
Abstract
KIT D816 mutations (KIT D816mut) are strongly associated with systemic mastocytosis (SM) but are also detectable in acute myeloid leukemia (AML), where they represent an adverse prognostic factor in combination with core binding factor (CBF) fusion genes. Here, we evaluated the clinical and molecular features of KIT D816mut/CBF-negative (CBFneg) AML, a previously uncharacterized combination. All KIT D816mut/CBFneg cases (n = 40) had histologically proven SM with associated AML (SM-AML). Molecular analyses revealed at least one additional somatic mutation (median, n = 3) beside KIT D816 (e.g., SRSF2, 38%; ASXL1, 31%; RUNX1, 34%) in 32/32 (100%) patients. Secondary AML evolved in 29/40 (73%) patients from SM ± associated myeloid neoplasm. Longitudinal molecular and cytogenetic analyses revealed the acquisition of new mutations and/or karyotype evolution in 15/16 (94%) patients at the time of SM-AML. Median overall survival (OS) was 5.4 months. A screen of two independent AML databases (AMLdatabases) revealed remarkable similarities between KIT D816mut/CBFneg SM-AML and KIT D816mut/CBFneg AMLdatabases (n = 69) with regard to KIT D816mut variant allele frequency, mutation profile, aberrant karyotype, and OS suggesting underlying SM in a significant proportion of AMLdatabases patients. Bone marrow histology and reclassification as SM-AML has important clinical implications regarding prognosis and potential inclusion of KIT inhibitors in treatment concepts.
Similar content being viewed by others


Introduction
According to the World Health Organization (WHO) classification, advanced systemic mastocytosis (advSM) comprises aggressive SM, SM with an associated hematologic neoplasm (SM-AHN), and mast cell leukemia [1,2,3]. SM-AHN is the most frequent subtype diagnosed in up to 80% of advSM patients [4]. The AHN is characterized in >90% of patients as a myeloid neoplasm, e.g., myelodysplastic/myeloproliferative neoplasm unclassifiable (SM-MDS/MPN-u), chronic myelomonocytic leukemia (SM-CMML), myeloproliferative neoplasm (SM-MPN), myelodysplastic syndrome (SM-MDS), or acute myeloid leukemia (SM-AML) [4].
In general, acquired mutations in KIT (usually KIT D816V) are detectable in >90% of patients with SM, acknowledged to be most relevant for disease pathogenesis [5]. In advSM, multi-lineage involvement (including non-mast-cell-lineage cells, e.g., monocytes, eosinophils, and others) of KIT mutations is frequently observed and the basis for the phenotype of SM-AHN [6,7,8]. Recent data have, however, also highlighted that the molecular pathogenesis of advSM is much more complex with the presence of one or more additional somatic mutations, e.g., in SRSF2, ASXL1, RUNX1, JAK2, TET2 [9,10,11]. These additional mutations are often acquired by neoplastic (stem) cells prior to KIT D816V thereby indicating a multi-mutated stem cell disease and a step-wise process of oncogenesis [12].
Core binding factor (CBF) positive AML (CBFpos AML) represents 5–8% of all AMLs and is defined by the presence of a t(8;21)(q22;q22) and the associated RUNX1–RUNX1T1 fusion gene, or an inv(16)(p13.1q22)/t(16;16)(p13.1;q22) with the resulting CBFB–MYH11 fusion gene. CBFpos AML is categorized to the genetically favorable risk group. However, KIT. mutations, most frequently at position D816 (KIT D816mut), are detectable in up to 45% of CBFpos patients and associated with adverse prognosis [13, 14]. The potential association of KIT D816mut/CBFpos AML with underlying SM has been described in various case reports, case-series, and/or literature reviews [15,16,17,18,19], however, there is little information available on KIT D816mut/CBFneg AML [20]. We therefore evaluated (a) clinical and molecular genetic characteristics, (b) response to treatment, and (c) survival and prognostic factors in 40 patients with KIT D816mut/CBFneg AML collected at 4 centers of the European Competence Network on Mastocytosis (ECNM). To further investigate whether KIT D816mut/CBFneg defines a distinct AML subtype associated with SM and poor prognosis, two independent AML databases (AMLdatabases, German/Austrian AML Study Group, Munich Leukemia Lab) were retrospectively screened for KIT D816mut/CBFneg AML patients (selection criteria were all AML patients with available status on CBF and KIT D816mut).
Methods
Diagnosis of SM-AML
The diagnosis of SM-AML was established according to the WHO classification [2, 21,22,23]. Bone marrow biopsies and smears were evaluated by reference pathologists of the ECNM (H-PH and K Sotlar). A total of 48 CBFneg SM-AML patients, diagnosed in 4 ECNM centers between 2003 and 2018, were included in this retrospective analysis. Eight patients negative for KIT D816 mutations (n = 5) or with unknown KIT D816 mutation status (n = 3) were excluded. Among all SM-AML patient from the 4 ECNM centers, one patient was KIT D816mut/CBFpos. The study design adhered to the tenets of the Declaration of Helsinki and was approved by the institutional review board of the Medical Faculty of Mannheim, Heidelberg University, as part of the “German Registry on Disorders of Eosinophils and Mast Cells”. All patients gave written informed consent.
Molecular analyses
Targeted next-generation sequencing (NGS) was either performed by 454 FLX amplicon chemistry (Roche, Penzberg, Germany) or library preparation based on the TruSeq Custom Amplicon Low Input protocol (Illumina, San Diego, CA, USA) and sequencing on the MiSeq instrument (Illumina) to investigate mutation status of KIT and the following 32 genes: ASXL1, BCOR, CALR, CBL, CSNK1A1, DNMT3A, ETNK1, ETV6, EZH2, FLT3, GATA1, GATA2, IDH1, IDH2, JAK2, KRAS, MLL, MPL, NPM1, NRAS, PHF6, PIGA, PTPN11, RUNX1, SETBP1, SF3B1, SRSF2, TET2, TP53, U2AF1, ZRSR2, and WT1 [9].
Subsequent to bcl2fastq and demultiplexing, alignment and variant calling were performed using JSI SeqNext v4.4.0 (JSI Medical Systems, Kippenheim, Germany) software with default parameters. Only basecalls with quality score of 30 or above were considered for further processing. In median ~1800 reads were aligned to the target region. All regions below the minimal coverage of 400 reads were rejected and resequenced for higher depth. Variants were called with a variant allele frequency (VAF) cutoff of 3% and each assessed manually for pathogenicity. Mutation assessment was performed using COSMIC (v78), dbSNP (v150), ClinVar (2018-07), gnomAD (r2.0.2 and dbNSFP v3.5).
Qualitative and quantitative assessments of KIT D816V and KIT D816V expressed allele burden, respectively, was performed using allele-specific quantitative real-time reverse transcriptase polymerase chain reaction analyses (qRT-PCR) as previously described [24]. Molecular analyses were performed at diagnosis of SM ± AHN and at diagnosis of SM-AML.
Conventional cytogenetic analysis and fluorescence in situ hybridization
Cytogenetic analyses of at least 20 Giemsa-banded bone marrow metaphases (24 h and/or 48 h culture) was performed and interpreted according to the International System for Human Cytogenetic Nomenclature [25]. If necessary, chromosome banding analysis was combined with fluorescence in situ hybridization according to the manufacturer's instructions (Metasystems, Altlussheim, Germany) [26].
Statistical analyses
Statistical analyses considered clinical, laboratory, or molecular parameters obtained at the time of diagnosis. Overall survival (OS) analysis was determined as time from date of diagnosis to date of death or last follow up. Pearson correlation analysis was performed for the correlation between two parameters. Differences in the distribution of continuous variables between categories were analyzed by Mann–Whitney test (for comparison of two groups). For categorical variables, Fisher’s exact test was used. OS probabilities were estimated with the Kaplan–Meier method and compared by the log-rank test in univariate analysis. For the estimation of hazard ratios (HRs) and multivariate analysis, the Cox proportional hazard regression model was used. P-values < 0.05 (two-sided) were considered significant. There was no adjustment for multiple testing as all analyses were explorative. SPSS version 22.0.0 (IBM Corporation, Armonk, NY, USA) was used for statistical analysis.
Results
Clinical and morphological characteristics
The median age of the 40 KIT D816mut/CBFneg SM-AML patients was 65 years (range 28–83, male 73%). The median percentage of mast cells in bone marrow trephine biopsies was 10% (range 5–65). Blood parameters analyzed in this study included: leukocytes (median 8.7 × 109/L, range 0.5–71.8), hemoglobin (median 8.3 g/dL, range 5.1–14.3; <10 g/dL in 79% of patients), platelets (median 40 × 109/L, range 5–412; <100 × 109/L in 88% of patients), eosinophils (0.2 × 109/L, range 0–16.7; > 1.0 × 109/L in 18% of patients), and monocytes (0.9 × 109/L, range 0.1–23.5; >1.0 × 109/L in 39% of patients). Median serum tryptase level (normal value < 11.4 µg/L) was 92 µg/L (range 13–885; >100 µg/L in 48% of patients). Signs of non-hematologic organ dysfunction included elevated alkaline phosphatase (AP, normal value < 130 U/L; median 145 U/L; range 52–1428; >150 U/L in 50% of patients), splenomegaly (64%), and ascites (25%) (Table 1a).
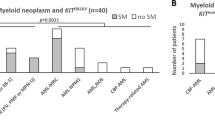
De novo SM-AML and secondary SM-AML
De novo SM-AML was diagnosed in 11/40 (28%) patients. Secondary SM-AML evolving from indolent SM (n = 5) or SM-AHN (n = 24) was observed in 29/40 (73%) patients with a median time to progression of 24 months (range 2–116). The 24 patients with AHN were classified as MDS/MPN-u (n = 8), CMML (n = 6), MDS (n = 5), or MPN associated with eosinophilia (MPN-eo) (n = 5) (Table 1a). The comparison between de novo and secondary AML revealed that patients with secondary AML were older, had a higher monocyte count, a higher AP level, and a lower serum tryptase level. However, there were no significant differences regarding OS (P = 0.2).
Somatic mutations
All patients were positive for KIT D816V with a median VAF of 36% (range 3–54). At the time of SM-AML, material for NGS analysis was available from 32/40 (80%) patients (Fig. 1a, Supplementary Table 1). All 32 patients had at least one additional somatic mutation (median 3, range 1–6) and 24/32 (75%) patients had ≥2 somatic mutations in addition to KIT D816V (Fig. 1b). There was a significant association between several pairs of mutations, specifically TET2/SRSF2, IDH1/2/SRSF2, IDH1/2/BCOR, and DNAMT3A/BCOR (P < 0.05) (Fig. 1c). The most frequently mutated genes were SRSF2 (n = 12, 38% of patients), RUNX1 (n = 11, 34%), TET2 (n = 11, 34%), ASXL1 (n = 10, 31%), NPM1 (n = 7, 22%), DNMT3A (n = 5, 16%), IDH1/2 (n = 5, 16%), N/KRAS (n = 4, 13%), BCOR (n = 3, 9%), SF3B1 (n = 3, 9%), SETBP1 (n = 2, 6%), TP53 (n = 2, 6%), and JAK2 (n = 2, 6%). CBL, EZH2, FLT3, MLL, MPL, PTPN11, and U2AF1 were less frequently affected (<5%) (Fig. 2b).

Mutational profile of 32 patients with KIT D816mut/CBFneg systemic mastocytosis associated with acute myeloid leukemia (SM-AML). a Alignment of gene mutations in 32 patients with SM-AML. Each column represents an individual patient, b distribution of number of affected genes, and c the co-occurrence and overall frequency of mutated genes represented by Circos diagram. Asterisk marks a significant association between several pairs of mutations. Supplementary Table 1 provides the variant allele frequency of all mutations

KIT D816 variant allele frequency (VAF), somatic mutations, and aberrant karyotype in KIT D816mut/CBFneg SM-AML in comparison to KIT D816mut/CBFneg AML from the two databases (AMLdatabases). a KIT D816 VAF, b relative frequency distribution of mutated genes, and c aberrant karyotype. Gray columns: KIT D816mut/CBFneg SM-AML and blue columns: KIT D816mut/CBFneg AMLdatabases. Asterisk represents targeted next-generation sequencing was performed in 32/40 SM-AML patients
At least one somatic mutation in SRSF2, ASXL1, and/or RUNX1 (S/A/Rpos) was identified in 21/32 (66%) patients. The rate of S/A/Rpos patients was significantly higher in secondary AML (20/23, 87%) as compared to de novo AML (1/9, 11%, P = 0.0001). Furthermore, there was a significant correlation between S/A/Rpos and age >60 years (P = 0.02).
To further evaluate whether KIT D816mut occurred in hematopoietic progenitor cells, we performed molecular analyses on DNA derived from CD34+ cells from 6 KIT D816V positive patients. KIT D816V was found in 1/6 (17%) patients while additional somatic mutations were detected in all 6 patients.
Cytogenetic analyses
At diagnosis of SM-AML, 19/40 (48%) patients had a normal and 21/40 (52%) patients an aberrant karyotype. All patients were CBFneg. Intermediate-risk and poor-risk AML karyotype were diagnosed in 7/21 (33%) and 14/21 (67%) patients, respectively (Fig. 2c, Supplementary Table 2) [27].
Longitudinal molecular and cytogenetic analyses in patients with secondary SM-AML
In 16/29 (55%) patients with secondary SM-AML, material from the time of diagnosis of SM ± AHN and from the time of diagnosis of secondary SM-AML was available for molecular and cytogenetic analyses. At the time of SM ± AHN, 11/16 (69%) patients were S/A/Rpos (Table 2). Acquisition of new somatic mutations and/or karyotype evolution at the time of secondary SM-AML was observed in 15/16 (94%) patients: 4 patients revealed acquisition of new somatic mutations (NPM1, n = 2; IDH2, n = 1; JAK2, n = 1) without karyotype evolution, 5 patients with karyotype evolution, and 6 patients with acquisition of new somatic mutations (TP53, n = 2; NPM1, n = 1; RUNX1, n = 1; ASXL1, n = 1; BCOR, n = 1; IDH1/2, n = 1) and karyotype evolution (Table 2, Supplementary Table 3).
Treatment modalities and response rate
Thirty-one of 40 (78%) patients were treated with intensive (induction) chemotherapy (n = 24, e.g., daunorubicin/cytarabine [DA, 7 + 3], mitoxantrone/cytarabine [S-HAM]) ± non-intensive therapy (hypomethylating agents, n = 8, ± cladribine, n = 2). The complete response (CR) rates after intensive induction chemotherapy and non-intensive therapy were 40% and 0, respectively. Two patients had cytarabine-based consolidation (without allogeneic stem cell transplantation [SCT]) and are alive 91 and 15 months, retrospectively, after diagnosis of SM-AML. Allogeneic SCT was performed in 12/40 (30%) patients with 4 patients being in CR prior to allogeneic SCT. A durable CR was achieved by 6/12 patients (50%). Nine of 40 (22%) patients received only best supportive care due to advanced age ± comorbidity.
S/A/Rpos ± presence of a poor-risk karyotype were negative predictive markers for response to treatment (intensive chemotherapy ± allogeneic SCT) with 10/11 (91%) non-responders presenting with S/A/Rpos ± poor-risk karyotype. On the other hand, 4/8 (50%) responders were S/A/Rpos ± poor-risk karyotype (P = 0.04) indicating that intensive treatment should not be withheld in this subgroup.
Comparison of KIT D816mut/CBFneg SM-AML with KIT D816mut/CBFneg AML from two independent databases
To further investigate whether KIT D816mut/CBFneg AML represents a distinct subtype which is associated with SM and poor prognosis, two independent AML databases (AMLdatabases) were retrospectively screened for KIT D816mut/CBFneg AML patients. Overall, 69 KIT D816mut/CBFneg AMLdatabases patients could be identified. Mutation profile and karyotype were available from all patients, detailed clinical characteristics from 17/69 patients (Tables 1b and 3).
This comparison revealed remarkable molecular and karyotype similarities between the KIT D816mut/CBFneg SM-AML and the KIT D816mut/CBFneg AMLdatabases cohort (Fig. 2a–c, Table 3): (a) The median KIT D816 VAF was 34% (range 3–54) and 29% (range 3–93), respectively, (b) with the exception of SRSF2 (38% vs. 18%), the frequency of the most frequently somatic mutations (RUNX1, TET2, ASXL1, NPM1, DNMT3A, IDH1/2) was highly similar between the two groups, (c) in contrast to de novo AML, the frequency of FLT3 aberrations was very low (3% and 7%, respectively), and (d) the frequency of an aberrant karyotype was 52% and 42, respectively, with a comparable rate of intermediate-risk and poor-risk karyotype.
The median OS of 40 KIT D816mut/CBFneg SM-AML and 17 evaluable KIT D816mut/CBFneg AMLdatabases patients was 5.4 (95% confidence interval, CI [1.7–9.1]) and 26.4 (95% CI [0–61.0]) months (P = 0.015), respectively. In the KIT D816mut/CBFneg SM-AML cohort, 16 patients received non-intensive therapy only, with a median OS of 2.7 months (95% CI [1.5–3.9]), while all 17 KIT D816mut/CBFneg AMLdatabases patients received intensive chemotherapy. Median OS was not significantly different (16.7 vs. 26.4 months, P = 0.4) between KIT D816mut/CBFneg SM-AML and KIT D816mut/CBFneg AMLdatabases patients who received intensive chemotherapy (Fig. 3a, b).

Kaplan–Meier estimates of overall survival (OS) of KIT D816mut/CBFneg SM-AML and AML from the databases (AMLdatabases). a OS of all KIT D816mut/CBFneg patients, b OS comparing the KIT D816mut/CBFneg SM-AML cohort with intensive chemotherapy (ICT) ± allogeneic stem cell transplantation (SCT) (yellow), the KIT D816mut/CBFneg AMLdatabases cohort with ICT ± allogeneic SCT (green), and the KIT D816mut/CBFneg SM-AML with non-intensive therapy (NIT)/best supportive care (BSC) (red), c OS of all KIT D816mut/CBFneg patients treated with ICT only (blue) or with allogeneic SCT (gray). CI confidence interval, n.s. non-significant. Asterisk refers to included patients with SM-AML and AMLdatabases
In a combined analysis of both cohorts, median OS was not significantly different between intensive chemotherapy (n = 22) only vs. intensive chemotherapy followed by allogeneic SCT (n = 19), 10.2 months (95% CI [5.9–14.4]) vs. 26.5 months (95% CI [0–58.5]), respectively (P = 0.3) (Fig. 3c). With exception of age (patients with allogeneic SCT were younger), no significant differences were observed between the two cohorts regarding clinical and molecular genetic characteristics (Table 4).
In univariate analyses (including age, hemoglobin, platelets, AML subtype, treatment modalities [non-allogeneic vs. allogeneic SCT], somatic mutations, and aberrant karyotype), only age >60 years, at least one additional somatic mutation in the S/A/R gene panel (S/A/Rpos) and a poor-risk karyotype were identified as poor prognostic variables regarding OS. In multivariate analysis, S/A/Rpos and a poor-risk karyotype remained the only independent adverse factors with regard to OS. Accordingly, a weighted score (based on the HR) of 1 was assigned to S/A/Rpos and poor-risk karyotype. Significantly different OS probabilities were observed for the comparisons S/A/Rneg + normal-/intermediate-risk karyotype (0 point, n = 14), S/A/Rpos or poor-risk karyotype (1 point, n = 23), and S/A/Rpos + poor-risk karyotype (2 points, n = 10) with median OS not reached vs. 14.0 [6.2–21.8] vs. 7.0 months [4.5–9.6] (P = 0.001). These results were independent of treatment modalities (Fig. 4a, b).

Kaplan–Meier estimates of overall survival (OS) of KIT D816mut/CBFneg SM-AML and AMLdatabases. OS of KIT D816mut/CBFneg patients treated with a non-intensive therapy (NIT)/best supportive care (BSC) or intensive chemotherapy (ICT) ± allogeneic stem cell transplantation (SCT) or b ICT ± allogeneic SCT. Depending on the SRSF2/ASXL1/RUNX1 (S/A/R) mutation status and karyotype, three different cohorts were identified: S/A/Rneg + normal-/intermediate-risk karyotype (green), S/A/Rpos or poor-risk karyotype (yellow), and S/A/Rpos + poor-risk karyotype (red). CI confidence interval, n.s. non-significant, S/A/Rpos at least one mutation in the S/A/R gene panel. Asterisk refers to included patients with SM-AML and AMLdatabases
Discussion
We report here on a large series of 40 patients with morphologically proven KIT D816mut/CBFneg SM-AML. Approximately 65% of patients evolved from other advSM subtypes. Similar to previous reports concerning the molecular profile of advSM, all patients with SM-AML had at least one additional somatic mutation, most frequently affecting TET2, SRSF2, ASXL1, RUNX1, and NPM1. In contrast to de novo AML, only one patient had a FLT3 mutation. The overall molecular profile of SM-AML therefore was more similar to the profile of advSM than to that of de novo AML [28].
Using CFU-GM-colonies and microdissected cells, we have previously shown that mast cells and AHN cells are not only positive for KIT D816V but also for additional somatic mutations, indicating that both derive from a common progenitor [12]. However, a significant proportion of colonies were positive for additional somatic mutations but negative for KIT D816V [12]. In line with this and other data demonstrating the absence of KIT D816V in myeloid blasts of 50% of SM-AML cases [20], we confirmed the absence of KIT D816V but the presence of additional somatic mutations in CD34+ cells in 5 of 6 SM-AML cases, indicating that the additional somatic mutations rather than KIT D816V are the driving force for progression to secondary AML. In addition, serial molecular genetic analyses revealed the acquisition of new somatic mutations, e.g., in NPM1, IDH1/2, RUNX1, with or without karyotype evolution in >90% of patients as further underlying mechanisms for progression to secondary SM-AML. This data is reminiscent of reports on progression in other myeloid neoplasms such as MDS or MDS/MPN, and our previous reports on progression of SM to advSM or progression within advSM subtypes, e.g., to secondary mast cell leukemia, in which somatic mutations in NPM1, IDH2, or RUNX1 were also identified as late events and drivers for disease progression [29,30,31,32,33,34,35].
SM-AHN is the most common subtype of advSM but the diagnosis is challenging because the mast cell infiltrate may obscure the AHN and vice versa [20, 36,37,38]. This is particularly true for AML where the morphological but not the histological examination of bone marrow has been established as a standard diagnostic tool. Recently reported data collected from deep targeted sequencing indicated that KIT D816 mutations can be identified in 1–6% of patients with various subtypes of myeloid neoplasms, e.g., MDS, MDS/MPN-u, CMML, polycythemia vera, essential thrombocythemia, or myelofibrosis [39,40,41,42,43,44]. However, many of these cases have not routinely been screened by histopathology for the presence of co-existing SM. Within our registry, all KIT D816Vmut patients, who had initially been diagnosed as myeloid neoplasms such as CMML, triple-negative MF, and others, in fact fulfilled the WHO-criteria for a diagnosis of SM-AHN.
We therefore sought to investigate the incidence of KIT D816mut/CBFneg in retrospective screens of two independent AML databases. Rather unexpectedly, 69 patients were identified which revealed remarkable similarities concerning the high KIT D816 VAF, the mutation profile and the aberrant karyotype (Table 2), suggesting that the vast majority of these AMLdatabases patients are likely to have SM-AML. Unfortunately, the lack of bone marrow trephine biopsies at initial diagnosis of AML has not allowed a definite re-evaluation of these cases and formal reclassification as SM-AML. However, based on our data, which are in line with previously published results, an underlying or concomitant SM can be diagnosed in most cases of KIT D816Vmut AML, when the bone marrow is investigated using standard histopathological and molecular studies.
The median OS of the 40 SM-AML patients was 5.4 months and thus even worse as compared to patients with mast cell leukemia, which is defined by the presence of ≥20% mast cells in a bone marrow smear [29]. No patient achieved a CR on treatment with hypomethylating agents and none of the patients was treated with midostaurin. Following intensive induction chemotherapy in eligible patients, the CR rate of 40% was significantly inferior as compared to the general CR rate of de novo AML (70–80%) [45] and median survival following intensive chemotherapy with or without allogeneic SCT was 17 months. In addition to the aforementioned similarities regarding the molecular genetic characteristics (KIT D816V VAF, additional somatic mutations, and aberrant karyotype), the poor median OS of 26 months in 17 KIT D816mut/CBFneg AML patients from the two independent AML databases adds further evidence that KIT D816mut/CBFneg AML may in fact represent SM-AML in the vast majority, if not all patients. Independently of treatment modalities and consistent with previous reports on other advSM subtypes, e.g., mast cell leukemia, mutations in S/A/R and a poor-risk karyotype conferred an adverse impact on response to treatment, disease progression, and OS [10, 29, 30].
Midostaurin, an orally administered multi-kinase/FLT3-/KIT-inhibitor improves survival in FLT3pos AML and achieves overall response rates of 60% in patients with advSM [46, 47]. Better survival is observed in advSM patients without additional somatic mutations in the S/A/R gene panel and a >25% reduction of the KIT D816V VAF at month 6 [30, 46,47,48,49]. If the presence of SM can be proven in KIT D816mut/CBFneg AML by bone marrow histology and elevated serum tryptase, KIT inhibitors (e.g., midostaurin, potentially avapritinib [BLU-285, Blueprint Medicines, Cambridge, MA, USA]) in combination with intensive chemotherapy and allogeneic SCT may help to improve the poor prognosis of this distinct AML subtype [50, 51].
We conclude that (a) progression to secondary AML from a preceding KIT D816mut SM-AHN is frequently observed and may be triggered by the acquisition of additional somatic mutations with or without karyotype evolution, (b) KIT D816mut/CBFneg AML is a distinct subtype with remarkable similarities compared to SM-AML cases concerning KIT D816 VAF mutation profile, aberrant karyotype, and poor prognosis, suggesting that a significant proportion of these AML patients may in fact have SM-AML, which is a strong argument to propose a new evaluation, (c) with its very high positive and negative predictive value, serum tryptase is an excellent screening marker for SM and should therefore be part of the diagnostic workflow in all AML patients. Cases with an elevated serum tryptase level should subsequently be screened for KIT D816mut, and (d) bone marrow histology is mandatory in KIT D816mut patients. This simple diagnostic procedure will allow reclassification to SM-AML and thus allow inclusion of KIT inhibitors in established treatment modalities of AML.
References
Valent P, Akin C, Escribano L, Fodinger M, Hartmann K, Brockow K, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37:435–53.
Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25:603–25.
Theoharides TC, Valent P, Akin C. mast Cells, mastocytosis, and related disorders. N Engl J Med. 2015;373:163–72.
Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612–24.
Gleixner KV, Mayerhofer M, Cerny-Reiterer S, Hormann G, Rix U, Bennett KL, et al. KIT-D816V-independent oncogenic signaling in neoplastic cells in systemic mastocytosis: role of Lyn and Btk activation and disruption by dasatinib and bosutinib. Blood. 2011;118:1885–98.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, Sanchez ML, Nunez R, Prados A, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108:2366–72.
Sotlar K, Marafioti T, Griesser H, Theil J, Aepinus C, Jaussi R, et al. Detection of c-kit mutation Asp 816 to Val in microdissected bone marrow infiltrates in a case of systemic mastocytosis associated with chronic myelomonocytic leukaemia. J Clin Pathol Mol Pathol. 2000;53:188–93.
Sotlar K, Colak S, Bache A, Berezowska S, Krokowski M, Bultmann B, et al. Variable presence of KITD816V in clonal haematological non-mast cell lineage diseases associated with systemic mastocytosis (SM-AHNMD). J Pathol. 2010;220:586–95.
Schwaab J, Schnittger S, Sotlar K, Walz C, Fabarius A, Pfirrmann M, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122:2460–6.
Jawhar M, Schwaab J, Schnittger S, Meggendorfer M, Pfirrmann M, Sotlar K, et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia. 2016;30:136–43.
Pardanani A, Lasho T, Elala Y, Wassie E, Finke C, Reichard KK, et al. Next-generation sequencing in systemic mastocytosis: Derivation of a mutation-augmented clinical prognostic model for survival. Am J Hematol. 2016;91:888–93.
Jawhar M, Schwaab J, Schnittger S, Sotlar K, Horny HP, Metzgeroth G, et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia. 2015;29:1115–22.
Kim HJ, Ahn HK, Jung CW, Moon JH, Park CH, Lee KO, et al. KIT D816 mutation associates with adverse outcomes in core binding factor acute myeloid leukemia, especially in the subgroup with RUNX1/RUNX1T1 rearrangement. Ann Hematol. 2013;92:163–71.
Yui S, Kurosawa S, Yamaguchi H, Kanamori H, Ueki T, Uoshima N, et al. D816 mutation of the KIT gene in core binding factor acute myeloid leukemia is associated with poorer prognosis than other KIT gene mutations. Ann Hematol. 2017;96:1641–52.
Hilmi FAI, Al-Sabbagh A, Soliman DS, Sabah HA, Ismail OM, Yassin M, et al. Acute myeloid leukemia with Inv(16)(p13q22) associated with hidden systemic mastocytosis: case report and review of literature. Clin Med Insights Blood Disord. 2017;10. https://doi.org/10.1117/1179545X17700858.
Escribano L, Garca-Montero A, Nunez-Lopez R, Lopez-Jimenez J, Almeida J, Prados A, et al. Systemic mastocytosis associated with acute myeloid leukemia: case report and implications for disease pathogenesis. J Allergy Clin Immunol. 2004;114:28–33.
Cornet E, Dumezy F, Roumier C, Lepelley P, Jouy N, Philippe N, et al. Involvement of a common progenitor cell in core binding factor acute myeloid leukaemia associated with mastocytosis. Leuk Res. 2012;36:1330–3.
Pullarkat V, Bedell V, Kim Y, Bhatia R, Nakamura R, Forman S, et al. Neoplastic mast cells in systemic mastocytosis associated with t(8;21) acute myeloid leukemia are derived from the leukemic clone. Leuk Res. 2007;31:261–5.
Pullarkat ST, Pullarkat V, Kroft SH, Wilson CS, Ahsanuddin AN, Mann KP, et al. Systemic mastocytosis associated with t(8;21)(q22; q22) acute myeloid leukemia. J Hematop. 2009;2:27–33.
Jawhar M, Schwaab J, Horny HP, Sotlar K, Naumann N, Fabarius A, et al. Imact of centralized evaluation of bone marrow histology in systemic mastocytosis. Eur J Clin Invest. 2016;46:392–7.
Horny HPAC, Metcalfe DD, Swerdlow SH, Campo E, Harris NL, et al. World Health Organization (WHO) classification of tumours. Mastocytosis (Mast cell disease). Pathology & genetics. Tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008. p. 54–63. Vol. 2.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;2391–405.
Pardanani A. Systemic mastocytosis in adults: 2017 update on diagnosis, risk stratification and management. Am J Hematol. 2016;91:1146–59.
Erben P, Schwaab J, Metzgeroth G, Horny HP, Jawhar M, Sotlar K, et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis. Ann Hematol. 2014;93:81–8.
Simons A, Shaffer LG, Hastings RJ. Cytogenetic nomenclature: changes in the ISCN 2013 compared to the 2009 Edition. Cytogenet Genome Res. 2013;141:1–6.
Schoch C, Schnittger S, Bursch S, Gerstner D, Hochhaus A, Berger U, et al. Comparison of chromosome banding analysis, interphase- and hypermetaphase-FISH, qualitative and quantitative PCR for diagnosis and for follow-up in chronic myeloid leukemia: a study on 350 cases. Leukemia. 2002;16:53–9.
Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Jawhar M, Schwaab J, Meggendorfer M, Naumann N, Horny HP, Sotlar K, et al. The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica. 2017;102:1035–43.
Jawhar M, Schwaab J, Naumann N, Horny HP, Sotlar K, Haferlach T, et al. Response and progression on midostaurin in advanced systemic mastocytosis: KIT D816V and other molecular markers. Blood. 2017;130:137–45.
Naumann N, Jawhar M, Schwaab J, Kluger S, Lubke J, Metzgeroth G, et al. Incidence and prognostic impact of cytogenetic aberrations in patients with systemic mastocytosis. Genes Chromosomes Cancer. 2018;57:252–9.
Mossner M, Jann JC, Wittig J, Nolte F, Fey S, Nowak V, et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood. 2016;128:1246–59.
Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–8.
Padron E, Abdel-Wahab O. Importance of genetics in the clinical management of chronic myelomonocytic leukemia. J Clin Oncol. 2013;31:2374–6.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27.
Bernd HW, Sotlar K, Lorenzen J, Osieka R, Fabry U, Valent P, et al. Acute myeloid leukaemia with t(8;21) associated with "occult" mastocytosis. Report of an unusual case and review of the literature. J Clin Pathol. 2004;57:324–8.
Pardanani A, Lim KH, Lasho TL, Finke C, McClure RF, Li CY, et al. Prognostically relevant breakdown of 123 patients with systemic mastocytosis associated with other myeloid malignancies. Blood. 2009;114:3769–72.
Wang SA, Hutchinson L, Tang G, Chen SS, Miron PM, Huh YO, et al. Systemic mastocytosis with associated clonal hematological non-mast cell lineage disease: clinical significance and comparison of chomosomal abnormalities in SM and AHNMD components. Am J Hematol. 2013;88:219–24.
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7.
Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376:536–47.
Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31:2428–36.
Tefferi A, Lasho TL, Finke CM, Elala Y, Hanson CA, Ketterling RP, et al. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016;1:105–11.
Tefferi A, Lasho TL, Guglielmelli P, Finke CM, Rotunno G, Elala Y, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1:21–30.
Lasho TL, Mudireddy M, Finke CM, Hanson CA, Ketterling RP, Szuber N, et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018;2:370–80.
Buchner T, Schlenk RF, Schaich M, Dohner K, Krahl R, Krauter J, et al. Acute Myeloid Leukemia (AML): different treatment strategies versus a common standard arm–combined prospective analysis by the German AML Intergroup. J Clin Oncol. 2012;30:3604–10.
Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N Engl J Med. 2016;374:2530–41.
DeAngelo DJ, George TI, Linder A, Langford C, Perkins C, Ma J, et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial. Leukemia. 2018;32:470–8.
Gotlib J, Berube C, Growney JD, Chen CC, George TI, Williams C, et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood. 2005;106:2865–70.
Gotlib J, George T, Akin C, Sotlar K, et al. Midostaurin (PKC412) Demonstrates a high rate of durable responses in patients with advanced systemic mastocytosis: results from the fully accrued global phase 2 CPKC412D2201 trial. Blood. 2014;124:21.
Falchi L, Verstovsek S. Kit mutations: new insights and diagnostic value. Immunol Allergy Clin North Am. 2018;38:411–28.
DeAngelo DJ, Quiery AT, Radia D, Drummond MW, Gotlib J, Robinson WA, et al. Clinical activity in a phase 1 study of BLU-285, a potent, highly-selective inhibitor of KIT D816V in advanced systemic mastocytosis. Blood. 2017;130:2.
Acknowledgements
This work was supported by the “Deutsche José Carreras Leukämie-Stiftung e.V.” (Grant no. DJCLS R 13/05), by the SEED program of the Medical Faculty Mannheim, Heidelberg University and by the Austrian Science Fund (FWF) SFB project F4701-B20 and F4704-B20.
Author information
Authors and Affiliations
Contributions
MJ, KD, JS, MM, NN, AF, NCPC, and AR performed the laboratory work for the study. MJ, KD, SK, JS, K Shoumariyeh, MM, LLFS, SF, BK, NvB, K Spiekermann, MH, GM, SK, HCK-N, TH, HD, WRS, PV, and AR provided patient material and information. H-PH and K Sotlar reviewed the bone marrow biopsies. MJ, KD, SK, JS, AF, NCPC, W-KH, PV, and AR wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
H-PH, K Sotlar, PV, and AR served as consultants in a global phase-II-study examining the effects of midostaurin in advanced systemic mastocytosis. H-PH, K Sotlar, K Shoumariyeh, PV, NCPC, and AR received honoraria and/or travel support from Novartis Pharmaceuticals. MJ and JS received travel support from Novartis Pharmaceuticals. TH has equity ownership of the MLL Munich Leukemia Laboratory. MM is employed by the MLL Munich Leukemia Laboratory. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jawhar, M., Döhner, K., Kreil, S. et al. KIT D816 mutated/CBF-negative acute myeloid leukemia: a poor-risk subtype associated with systemic mastocytosis. Leukemia 33, 1124–1134 (2019). https://doi.org/10.1038/s41375-018-0346-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0346-z
This article is cited by
-
Allogeneic haematopoietic cell transplantation for advanced systemic mastocytosis: Best practice recommendations on behalf of the EBMT Practice Harmonisation and Guidelines Committee
Leukemia (2024)
-
Response and resistance to cladribine in patients with advanced systemic mastocytosis: a registry-based analysis
Annals of Hematology (2023)
-
Systemic mastocytosis with acute myeloid leukemia occurs from mutually exclusive clones expressing KITD816V and FLT3-ITD
Leukemia (2021)
-
Detection of the KIT mutation in myelodysplastic and/or myeloproliferative neoplasms and acute myeloid leukemia with myelodysplasia-related changes predicts concurrent systemic mastocytosis
Modern Pathology (2020)
-
CEBPE expression is an independent prognostic factor for acute myeloid leukemia
Journal of Translational Medicine (2019)
