ T cells, and an excessive number of Tfh cells. san/+ mice show many features of human AITL such as hypergammaglobulinemia [93], and their analysis has contributed to recognition that enhanced ICOS signaling can promote AITL tumorigenesis. There are, however, no reports that ICOS overexpression per se causes AITL/Tfh lymphoma, and thus, it remains uncertain whether other factors downstream of Roquinsan contribute to AITL/Tfh lymphoma in concert with aberrant ICOS expression.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), one of ten enzymes functioning in aerobic glycolysis, has additional functions [96, 97]. It was recently reported that mice with GAPDH overexpression in T cells develop Tfh lymphoma with AITL-like features by the age of 18 months or older [98]. GAPDH reportedly activates the non-canonical nuclear factor kappa B (NFκB) pathway, a phenotype seen in these mice. As a consequence, CD4+ T cells undergo Tfh differentiation and express PD1. In GC B cells, the non-canonical NFκB pathway is activated through PD1–PDL1 ligation, which likely contributes to AITL-like lymphoma development in GAPDH-overexpressing mice. Non-canonical NFκB signaling is also activated in human AITL [98], although it is uncertain whether PD1–PDL1 ligation triggers such an activation in patients. GAPDH-overexpressing mice have also been used to test efficacy of an inhibitor of NFκB-inducing kinase (NIK), which activates non-canonical NFκB signaling. The efficacy of NIK inhibitors as treatment for AITL should be elucidated in clinical trials.
Microenvironmental cells derived from clonal hematopoiesis
With age, healthy individuals increasingly exhibit clonal blood cells harboring driver mutations, most frequently in DNMT3 or TET2 [99, 100]. Given that disruption of either Tet2 [20, 62] or Dnmt3a [101] in mice enhances HSC self-renewal, comparable loss-of-function mutations likely enhance self-renewal of human HSCs.
In AITL and other Tfh lymphomas, TET2 (and DNMT3) mutations are often (7 of 10 cases by 1 group [20, 21] and 4 of 7 cases by another [29]) identified in normal-appearing bone marrow cells, CD34+ cells, and colony-forming blood progenitor cells. These mutations are also reported in multiple lineages of tumor-infiltrating cells by two groups (16 of 17 AITL and PTCL cases by 1 group [67] and 6 of 10 AITL cases by another [102]). Therefore, it is anticipated that most cases of AITL show clonal hematopoiesis, with most carrying either TET2 or TET2/DNMT3A mutations. It remains a fundamental question whether TET2 and DNMT3A mutations function in multiple lineages of HSC progeny to drive AITL development. This possibility was discussed in part in the previous comparison of three AITL mouse models and is addressed in greater detail below.
B cells
Mice mutant in Tet2 in B cells, when immunized with SRBC, exhibit GC hyperplasia, possibly due to impaired exit of GC B cells from the GC light zone following failure to initiate cell-autonomous gene expression patterns [82]. Thus, GC B cells are retained longer in the GC center and likely have increased opportunities to interact with Tfh cells (Fig. 2b). When typical AITL is initiated, either a single Tfh cell or a CD4+ T cell specified to become a Tfh cell due to TET2 mutations likely acquires a RHOAG17V mutation and proliferates. Eventually, Tfh cells harboring both mutations interact with TET2-mutant activated antigen-specific B cells. Then, among scenarios proposed to explain AITL histopathology, follicles may burst for unknown reasons (Fig. 3). In analysis of B-cell-specific Tet2-deficient mice, differentiation of B cells to plasma cells is impaired via downregulation of the gene encoding the transcription factor Blimp-1 [82] (Fig. 2b). However, polyclonal gammopathy is an AITL hallmark. Indeed, plasma cells are often abundant in AITL tissue, and polyclonal plasmacytosis is sometimes seen in AITL patients [103, 104]. Therefore, a plasma cell differentiation impairment, which is seen in mice mutant in Tet2 in B cells, may be reversed in AITL (Fig. 3). Or, TET2 deficiency due to somatic mutation in AITL may affect the plasma cell differentiation differently from the Tet2-deficient mouse model, in which Tet2 is disrupted at the B-cell development during embryogenesis.
DCs and macrophages
DCs and macrophages are among infiltrating myeloid cells in AITL [47]. Tet2 activity resolves inflammation by actively and selectively repressing Il-6 expression in innate myeloid cells, including DCs and macrophages [105]. In this case, Il-6 repression is independent of 5-mC dioxygenase activity: Tet2 recruits histone deacetylase 2 at the late phase of inflammation in response to innate immune stimulation [105]. Although this activity is not directly related to AITL development, elevated Il-6 production is seen in a mouse AITL model [81]. That excess IL-6 may function in AITL development and be secreted by DCs and macrophages carrying TET2 mutations (Fig. 2c).
When cultured with low-density lipoprotein (LDL), macrophages from bone marrow of Tet2 knockout mice express higher levels of mRNAs encoding chemokines and cytokines such as Cxcl1, Cxcl2, Cxcl3, platelet factor 4 (Pf4), Il-1b, and Il-6 [106]. Indeed, mice with mature myeloid cell-specific Tet2 deficiency show enhanced atherosclerosis [106]. Macrophage and DC hyperactivity may not be confined to LDL stimulation: some microenvironmental stimulation may recapitulate activity of TET2-mutant macrophages and DCs and contribute to AITL development in lymph nodes.
T cells
A variety of T-cell subsets are also present in the microenvironment, and a substantial proportion of each subset is presumed to carry TET2 mutations or TET2 plus DNMT3A mutations. As expected, TET2 mutations are acquired before the TCRB rearrangement and multiple TCRB clones are produced from a progenitor carrying a single TET2 mutation [107]. T-cell functions are highly variable depending on the subset and the context. These variations may be further complicated by the TET2 and DNMT3A mutations, details of which remain to be clarified.
Other genetic or genomic abnormalities seen in B cells in AITL
EBV infection
In AITL cases with EBV+ B cells (which accounts for 66–91% of AITL cases [37, 40,41,42, 49]), EBER-ISH often detects virus in blastoid B cells or large B cells resembling Hodgkin/Reed–Sternberg (HRS) cells [108, 109]. Monoclonal or oligoclonal immunoglobulin (Ig) gene rearrangement is detected in up to one-third of AITL cases [47]. EBV infection and clonal expansion of B cells in AITL are often associated with each other. Such clonal B-cell growth may be further associated with development of diffuse large B-cell lymphoma (DLBCL) and other B-cell lymphomas, which are sometimes observed concomitantly with AITL (as a composite lymphoma [110]) or at a later phase of AITL. Indeed, tumor cells in B-cell lymphoma developing in association with AITL are often infected with EBV. Interestingly, EBV+ B cells reportedly lose functional Ig due to destructive hypermutations in most AITL cases accompanied by EBV+ B cells [108], indicating that B-cell proliferation in these cases does not require B-cell receptor (BCR) signaling but rather is driven by a different pathway (Fig. 3).
In de novo DLBCL, the frequencies of TET2 mutations were reported to be 5/48 (10.4%) and 9/27 (33%), and those of DNMT3A mutations 0/48 (0%) and 3/27 (11.1%) (p < 0.05 in both comparisons) in EBV− and EBV+ DLBCL, respectively [111]. All the three cases with DNMT3A mutations had TET2 mutations, the situation very similar to AITL. TET2 and DNMT3A mutations were the only ones which showed significant positive correlation with EBV positivity, while multiple gene mutations showed positive correlation with EBV negativity in DLBCL. It is thus reasonable to speculate that TET2 (and DNMT3A) mutations cooperate with EBV for DLBCL development, irrespectively of whether it is de novo or associated with AITL.
B cell-specific somatic mutations in AITL/PTCL-NOS
While HRS-like cells in AITL [109] and tumor cells in DLBCL, which is associated with AITL [112] are often infected with EBV, HRS-like cells in some AITL cases and tumor cells in some AITL-associated DLBCL cases can be EBV-negative. In these cases, it is reasonable to assume acquisition of genetic hits that recapitulate abnormal B-cell activities by EBV infection.
Somatic mutations identified by targeted sequencing in whole AITL and PTCL-NOS tissues were separately analyzed in a CD19+ cell fraction enriched for B cells and a PD1+ cell fraction enriched for tumor cells, both isolated by microdissection of individual samples [67]. Several mutations in TET2, DNMT3A, and other genes were identified in both fractions. RHOAG17V and IDHR172 mutations plus other mutations were exclusively identified in the PD1+ cell fraction. Interestingly, patient-specific NOTCH1 mutations were identified only in the CD19+ cell fraction in the three samples. In multiple B-cell malignancies such as chronic lymphocytic leukemia [113], mantle cell lymphoma [114], and DLBCL [115], activating NOTCH1 mutations are among major drivers. Targets of NOTCH signaling in this context are associated with BCR and cytokine signaling, as well as MYC pathways [116]. NOTCH1 mutations may be acquired when TET2 mutation-carrying clonal hematopoiesis cells differentiate into B cells and subsequently may enhance effects of TET2 mutations. Interactions of B cells harboring TET2 and NOTCH1 mutations with Tfh cells harboring TET2 and RHOA mutations likely mediate bidirectional signaling necessary for AITL development (Fig. 3).
B-cell-specific mutations and EBV infection may be important in AITL development. The relationship between the mutations and EBV infection, however, is still not known.
Non-hematopoietic microenvironmental cells in AITL
FDCs and HEVs
Increases in FDCs and HEVs are unique to AITL; thus, these two cell types are likely involved in AITL pathophysiology [117]. Both cell types are distinct from other immune cells, which are HSC progeny; FDCs are mesenchymal in origin and HEVs are endothelial.
In a normal GC reaction, FDCs provide antigens to GC B cells at the light zone, an activity required for affinity maturation of GC B cells [118]. GCs cannot be maintained without FDCs [19]. Mechanisms underlying abnormal FDC growth in AITL are not known. FDCs produce abundant IL-21, which may contribute to AITL development. At least in AITL cases with EBV infection, FDCs likely do not provide antigens to B cells, given that proliferation of EBV-infected B cells is independent of BCR signaling [108], as discussed. It is demonstrated that ICOSL-ICOS- phosphatidyl inositol 3-kinase (PI3K) pathway plays an important role for the growth of neoplastic Tfh cells in the AITL mouse model [77]. Because ICOSL is highly expressed in FDCs as well as B cells, activated monocytes, and mDCs [77], one of the potential functions of FDCs in AITL could be to stimulate ICOS in neoplastic Tfh cells.
VEGF overproduction is likely associated with the increase in HEVs, although how HEVs impact AITL establishment is obscure. Typically, HEVs, FDCs, and neoplastic Tfh cells localize near each other [47, 50]. Thus, cell-cell contact among them may be important for AITL establishment and development. Understanding the precise roles of FDCs and HEVs awaits development of new research tools, such as use of single cell analysis (Fig. 3).
AITL as an “immunodysplastic” syndrome
AITL was originally designated an immunodysplastic disease [4], and “immunodysplastic syndrome (LDS)” was proposed as a name for this disease [119]. The term “dysplastic” appears to have indicated both morphologic and functional features. The former indicated appearance of multiple kinds of abnormal cells in tumorous lymph nodes, including immunoblasts and clear cells (and may be HRS-like cells), as well as increased HEVs and FDCs. “Dysplastic” function was used to describe hypergammaglobulinemia and immunodeficiency. LDS was proposed to emphasize the contrast with myelodysplastic syndrome(s) (MDS). If myeloblasts are regarded as tumor cells in MDS, then low tumor cell burden is common to both MDS and AITL. In both diseases, most cellular components are mature HSC-derived non-tumor cells, including myeloid cells in MDS and both lymphoid and myeloid cells in AITL. These cells, originating from clonal hematopoiesis and often harboring TET2 mutations, provide an abnormal tumor microenvironment and likely function in development of both diseases.
Budding of mechanism-based treatments
The prognosis of AITL is generally poor as already discussed, though with some long-term survivors. There have been no satisfactory strategies including a variety of combination chemotherapies.
Introduction of rituximab
Based on the idea that environmental B cells could be an important contributor for AITL promotion and maintenance, a clinical trial of combination of rituximab and CHOP was performed [44]. In this phase II study (n = 25), an overall response rate was as high as 80% but the median response duration was 25 months in patients achieving a complete or unconfirmed complete response. It was thus concluded that the addition of rituximab to the standard CHOP did not show a clear benefit. This result, however, may not exclude the possibility of targeting B cells in the future clinical trials if suitable candidates for combination emerge.
Immune checkpoint inhibitors
Another one of the interests is immune checkpoint inhibitors. In the vast majority of AITL, the neoplastic Tfh cells express PD1, which may play a role for the Tfh cell migration [59] or NFκB activation in PDL1-expressing B cells [98]. Therefore, the significance of the blockade of PD1–PDL1 ligation in AITL may be different from the usual immune checkpoint inhibition in other neoplasms. A phase II clinical trial testing nivolmab, an anti-PD1 antibody, in patients with PTCL was halted because 4 of 12 patients evaluated in the interim analysis showed hyperprogressive disease (rapid progression within 1 cycle of treatment) [120]. Six of the 12 patients were those with AITL, among whom complete response was achieved in one. Hyperprogressive disease was indeed seen in at least one AITL patient [121]. This could indicate that PD1–PDL1 ligation is suppressing AITL, despite the speculation that PD1 plays a tumor promoting role. Studies in animal models might convey further information.
Hypomethylating agents
Hypomethylating agents (HMAs), 5-azacytidine (Aza), and decitabine, are approved for the treatment of MDS in many countries. HMAs counterbalance the DNA hypermethylation status caused by mutations in epigenetic regulators, such as TET2 and IDH2. Theoretically, therefore, HMAs can be effective in diseases carrying TET2 mutations. Actually, Aza is shown to be significantly more effective for MDS with TET2 mutations than for MDS without [122, 123]. Because the frequencies of TET2 mutations are much higher in AITL than in MDS, HMAs can be a good candidate as a medicine to treat AITL. Aza alone indeed showed a promising result in a retrospective study [124]; all 12 patients had TET2 mutations and 9 of the 12 responded to Aza. The response was sustained for >23 months in all the five patients achieving a complete response. The result of a phase II clinical trial designing a combination of oral Aza and romidepsin, a histone deacetylase inhibitor, was also promising for PTCL patients (overall response in 8 of 11; median duration of response not reached at the median follow-up period of 15.3 months), including some with AITL [125].
Dasatinib
As a mechanism how RHOAG17V mutations drive AITL, hyperphosphorylation of VAV1 [68] is already described. A multi-kinase inhibitor dasatinib inhibits the Src family tyrosine kinases including FYN and LCK, probable candidates responsible for VAV1 phosphorylation at the downstream of TCR. Based on the results showing the effect of dasatinib on the AITL mouse model, phase I clinical trial of dasatinib in AITL patients were performed and showed a promising result (4 of 5 responded to dasatinib) [81]. The efficacy should be shown in the next-step clinical trials.
Other possibilities
NIK inhibitors are already referred to [98]. Duvelisib, a PI3Kδ inhibitor that blocks ICOS-PI3K pathway, may be a candidate [77]. Many other possible agents, antibodies, etc. may emerge; they could target neoplastic Tfh cells, environmental cells, and both. Inhibition of cell-to-cell interaction can also be a right target.
Future perspectives
New technologies, such as single cell RNA sequencing and imaging clarifying cell identity and cell-to-cell communication in situ, will further dissect the pathophysiology of AITL. Such innovative research will eventually provide new treatment modalities, with which we can control this intractable disease better.
References
Grogg KL, Attygalle AD, Macon WR, Remstein ED, Kurtin PJ, Dogan A. Angioimmunoblastic T-cell lymphoma: a neoplasm of germinal-center T-helper cells? Blood. 2005;106:1501–2.
Dupuis J, Boye K, Martin N, Copie-Bergman C, Plonquet A, Fabiani B, et al. Expression of CXCL13 by neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL): a new diagnostic marker providing evidence that AITL derives from follicular helper T cells. Am J Surg Pathol. 2006;30:490–4.
De Leval L, Rickman DS, Thielen C, De Reynies A, Huang YL, Delsol G, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109:4952–63.
Suchi T. Atypical lymph node hyperplasia with fatal outcome—a report on the histopathological, immunological and clinical investigations of the cases. Recent Adv RES Res. 1974;14:13–34.
Frizzera G, Moran EMRH. Angioimmunoblastic lymphoadenopathy with dysproteinemia. Lancet. 1974;303:1070–3.
Lukes RJ, Tindle BH. Immunoblastic lymphadenopathy a hyperimmune entity resembling Hodgkin’s disease. N Engl J Med. 1975;292:1–8.
Nathwani BN, Rappaport H, Moran EM, Pangalis GA, Kim H. Immunoblastic lymphadenopathy. Cancer. 1978;41:578–606.
Shimoyama M, Minato K, Saito H, Takenaka T, Watanabe S, Nagatani T, et al. Immunoblastic lymphadenopathy (IBL)-like T-cell lymphoma. Jpn J Clin Oncol. 1978;9:347–56.
Stansfeld AG, Diebold J, Kapanci Y, Kelényi G, Lennert K, Mioduszewska O, et al. Updated Kiel classification for lymphomas. Lancet. 1988;331:292–3.
Suchi T, Lennert K, Tu LY, Kikuchi M, Sato E, Stansfeld AG, et al. Histopathology and immunohistochemistry of peripheral T cell lymphomas: a proposal for their classification. J Clin Pathol. 1987;40:995–1015.
Feller AC, Griesser H, Schiller CV, Wacker HH, Dallenbach F, Bartels H, et al. Clonal gene rearrangement patterns correlate with immunophenotype and clinical parameters in patients with angioimmunoblastic lymphadenopathy. Am J Pathol. 1988;133:549–56.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JKC, Cleary ML, et al. A revised European-American classification of lymphoid neoplasms. Blood. 1994;84:1361–92.
Jaffe ES, Ralfkiaer E. Angioimmunoblastic T-cell lymphoma. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. Pathology and genetics of tumours of haematoloietic and lymphoid tissues. WHO classification of tumours. 3rd ed. Lyon: IARC Press; 2001.
Dogan A, Gaulard P, Jaffe ES, Ralfkiaer E, Muller-Hermelink HK. Angioimmunoblastic T-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press; 2008.
Dogan A, Gaulard P, Jaffe ES, Muller-Hermelink HK, de Leval L. Angioimmunoblastic T-cell lymphoma and other nodal lymphomas of T follicular helper cell origin. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th ed. Lyon: IARC Press; 2017.
Nathwani BN, Jaffe ES. Angioimmunoblastic lymphadenopathy (AILD) and AILD-like T-cell lymphomas. In: Jaffe ES, editor. Surgical pathology of the lymph nodes and related organs. Philadelphila: Saunders; 1995. pp 390–412.
Attygalle A, Al-Jehani R, Diss TC, Munson P, Liu H, Du MQ, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627–33.
Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol. 2005;5:853–65.
Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50:1132–48.
Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38.
Couronné L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366:95–6.
Cairns RA, Iqbal J, Lemonnier F, Kucuk C, De Leval L, Jais JP, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–3.
Ito S, Dalessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Nangalia J, Green TR. The evolving genomic landscape of myeloproliferative neoplasms. Hematology. 2014;2014:287–96.
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171–5.
Palomero T, Couronné L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166–70.
Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293–6.
Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood. 2015;126:1741–52.
Heavican TB, Bouska A, Yu J, Lone W, Amador C, Gong Q, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood. 2019;133:1664–76.
Vallois D, Dobay MPD, Morin RD, Lemonnier F, Missiaglia E, Juilland M, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood. 2016;128:1490–502.
Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:371–5.
Lee SH, Kim JS, Kim J, Kim SJ, Kim WS, Lee S, et al. A highly recurrent novel missense mutation in CD28 among angioimmunoblastic T-cell lymphoma patients. Haematologica. 2015;100:e505–7.
Dobay MP, Lemonnier F, Missiaglia E, Bastard C, Vallois D, Jais JP, et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica. 2017;102:e148–51.
Watatani Y, Sato Y, Miyoshi H, Sakamoto K, Nishida K, Gion Y, et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia. 2019;33:2867–83.
Vose JM, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes international T-cell lymphoma project. J Clin Oncol. 2008;26:4124–30.
de Leval L, Parrens M, Le Bras F, Jais J-P, Fataccioli V, Martin A, et al. Angioimmunoblastic T-cell lymphoma is the most common T-cell lymphoma in two distinct French information data sets. Haematologica. 2015;100:e361–4.
Mourad N, Mounier N, Brière J, Raffoux E, Delmer A, Feller A, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the groupe d’etude des lymphomes de i’adulte (GELA) trials. Blood. 2008;111:4463–70.
Tokunaga T, Shimada K, Yamamoto K, Chihara D, Ichihashi T, Oshima R, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837–43.
Federico M, Rudiger T, Bellei M, Nathwani BN, Luminari S, Coiffier B, et al. Clinicopathologic characteristics of angioimmunoblastic t-cell lymphoma: Analysis of the international peripheral t-cell lymphoma project. J Clin Oncol. 2013;31:240–6.
Delfau-Larue MH, de Leval L, Joly B, Plonquet A, Challine D, Parrens M, et al. Targeting intratumoral B cells with rituximab in addition to CHOP in angioimmunoblastic T-cell lymphoma. A clinicobiological study of the GELA. Haematologica. 2012;97:1594–602.
Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123:2915–23.
Park SI, Horwitz SM, Foss FM, Pinter-Brown LC, Carson KR, Rosen ST, et al. The role of autologous stem cell transplantation in patients with nodal peripheral T-cell lymphomas in first complete remission: report from COMPLETE, a prospective, multicenter cohort study. Cancer. 2019;125:1507–17.
de Leval L. Approach to nodal-based T-cell lymphomas. Pathology. 2020;52:78–99.
Merchant SH, Amin MB, Viswanatha DS. Morphologic and immunophenotypic analysis of angioimmunoblastic T-cell lymphoma: emphasis on phenotypic aberrancies for early diagnosis. Am J Clin Pathol. 2006;126:29–38.
Hsi ED, Horwitz SM, Carson KR, Pinter-Brown LC, Rosen ST, Pro B, et al. Analysis of Peripheral T-cell Lymphoma Diagnostic Workup in the United States. Clin Lymphoma Myeloma Leuk. 2017;17:193–200.
De Leval L, Gisselbrecht C, Gaulard P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br J Haematol. 2010;148:673–89.
Karube K, Aoki R, Nomura Y, Yamamoto K, Shimizu K, Yoshida S, et al. Usefulness of flow cytometry for differential diagnosis of precursor and peripheral T-cell and NK-cell lymphomas: analysis of 490 cases. Pathol Int. 2008;58:89–97.
Singh A, Schabath R, Ratei R, Stroux A, Klemke C-D, Nebe T, et al. Peripheral blood sCD3(−) CD4(+) T cells: a useful diagnostic tool in angioimmunoblastic T cell lymphoma. Hematol Oncol. 2014;32:16–21.
Ree HJ, Kadin ME, Kikuchi M, Ko YH, Suzumiya J, Go JH. Bcl-6 expression in reactive follicular hyperplasia, follicular lymphoma, and angioimmunoblastic T-cell lymphoma with hyperplastic germinal centers: heterogeneity of intrafollicular T-cells and their altered distribution in the pathogenesis of angioimmuno. Hum Pathol. 1999;30:403–11.
Amé-Thomas P, Hoeller S, Artchounin C, Misiak J, Braza MS, Jean R, et al. CD10 delineates a subset of human IL-4 producing follicular helper T cells involved in the survival of follicular lymphoma B cells. Blood. 2015;125:2381–5.
Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–6.
Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–10.
Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–68.
Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–46.
Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 controls follicular T helper cell positioning and function. Immunity. 2018;49:264–74.e4.
Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–56.
Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24.
Muto H, Sakata-Yanagimoto M, Nagae G, Shiozawa Y, Miyake Y, Yoshida K, et al. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J. 2014;4:e264.
Nagata Y, Kontani K, Enami T, Kataoka K, Ishii R, Totoki Y, et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood. 2016;127:596–604.
Rohde M, Richter J, Schlesner M, Betts MJ, Claviez A, Bonn BR, et al. Recurrent RHOA mutations in pediatric Burkitt lymphoma treated according to the NHL-BFM protocols. Genes Chromosomes Cancer. 2014;53:911–6.
Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet. 2014;46:583–7.
Nguyen TB, Sakata-Yanagimoto M, Asabe Y, Matsubara D, Kano J, Yoshida K, et al. Identification of cell-type-specific mutations in nodal T-cell lymphomas. Blood Cancer J. 2017;7:1–10.
Fujisawa M, Sakata-Yanagimoto M, Nishizawa S, Komori D, Gershon P, Kiryu M, et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia. 2018;32:694–702.
Tybulewicz VLJ. Vav-family proteins in T-cell signalling. Curr Opin Immunol. 2005;17:267–74.
Saveliev A, Vanes L, Ksionda O, Rapley J, Smerdon SJ, Rittinger K, et al. Function of the nucleotide exchange activity of vav1 in T cell development and activation. Sci Signal. 2009;2:ra83.
Han J, Das B, Wei W, Van Aelst L, Mosteller RD, Khosravi-Far R, et al. Lck regulates Vav activation of members of the Rho family of GTPases. Mol Cell Biol. 1997;17:1346–53.
Bertagnolo V, Brugnoli F, Marchisio M, Celeghini C, Carini C, Capitani S. Association of PI 3-K with tyrosine phosphorylated Vav is essential for its activity in neutrophil-like maturation of myeloid cells. Cell Signal. 2004;16:423–33.
Huang J, Tilly D, Altman A, Sugie K, Grey HM. T-cell receptor antagonists induce Vav phosphorylation by selective activation of Fyn kinase. Proc Natl Acad Sci USA. 2000;97:10923–9.
Amarasinghe GK, Rosen MK. Acidic region tyrosines provide access points for allosteric activation of the autoinhibited Vav1 Db1 homology domain. Biochemistry. 2005;44:15257–68.
Ditoro D, Winstead C, Pham D, Witte S, Andargachew R, Singer JR, et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science. 2018;361:eaao2933.
Zang S, Li J, Yang H, Zeng H, Han W, Zhang J, et al. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J Clin Investig. 2017;127:2998–3012.
Cortes JR, Ambesi-Impiombato A, Couronné L, Quinn SA, Kim CS, da Silva Almeida AC, et al. RHOA G17V induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell. 2018;33:259–73.e7.
Ng SY, Brown L, Stevenson K, DeSouza T, Aster JC, Louissaint A, et al. RhoA G17V is sufficient to induce autoimmunity and promotes T-cell lymphomagenesis in mice. Blood. 2018;132:935–47.
Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity. 2015;42:239–51.
Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz LJ, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating krüppel-like factor 2. J Exp Med. 2015;212:217–33.
Nguyen TB, Sakata-Yanagimoto M, Fujisawa M, Nuhat ST, Miyoshi H, Nannya Y, et al. Dasatinib is an effective treatment for angioimmunoblastic T-cell lymphoma. Cancer Res. 2020;80:1875–84.
Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Béguelin W, Fontán L, et al. TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes b-cell lymphomagenesis. Cancer Discov. 2018;8:1633–53.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44.
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–8.
Lemonnier F, Cairns RA, Inoue S, Li WY, Dupuy A, Broutin S, et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci USA. 2016;113:15084–9.
Boddicker RL, Razidlo GL, Dasari S, Zeng Y, Hu G, Knudson RA, et al. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood. 2016;128:1234–45.
Yoo HY, Kim P, Kim WS, Lee SH, Kim S, Kang SY, et al. Frequent CTLA4-CD28 gene fusion in diverse types of T-cell lymphoma. Haematologica. 2016;101:757–63.
Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga JI, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47:1304–15.
Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016;48:1014–23.
Scourzic L, Couronne L, Pedersen MT, Della Valle V, Diop M, Mylonas E, et al. DNMT3A(R882H) mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia. 2016;30:1388–98.
Auguste T, Travert M, Tarte K, Ame-Thomas P, Artchounin C, Martin-Garcia N, et al. ROQUIN/RC3H1 alterations are not found in angioimmunoblastic T-cell lymphoma. PLoS ONE. 2013;8:e64536.
Ellyard JI, Chia T, Rodriguez-Pinilla S-M, Martin JL, Hu X, Navarro-Gonzalez M, et al. Heterozygosity for Roquinsan leads to angioimmunoblastic T-cell lymphoma-like tumors in mice. Blood. 2012;120:812–21.
Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–8.
Di YU, Tan AHM, Hu X, Athanasopoulos V, Simpson N, Silva DG, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature. 2007;450:299–303.
Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–97.
Jacquin MA, Chiche J, Zunino B, Bénéteau M, Meynet O, Pradelli LA, et al. GAPDH binds to active Akt, leading to Bcl-xL increase and escape from caspase-independent cell death. Cell Death Differ. 2013;20:1043–54.
Mondragón L, Mhaidly R, De Donatis GM, Tosolini M, Dao P, Martin AR, et al. GAPDH overexpression in the T cell lineage promotes angioimmunoblastic T cell lymphoma through an NF-κB-dependent mechanism. Cancer Cell. 2019;36:268–87.e10.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31.
Schwartz FH, Cai Q, Fellmann E, Hartmann S, Mäyränpää MI, Karjalainen-Lindsberg ML, et al. TET2 mutations in B cells of patients affected by angioimmunoblastic T-cell lymphoma. J Pathol. 2017;242:129–33.
Ahsanuddin AN, Brynes RK, Li S. Peripheral blood polyclonal plasmacytosis mimicking plasma cell leukemia in patients with angioimmunoblastic T-cell lymphoma: report of 3 cases and review of the literature. Int J Clin Exp Pathol. 2011;4:416–20.
Nagoshi H, Kuroda J, Kobayashi T, Maegawa S, Chinen Y, Kiyota M, et al. Clinical manifestation of angioimmunoblastic T-cell lymphoma with exuberant plasmacytosis. Int J Hematol. 2013;98:366–74.
Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–93.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–21.
Yao WQ, Wu F, Zhang W, Chuang SS, Thompson JS, Chen Z, et al. Angioimmunoblastic T-cell lymphoma contains multiple clonal T-cell populations derived from a common TET2 mutant progenitor cell. J Pathol. 2020;250:346–57.
Bräuninger A, Spieker T, Willenbrock K, Gaulard P, Wacker HH, Rajewsky K, et al. Survival and clonal expansion of mutating ‘forbidden’ (immunoglobulin receptor-deficient) Epstein–Barr virus-infected B cells in angioimmunoblastic T cell lymphoma. J Exp Med. 2001;194:927–40.
Nicolae A, Pittaluga S, Venkataraman G, Vijnovich-Baron A, Xi L, Raffeld M, et al. Peripheral T-cell lymphomas of follicular T-helper cell derivation with Hodgkin/Reed–Sternberg cells of B-cell lineage: both EBV- positive and EBV-negative variants exist. Am J Surg Pathol. 2013;37:816–26.
Suefuji N, Niino D, Arakawa F, Karube K, Kimura Y, Kiyasu J, et al. Clinicopathological analysis of a composite lymphoma containing both T- and B-cell lymphomas. Pathol Int. 2012;62:690–8.
Kataoka K, Miyoshi H, Sakata S, Dobashi A, Couronné L, Kogure Y, et al. Frequent structural variations involving programmed death ligands in Epstein–Barr virus-associated lymphomas. Leukemia. 2019;33:1687–99.
Willenbrock K, Bräuninger A, Hansmann M-L. Frequent occurrence of B-cell lymphomas in angioimmunoblastic T-cell lymphoma and proliferation of Epstein–Barr virus-infected cells in early cases. Br J Haematol. 2007;138:733–9.
Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer. 2016;16:145–62.
Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119:1963–71.
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-Cell lymphoma. N Engl J Med. 2018;378:1396–407.
Ryan RJH, Petrovic J, Rausch DM, Zhou Y, Lareau CA, Kluk MJ, et al. A B cell regulome links notch to downstream oncogenic pathways in small B cell lymphomas. Cell Rep. 2017;21:784–97.
Gaulard P, de Leval L. The microenvironment in T-cell lymphomas: emerging themes. Semin Cancer Biol. 2014;24:49–60.
Allen CDC, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202.
Nanba K, Sasaki N. Pathology of diffuse lesion of the lymph node with cellular atypism-a proposal for diffuse lymphoid dysplasia or lymphodysplastic syndrome. Nihon Ketsueki Gakkai Zasshi. 1987;50:1635–43.
Bennani NN, Pederson LD, Atherton P, Micallef I, Colgan JP, Thanarajasingam G, et al. A phase II study of nivolmab in patients with relapsed or regractory peripheral T-cell lymphoma. Blood. 2019;134 (supplement_1):467.
Bennett C. Hyperprogression halts study of nivolman in peripheral T-cell lymphoma. Cancer Ther. Advis. 2019. https://www.cancertherapyadvisor.com/home/news/conference-coverage/american-society-of-hematology-ash/ash-2019/nivolumab-tcell-lymphoma-hyperprogression-halts-study/. Accessed 17 Jun 2020.
Lin Y, Lin Z, Cheng K, Fang Z, Li Z, Luo Y, et al. Prognostic role of TET2 deficiency in myelodysplastic syndromes: a meta-analysis. Oncotarget. 2017;8:43295–305.
Cedena MT, Rapado I, Santos-Lozano A, Ayala R, Onecha E, Abaigar M, et al. Mutations in the DNA methylation pathway and number of driver mutations predict response to azacitidine in myelodysplastic syndromes. Oncotarget. 2017;8:106948–61.
Lemonnier F, Dupuis J, Sujobert P, Tournillhac O, Cheminant M, Sarkozy C, et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood. 2018;132:2305–9.
O’Connor OA, Falchi L, Lue JK, Marchi E, Kinahan C, Sawas A, et al. Oral 5-azacytidine and romidepsin exhibit marked activity in patients with PTCL: a multicenter phase 1 study. Blood. 2019;134:1395–405.
Acknowledgements
We thank N. Nakamura and Y. Hashimoto for helpful discussions about AITL history. The Japanese Society of Hematology is appreciated for providing the registry data.
Funding
A grant from MEXT, Japan (KAKENHI #19H03684) and grants from AMED, Japan (cm0106505h and lm0203097h) to SC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors are conducting a phase II clinical trial of dasatinib in AITL and other Tfh lymphoma patients with a drug supply from Bristol-Meyer Squibb that sells dasatinib.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chiba, S., Sakata-Yanagimoto, M. Advances in understanding of angioimmunoblastic T-cell lymphoma. Leukemia 34, 2592–2606 (2020). https://doi.org/10.1038/s41375-020-0990-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-0990-y
This article is cited by
-
Lymphoma-associated hemophagocytic lymphohistiocytosis (LA-HLH): a scoping review unveils clinical and diagnostic patterns of a lymphoma subgroup with poor prognosis
Leukemia (2024)
-
Allogenic and autologous anti-CD7 CAR-T cell therapies in relapsed or refractory T-cell malignancies
Blood Cancer Journal (2023)
-
Deciphering the spectrum of cutaneous lymphomas expressing TFH markers
Scientific Reports (2023)
-
DNMT3AR882H accelerates angioimmunoblastic T-cell lymphoma in mice
Oncogene (2023)
-
Involvement of spleen is associated with shorter survival in patients with angioimmunoblastic T cell lymphoma
Journal of Cancer Research and Clinical Oncology (2023)
