
Abstract
Increasing evidence suggests that the immune system affects prognosis of chronic myeloid leukemia (CML), but the detailed immunological composition of the leukemia bone marrow (BM) microenvironment is unknown. We aimed to characterize the immune landscape of the CML BM and predict the current treatment goal of tyrosine kinase inhibitor (TKI) therapy, molecular remission 4.0 (MR4.0). Using multiplex immunohistochemistry (mIHC) and automated image analysis, we studied BM tissues of CML patients (n = 56) and controls (n = 14) with a total of 30 immunophenotype markers essential in cancer immunology. CML patients’ CD4+ and CD8+ T-cells expressed higher levels of putative exhaustion markers PD1, TIM3, and CTLA4 when compared to control. PD1 expression was higher in BM compared to paired peripheral blood (PB) samples, and decreased during TKI therapy. By combining clinical parameters and immune profiles, low CD4+ T-cell proportion, high proportion of PD1+TIM3−CD8+ T cells, and high PB neutrophil count were most predictive of lower MR4.0 likelihood. Low CD4+ T-cell proportion and high PB neutrophil counts predicted MR4.0 also in a validation cohort (n = 52) analyzed with flow cytometry. In summary, the CML BM is characterized by immune suppression and immune biomarkers predicted MR4.0, thus warranting further testing of immunomodulatory drugs in CML treatment.
Similar content being viewed by others


Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disease driven by the BCR-ABL1 oncokinase as a result of chromosomal translocation t(9;22) in myeloid stem cells of the bone marrow (BM). Following the development of tyrosine kinase inhibitors (TKI), the prognosis of chronic-phase (CP) CML has improved. The principal aim of TKI treatment is to achieve and maintain a deep molecular response (MR) to prevent disease progression. Such responses enable therapy discontinuation in a marked proportion of patients, thus sparing potential side effects, decrease in the quality of life, and escalating costs of treatment [1, 2].
The BM constitutes the home niche for leukemic cells. Its specific cellular and molecular structure remains unknown, although increasing evidence suggests that the immune system impacts the pathogenesis and prognosis of CML patients [3,4,5]. Furthermore, TKIs have been shown to induce immunomodulatory changes both in vivo and in vitro [6]. A deep MR correlates with increased NK-cell and CD8+ T-cell counts and cytotoxicity, and decreased myeloid-derived suppressor cell (MDSC) counts in the peripheral blood (PB) of CML patients [7, 8]. In addition, high CD62L+ T cells and low sCD62L+ have been suggested to predict optimal treatment response to nilotinib [9]. Although it is difficult to prove the direct causal relationship of the immune system and therapy response in patients, data from TKI discontinuation studies suggest that immune response is one of the important biological factors in successful treatment-free remission (TFR) [1, 10,11,12,13,14,15,16]. For example, the high proportion of CD56bright NK-cells, low CD56dim NK-cell TNF-α/IFN-γ cytokine level, and high expression of CD86+ in plasmacytoid dendritic cells (pDC) have been suggested as biomarkers predicting relapse after TKI discontinuation [1, 10,11,12, 14].
Despite long-term remissions in many solid tumor types, the therapeutic potential of immune checkpoint inhibitors in CML, or in other hematological malignancies, has not been fully evaluated. Here we present a comprehensive analysis of immune cells in the BM of CP CML patients at diagnosis using multiplexed immunohistochemistry (mIHC). In addition, T-cell exhaustion status was determined at diagnosis and after 1, 3, and 6 months of TKI treatment in both PB and BM with flow cytometry (FC). Based on the results, we developed a novel risk stratification model predicting the current treatment goal of TKI therapy, molecular response 4.0 (MR4.0), and compared that to stratification based on the BCR-ABL International scale (IS) transcript level <10% at 3 months [17]. The model was validated using FC. Together, the findings provide a detailed understanding of the immune landscape in CML BM and its clinical significance in predicting treatment responses.
Materials and methods
Study design
IHC cohort
To examine immune cell numbers and their immunophenotype using mIHC, we collected all available diagnostic-phase, pretreatment BM biopsies of CP CML patients treated in the Department of Hematology, Helsinki University Central Hospital (HUCH) between 2005 and 2015 (n = 56). Patients with previous hematologic malignancy were excluded. In addition, we recruited control subjects with a BM biopsy taken in 2010 in HUCH (n = 14). The most common biopsy indication was prolonged mild thrombocytosis or anemia noticed at routine check-up (Table S1). However, subjects presented no symptoms and had neither previously or during 6 years of later follow-up any hematologic malignancy, chronic infection nor autoimmune disorder. All study subjects gave written informed research consent to the study and to the Finnish Hematology Registry. The study complied with the Declaration of Helsinki and the HUCH ethics committee (DNRO 303/13/03/01/2011).
FC validation cohort
To validate the covariates of the prediction model, we recruited CML patients with a BM aspirate taken before TKI treatment and from which T-cell proportions had been analyzed with FC (n = 52). A more detailed T-cell immunophenotype was not available as immune exhaustion markers were not part of the clinical panel.
FC longitudinal cohort
To examine longitudinal changes in immune cell profiles, we analyzed PB and BM aspirates of additional CML patients (n = 16) during TKI therapy with FC. Immunoprofiling experiments were performed once.
In total, three cohorts included 86 individual CML patients.
Clinical data
In the ‘IHC cohort’, we assessed in total 47 clinical diagnostic-phase baseline variables including pretreatment laboratory exam values, spleen size, patient characteristics, and medical history (Table S2). In addition, we used complete cytogenetic response (CCyR), major molecular response (MMR) and MR4.0 as treatment endpoints according to ELN recommendations [18, 19]. The observation time in the cohorts was the interval between treatment start to treatment response or last follow-up date. Patients without treatment response were censored at the last follow-up date. Competing risk analysis was not required, as there were no frequent competing events.
Methods
Tissue microarrays (TMAs)
According to standard clinical procedure, fresh BM biopsies were formalin-fixed and paraffin-embedded (FFPE) in the Department of Pathology, HUSLAB at HUCH. Using hematopathologic expertize, we constructed FFPE TMA blocks from duplicate 1 mm punches from areas with highest leukemic cell infiltration in the BM biopsy (Fig. 1a). Control spots were selected from the most representative region.

a Overview of the analysis pipeline. Tissue microarrays (TMAs) were constructed from duplicate bone marrow (BM) biopsy punches of regions with high-leukemic infiltrates. TMA slides were stained with multiplexed immunohistochemistry (mIHC) consisting of 5-plex fluorescent (PD1 [white], TIM3 [red], CD8 [green], CD4 [blue], DAPI [gray]; left panel) and 3-plex chromogenic (CD3 [green], FOXP3 [red] and hematoxylin counterstain; right panel) stainings. For different antibody panels see Tables S3 and S4. Scanned histological images are registered enabling analysis of marker localization. Cells are segmented based on differential spatial intensity using Otsu thresholding method. The intensity of each marker is quantified in segmented cells and then classified. The various immunophenotypes are aggregated for more detailed characterization and survival analyses. b Heatmap visualizing quantified immune cells (proportion of all cells in a TMA spot) and their immunophenotypes (proportion of the parent immune cell) organized by hierarchical clustering using Spearman correlation distance and Ward linkage (ward.D2) method. c The CML-to-control ratios of significantly varying (q < 0.05) median values of each variable were transformed by twofold logarithmic transformation and annotated according to the literature as anti-cancer immunity (green) or immunosuppression marker (orange)
Multiplexed immunohistochemistry (mIHC)
The mIHC method combines 5-plex fluorescence and 3-plex chromogenic IHC. For antibody panels, see Tables S3 and S4. For original protocol, see Blom et al.[20] technical protocol and Supplementary Methods.
Image analysis
Cell masks were segmented with parent immune cell markers (e.g., CD3 for T cells) using adaptive Otsu thresholding, and individual cells separated from clumps using intracellular intensity patterns. We used the image analysis platform CellProfiler 2.1.2 for cell segmentation, intensity measurements, and immune cell classification (Fig. 1a) [21,22,23]. As CD34 is also an endothelial marker, vessel-like structures were omitted from analysis. We computed total cell numbers for each TMA spot from the total area of binary DAPI images using Fiji. We computed marker co-localization and cell classification with integrated intensity using single-cell analysis (FlowJo v10; SI).
Spots with <1000 cells were excluded from analysis. To exclude any bias due to varying cell numbers in different TMA spots, different cell types were quantified as either proportion to all cells (e.g., number of CD3+CD8+ T cells to all cells in a TMA spot) or proportion of an immunophenotype defined by 1–2 markers to the cell type of interest (e.g., CD3+CD8+/PD1+TIM3+ T cells of all CD3+CD8+ T cells). Two TMA spots from each CML and control subjects were analyzed, and joined by the mean value of each cell types and their immunophenotype.
Flow cytometry
FC validation cohort
Fresh diagnostic-phase BM aspirates (n = 52) were analyzed for CD3+, CD3+CD4+, and CD3+CD8+ cells (Supplementary Methods). Their proportions were defined from the remaining non-debris BM cells similarly as with mIHC data.
FC longitudinal cohort
Vitally frozen mononuclear cells from PB and BM samples were stained for longitudinal analysis (Supplementary Methods).
Statistical analysis
To compare two groups of continuous variables, we used Mann–Whitney U-test (unpaired, two-tailed), and Kruskal–Wallis test for ≥3 groups. We excluded outliers from comparison analyses with Grubbs’ test (alpha-value 0.2), and adjusted p-values using Benjamini&Hochberg’s false discovery rate (FDR) correction (q < 0.05 corresponding to p < 0.032 defined statistical significance) [24]. To compare continuous variables of two matched samples, we used Wilcoxon signed-rank test (two-tailed). Statistical associations between two continuous variables were assessed with Spearman’s rank correlation coefficient. For clustering analyses, data were z-scored and clustered by Spearman correlation distance and Ward linkage (ward.D2) methods.
To develop a model predicting TKI treatment efficacy with minimal false discovery rate, we filtered first variables predicting MR4.0 using univariate Cox proportional-hazards analysis. Significant variables (p < 0.05, log-rank test) were included to an L1-penalized elastic net regularized regression analysis method that performs both model shrinkage and variable selection [25, 26]. The optimal shrinkage parameter lambda (λ) was computed by the mean of iterated threefold crossvalidated (number of iterations: 100) lambda.min (λ = 0.182) and lambda.1se (λ = 0.318). The final model consisted of significant (log-rank, p < 0.05) variables in multivariate model after adjusting for TKI treatment (1st vs. 2nd generation TKI). The proportional-hazards assumption of the model was ensured with scaled Schoenfeld residuals. Models were compared with area under the receiver operating characteristic (AUROC) curve by bootstrapping (number of bootstrap replicates: 4000), C-statistic and time-dependent AUC (IPCW approach) [27]. We performed the statistical analyses with Prism v6.00 (GraphPad Software Inc.), and R v3.3.3 [28].
Results
Patient characteristics
The characteristics of the study patients are presented in Table 1. The ‘IHC cohort’, the ‘FC validation cohort’ and the ‘FC longitudinal cohort’ were indifferent in terms of patient age, gender, and prevalence of Sokal and Hasford risk groups when compared to the ELN/EUTOS CML registry ‘Out-study cohort’ used here as a reference CML patient cohort [29]. The median age of control subjects was 51 years (range 13–65 years) and 57% were male and is, thus, comparable with CML patients in the ‘IHC cohort’. Although median age of control subjects is 6 years lower than CML patients, no correlation between age and immunophenotype markers could be found. The EUTOS low-risk group was overrepresented in the ‘FC longitudinal cohort’ and the ELTS low-risk group in both FC cohorts when compared to the ‘Out-study cohort’.
The first-line TKI varied between cohorts due to difference in participation in clinical studies comparing first-line TKI and not to difference in disease burden. Time from treatment start to MR4.0 was shorter in the ‘FC longitudinal cohort’ than in other cohorts. The prevalence of BCR-ABL <10% IS was equivalent in IHC and FC cohorts.
Multiplexed immunohistochemistry reveals an immune suppression-skewed immune profile in CML BM
To visualize integrated immune profiles of CML vs. control BM, we mapped all immune cell subsets and their single-marker phenotypes analyzed from the ‘IHC cohort’ (Fig. 1a, b). Clustering analysis demonstrated that immune cell subtypes associated with pro-inflammatory anti-cancer immune response (e.g., proportion of CD3+ T cells, OX40+ and granzyme B producing (GrB+) CD8+ T cells) were decreased and immunosuppressive markers (e.g., CTLA4+ CD4+ and CD8+ T cells, MDSC-like cells and M2-type macrophages) increased in CML BM, grouping CML patients and control subjects distinctly from each other.
Next, to identify most significant immunological parameters, we compared the expression levels of markers defining either immune cell exhaustion or anti-cancer immunity, and differing significantly (Mann–Whitney U-test, q < 0.05) between CML vs. control BM subsets (Fig. 1c). Immune cell subgroups and immunophenotype markers associated with immune exhaustion were higher and anti-cancer immunity markers lower in CML patients than in control subjects.
T cells in CML BM display an exhausted phenotype that partially resolves during TKI treatment
To further dissect the various CML immune profiles, we focused on individual immune cell subsets and surface marker profiles reflecting their functional states (Fig. 2a). Due to myeloid expansion, lower proportion of CD3+, CD3+CD4+, and CD3+CD8+ T cells from all cells were observed in CML vs. control BM (Fig. 2b). In contrast, the proportion of FOXP3+CD4+ regulatory T cells (Tregs) and CD3–CD20+ B cells were enriched in CML vs. control BM (Supplementary Figure 1A, and Fig. 3c). Moreover, T cells were more frequently CD27− and CD45RO+ in CML as compared to control suggesting increased differentiation toward an effector memory phenotype (Fig. 2c and Supplementary Figure 1C).

The chronic myeloid leukemia (CML) bone marrow (BM) is characterized by lymphoid-lineage immunosuppression. a Representative example of cell gating based on marker intensity in ‘IHC cohort’. Levels of b CD3+ T cells, CD4+ T cells, and CD8+ T cells, c CD25 and CD27 expression in CD4+ and CD8+ T cells, and d PD1, CTLA4, OX40, and TIM3 expression in CD4+ and CD8+ T cells were compared with Mann–Whitney U-test after outlier exclusion (Grubb’s test, alpha = 0.20), and p-values adjusted with Benjamini-Hochberg procedure to reduce false discovery rate (q-values), and results displayed with scatter plots. e Pie charts visualizing PD1 and TIM3 expression in T-cell subgroups. f, g PD1, LAG3, and TIM3 expression and percentages of T-cell subsets in CD4+ and CD8+ T cells of paired BM and peripheral blood (PB) samples measured (n = 11) by flow cytometry (FC). We defined CD45RA–CCR7– as effector memory (EM) T cells, CD45RA+CCR7− as terminally differentiated effector memory (TEMRA), CD45RA–CCR7+ as central memory (CM) T cells, and CD45RA+CCR7+ as naive T cells. P-values are calculated with Wilcoxon signed-rank test. h Representative FC scatter plots of PD1 expression in CD3+CD8+ T cells from paired BM and PB samples. i Heatmap of median percentage of PD1+ cells out of indicated T-cell subsets in CML patients treated with imatinib (n = 7) or dasatinib (n = 4). Rows are clustered using Spearman correlation distance and Ward linkage (ward.D2) method. j Expression of PD1 on CD4+ and CD8+ T cells in the BM at diagnosis and at 1 month (imatinib, dasatinib) or 3 months (nilotinib) after the start of TKI treatment in paired FC samples in CML patients (n = 16). P-values are calculated with Wilcoxon signed-rank test. k Expression of PD1 in CD4+ and CD8+ T cells in the BM at diagnosis and different time points during TKI treatment with imatinib (n = 7), dasatinib (n = 4) or nilotinib (n = 4) in CML patients. One dasatinib-treated patient was excluded for missing data at 6 months. Points indicate median and error bars represent interquartile ranges. * <0.05, ** <0.01, *** <0.001

The chronic myeloid leukemia (CML) bone marrow (BM) is characterized by myeloid-lineage immunosuppression. Levels of (a) CD68+ monocytes and CD33+CD11b+HLA–DR– myeloid-derived suppressor cells, b CD68+/pSTAT1+cMAF– M1-macrophages and CD68+/pSTAT1–cMAF+ M2-macrophages, c CD11c+BDCA1+ and CD11c+BDCA3+ myeloid dendritic cells type 1 (mDC1) and type 2 (mDC2), respectively, and CD3–CD20+ B cells, d HLA-DRhigh and CD11b+ expression in mDC1 and mDC2 cells, e HLA-ABC expression in all BM cells and CD34+ stem cells, f and HLAG, PDL1 and PDL2 expression in all BM cells of CML patients (n = 56) and controls (n = 14) were compared with Mann–Whitney U-test after outlier exclusion (Grubb’s test, alpha = 0.20), and p-values adjusted with Benjamini-Hochberg procedure to compute false discovery rate (q-values), and results displayed with scatter plots. g PDL1 and PDL2 expression on CD34+ cells of paired BM and PB samples of CML patients (n = 5) measured by flow cytometry. P-values are calculated with Wilcoxon signed-rank test. PDL1 (h) and PDL2 (i) expression on CD34+ cells of unpaired BM and PB samples of CML patients (n = 11) and controls (n = 9) measured by flow cytometry. * <0.05, ** <0.01, *** <0.001
We next studied the expression of inhibitory (PD1, TIM3, CTLA4, and LAG3) and activating (OX40) immune checkpoint molecules in T cells. Higher PD1, TIM3, and especially CTLA4 were observed in T cells of the CML vs. control BM (Fig. 2d). Of particular interest, higher levels of PD1+TIM3− in both CD4+ and CD8+ T cells (10.3% IQR [7.0–15.6%] vs. 1.7% [0.95–3.0%], q< 0.0001, and 8.8% [5.6–14.0%] vs. 2.0% [1.0–5.1%], q < 0.0001, respectively) and PD1+ TIM3+ (Fig. 2e) were observed in CML vs. control BM. However, lower proportion of LAG3 was detected in both CD4+ and CD8+ T cells in CML as compared to control BM (Supplementary Figure 1D). Lower expression of the secondary co-stimulatory molecule OX40 was noted in CD8+ T cells (9.0% [6.7––14.8%] vs. 16.0% [11.8–18.3%], q = 0.002), but not in CD4+ T cells in CML vs. control BM. Finally, we found decreased production of the cytolytic protein GrB and CD57 in T cells of CML patients, suggesting that the exhausted surface marker phenotype is linked to decreased cytolytic activity (Supplementary Figure 1A and B).
To test whether the BM represents a distinct milieu in regard to T-cell phenotype and differentiation, we studied paired BM and PB samples in CML patients of the ‘longitudinal FC cohort’. We found that both CD8+ and CD4+ T cells had more pronounced PD1 expression in BM vs. PB (26.9% vs. 12.7%, p = 0.001 and 18.9% vs. 10.4%, p = 0.001, respectively; Fig. 2f–h). This difference was significant in all T-cell subsets, including CD45RA-CCR7- effector memory (TEM), CD45RA+CCR7− terminally differentiated (TEMRA), CD45RA–CCR7+ central memory (TCM) and CD45RA+CCR7+ naive, with the highest PD1 positivity in TEM and TEMRA cells (Fig. 2i). Moreover, the number of central memory CD4+ T cells and naive CD8+ T cells was lower in BM (Fig. 2f, g).
Next, we evaluated PD1 expression on T cells during TKI treatment in follow-up BM aspirates. We observed a decrease in PD1+CD8+ but not CD4+ T cells at 1 and 3 months after the start of TKI therapy compared to diagnosis (27.1% vs. 21.6%, p = 0.009; Fig. 2j). The trend was similar with all TKIs (imatinib, dasatinib, and nilotinib), although statistical significance was not reached when considering the TKIs individually due to sample size (Fig. 2k). The reduction in PD1 expression was most prominent in CD8+ TEM cells (Fig. 2i–k). Together, these findings indicate that in CML patients, particularly the BM milieu is associated with changes in T-cell populations, and that an exhausted T-cell phenotype is partially resolved during TKI therapy.
Increased immunosuppressive myeloid cells and decreased class I HLA in CML BM
As the BM of TKI-naive CML patients demonstrated immunosuppression in the lymphoid lineage, we investigated next the composition and phenotype of myeloid cells. Higher proportion of CD68+ myeloid cells from all cells was observed in the CML BM compared to control subjects (Fig. 3a). Interestingly, only the proportion of immunosuppressive cMAF+pSTAT1−CD68+ cells (M2-macrophages) was increased whereas cMAF-pSTAT1+CD68+ cells (M1-macrophages) remained equal in CML vs. control BM (Fig. 3b). As MDSCs might be difficult to discern from a CML background, we have annotated MDSC-like cells as CD11b+CD33+HLADR- [30]. The proportion of MDSC-like cells from all cells were elevated in CML vs. control BM (Fig. 3a). Myeloid dendritic cells type 1 (mDC1) and type 2 (mDC2) were characterized as CD11c+BDCA1+ and CD11c+BDCA3. To study dysfunction in antigen presentation and mobility, we measured both the expression level of class II HLA-DR and CD11b, respectively. Intriguingly, both mDC subpopulations, as well as their expression of HLA-DR and CD11b were reduced in CML vs. control BM (Fig. 3c, d).
At diagnosis, the majority of cells occupying the CP CML BM are differentiated leukemic cells. However, following successful treatment only few CD34+ blasts remain and normal hematopoiesis recovers. Thus, we compared the expression of HLA class I molecules HLA-A, -B, and -C (HLA-ABC) and inhibitory immune checkpoint ligands PDL1/2 separately in CD34+ and all cells. We observed an overall lower production of HLA-ABC in all BM cells but higher HLA-ABC level in CD34+ cells when comparing CML BM to control (Fig. 3e). Interestingly, HLA-ABC expression was lower in CD34+ cells than in all BM cells in both CML and control BM. Moreover, the expression of PDL1/2 and the inhibitory class I HLA-G were higher in all cells in CML vs. control BM (Fig. 3f). Analyzed by FC, PD-L1 expression showed a trend toward elevated expression in BM CD34+ cells compared to peripheral circulation, whereas no statistically significant difference to control CD34+ was observed (Fig. 3g–i).
The proportion of helper T cells and PD1+TIM3– cytotoxic T cells, and neutrophil count predict MR4.0
Given the observed immune dysregulation associated with CML, we aimed to understand the relation of the immune profile to treatment responses and whether immunological parameters can be used to predict responses. To stratify CML patients by predisposition to reach MR4.0, we combined PB and BM laboratory exam values, spleen size, patient demographics, and mIHC profiling data. To model the coordinated interactions among multiple variables, we performed an L1-penalized Cox regression analysis on variables found significant in univariate Cox regression (log-rank p < 0.05), and visualized their similarities and dissimilarities using unsupervised, principal component analysis (PCA) and hierarchical clustering analysis (Table S5, Fig. 4a, b). Interestingly, patients with high CD4+ T-cell proportion in the BM were more inclined to achieve MR4.0 (Fig. 4b). In subjects with low CD4+ T-cell proportion, HLA–ABC+ proportion in blasts correlated inversely with failure to reach MR4.0, while markers of T-cell exhaustion, e.g., PD1+TIM3– CD8+ T cells and PD1+OX40+CD4+ T cells, and of disease burden, e.g., PB neutrophil count, correlated positively with failure to reach MR4.0.

Introduction of the chronic myeloid leukemia immunology (CMLi) model. a Principal component analysis (PCA) plot of variables selected with L1-penalized Cox regression model with top prediction of molecular response 4.0 (MR4.0). b Significant covariates after adjustment with TKI generation (imatinib vs. dasatinib or nilotinib) are organized by hierarchical clustering using Spearman correlation distance and Ward linkage (ward.D2) method. c Summary of the CMLi model computed with multivariate Cox regression analysis (log-rank test). d The CMLi model was categorized into three even-sized subgroups and computed to predict (i) MR4.0, (ii) major molecular response (MMR), and (iii) complete cytogenic remission (CCyR) using Cox regression analysis. e The prediction power of the uncategorized CMLi model to stratification by BCR-ABL < 10% IS at 3 months and their combination was measured with area under the receiver operating characteristic curve (AUROC). The combined model outperformed stratification by BCR-ABL < 10% IS at 3 months (* <0.05, bootstrap method) HR hazard ratio. 95% CI 95% confidence interval, Coef correlation coefficient
In the treatment of CP CML patients, 2nd generation TKIs have been shown to be superior to imatinib in terms of both treatment response and progression-free survival [31,32,33]. Thus, we adjusted significant variables for TKI therapy (1st vs. 2nd generation TKI) in multivariate Cox regression analysis. The “CML immunology” model (“CMLi”) was categorized to even-sized groups based on tertiles, e.g. the low-risk, intermediate-risk (HR 2.8, p = 0.009, 95% CI [1.3–6.0]), and high-risk (HR 12.8, p < 0.0001, 95% CI [5.2–31.6]; Fig. 4c, d). The R2 of the categorized model was 0.45 (p < 0.0001). The CMLi model predicted MR4.0, MMR, and CCyR using Cox regression analysis (log-rank test, Fig. 4d). CMLi predicted TKI response superiorly than stratification by BCR-ABL < 10% IS at 3 months using time-dependent ROC (IPCW approach) and C-statistic, and with a similar but non-significant trend using AUROC (bootstrap method; Fig. 4e and Supplementary Figure 2). Interestingly, the combination of both models and exclusion of neutrophil count due to covariate insignificance improved prediction performance further. The CMLi model also predicted MR4.0 with higher confidence than Sokal, Hasford, and EUTOS scores using AUROC, but it should be noted that these scores have not been developed to predict MR4.0 (Supplementary Figure 3).
To dissect the components of the CMLi model, we computed univariate Cox regression analyses of individual covariates using median as cutoff (Supplementary Figure 4). In addition, we studied the Spearman correlation of the covariates of the CMLi model and clinical CML scores, as well as with complete blood count (Supplementary Figure 5A). PB neutrophil count clustered with leukocyte count and spleen size embodying disease burden. CD4+ T cells correlated negatively with disease burden and positively with the proportion of PB neutrophils and lymphocytes, and with hemoglobin shown earlier to relate with leukemic stem cell burden [34]. In addition, the proportion of PB eosinophils and basophils, platelet count, and patient age defined a distinct group correlating weakly with other variables. We identified lower T-cell proportion, higher PD1+ and OX40+ T cells (e.g., immune exhaustion and inflammation markers), as well as higher PB leukocyte count and lactate dehydrogenase, (e.g., biomarkers of disease burden) as clinicoimmunological variables associated with higher CMLi risk group (Supplementary Figure 5B).
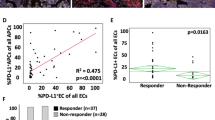
To validate the role of CD4+ T cells and neutrophils, we analyzed fresh BM aspirates of the ‘FC validation cohort’. Even in a small cohort of 52 patients, the proportion of CD4+ T cells and low neutrophil count significantly predicted faster and more frequent MR4.0 (Fig. 5a–c). Interestingly, the trend was similar with high proportion of CD3+ (HR 2.74, p = 0.003, log-rank test; CI 95% [1.38–5.44]), but not with CD8+ T cells (HR 1.53, p = 0.17; CI 95% [0.82–2.85]) and reaching MR4.0 (Supplementary Figure 6).

Prediction of MR4.0 in the ‘FC validation cohort’. a–c Summary and survival curves of univariate Cox regression analysis (log-rank test) of CD4+ T cells analyzed from bone marrow (BM) aspirates and peripheral blood (PB) neutrophils of the ‘FC validation cohort’ (n = 52). HR hazard ratio, 95% CI 95% confidence interval
Discussion
Our results suggest that the CML BM is analogous to solid tumor microenvironment in having an immune landscape with severe myeloid and lymphoid cell-mediated immunosuppression, which may partially resolve with successful TKI treatment. In addition, we developed a novel risk stratification model predicting TKI treatment response.
We aimed to clarify the clinical significance of the immune landscape of CP CML patients, and therefore focused on immune cells and checkpoint molecules with known therapeutic potential in clinical trials. We applied a novel mIHC technique combining six antibodies on a single TMA slide. The major advantage of mIHC over conventional FC is the possibility to analyze hundreds of samples in parallel simultaneously and rapidly without cryopreservation, which might affect cell number, viability and phenotype [35]. We supplemented mIHC with automated image analysis to objectively classify and quantify large numbers of immune cells, which would not have been possible manually. As follow-up BM biopsies are not sampled routinely, mIHC is not suited for routine hematological clinical work, but may substitute FC in academic retrospective studies. Potentially, it can be applied in oncology for biomarker discovery improving risk stratification in clinical studies.
Consistent with previous studies, MDSC-like cells and M2-macrophages were more numerous in CML patients [7, 36, 37]. In addition, CD4+ and CD8+ T cells exhibited higher levels of immune checkpoint receptors PD1, CTLA4, and TIM3 in CML than in control BM. PD1 expression was elevated in the BM T-cells compared to paired PB samples. In addition, PD1 expression decreased in CD8+ T cells during TKI treatment. The exhausted phenotype was associated with lower expression of co-stimulatory marker CD27, cytotoxicity marker GrB and activation marker OX40 in CML vs. control BM suggesting weaker CD8+ T-cell cytotoxicity and activation, and elevated antigen-experienced memory CD45RO+ T cells in CML.
Although these findings suggest marked immunosuppression in CML BM, it is unclear, whether they reflect a direct adaptive immune response to leukemia cells or chronic cytokine-induced inflammation by the myeloid cells [38, 39]. Previously, PD1 and CTLA4 expression in T cells have been shown to be increased at diagnosis and PD1 to decrease with deepening MR status in PB samples of untreated CML patients as elegantly demonstrated by Hughes et al. [7, 36]. Moreover, in a murine CML model the PD1 status of T cells has been associated with weaker cytotoxic activity and cytokine production in vitro, and blocking the PD1/PDL1 signaling improved survival suggesting T-cells counteract disease progression [40]. This study also suggests lower class I HLA expression to desensitize CML stem cells from being identified by T cells.
Yet, the role of IFNγ secreted by activated lymphocytes is paradoxical in CML. It has been shown that IFNγ promotes myeloid expansion via the JAK2/STAT5 and ERK1/2 pathways and decreases apoptosis by upregulating RUNX1 [41]. Moreover, therapeutic adoptive T-cell infusion alone increases CD34+ stem cell proliferation via a IFNγ-mediated mechanism [42]. However, this effect could be overrun by simultaneously blocking PD1/PDL1 signaling emphasizing the protective role immune checkpoint molecules confer on CML stem cells [43].
Most studies on CML immunology have been conducted with PB samples. Our combined mIHC and FC analyses suggest that the BM is a particularly immunosuppressive microenvironment in CML, and should be the standard tissue to study in future immuno-oncological studies.
Using L1-penalized regression modeling, we propose low CD4+ T-cell proportion, high PD1+TIM3−CD8+ T-cell proportion, and high PB neutrophil count as predictors of weaker TKI treatment response. While the CMLi model composed of baseline covariates might predict MR4.0 with higher confidence than stratifying patients by BCR-ABL < 10% at 3 months, the combined model improves prediction even further suggesting synergy. In addition, patients of the CMLi high-risk group were associated with markers of T-cell immunosuppression and greater tumor burden defined by clinical parameters. Given the lack of immune exhaustion markers in the ‘FC validation cohort’, the multivariate CMLi model could not be validated completely. However, we confirmed both CD4+ T cell and PB neutrophil counts as novel predictive biomarkers for MR4.0 using fresh diagnostic-phase BM aspirate samples analyzed by FC.
The immune system of CML patients with deep immunosuppression might be incapable of killing cancer cells. We have shown that TKI treatment responses are more potent in patients with less exhausted T cells. While immune dysfunction might reflect disease aggressiveness, and not inflict relative insensitivity to TKIs, the findings could alternatively be explained by off-target immunomodulatory effects of TKIs boosting pre-existing anti-leukemic immune responses or dampening immunosuppressive cells such as Tregs [6, 44]. Imatinib has for example been shown to suppress the levels of immunosuppressive IDO expression in gastrointestinal stromal tumors, and dasatinib to increase the number of cytotoxic cells in the circulation and inside the tumors [6, 45,46,47,48].
In most solid tumors, T cells have been associated with positive prognosis. This notion has led to the development of the prognostic immunohistochemical tool ‘Immunoscore’, which cannot be directly applied for leukemic patients, as the BM lacks a clear invasive margin and central tumor [49, 50]. Thus, we suggest that the combination of clinical and immune cell parameters can be used as an analogous prediction tool in CML.
In summary, the CMLi model highlights the role of T cells and their immunophenotype, as well as disease burden as critical predictive factors of TKI treatment response in CP CML. Immunotherapeutic options should be considered in CML treatment regimens as they might enable molecular remission and treatment discontinuation. However, this hypothesis and CMLi cutoff values have to be further validated in a multicenter study using FC.
References
Saussele S, Richter J, Hochhaus A, Mahon F-X. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia. 2016;30:1638–47.
Saussele S, Richter J, Guilhot J, Gruber FX, Hjorth-Hansen H, Almeida A, et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): a prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018;2045:1–11.
Hughes A, Yong ASM. Immune effector recovery in chronic myeloid leukemia and treatment-free remission. Front Immunol. 2017;8:1–12.
Molldrem JJ, Lee PP, Kant S, Wieder E, Jiang W, Lu S, et al. Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J Clin Invest. 2003;111:639–47.
Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE, et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med. 2000;6:1018–23.
Kreutzman A, Porkka K, Mustjoki S. Immunomodulatory effects of tyrosine kinase inhibitors. Int Trends Immun. 2013;1:17–28.
Hughes A, Clarson J, Tang C, Vidovic L, White DL, Hughes TP, et al. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood. 2017;129:1166–76.
Sopper S, Mustjoki S, Gjertsen BT, Giles F, Hochhaus A, Janssen JJWM, et al. NK cell dynamics and association with molecular response in early chronic phase chronic myelogenous leukemia (CML-CP) patients treated with nilotinib. Leukemia. 2017;31:2264–7.
Sopper S, Mustjoki S, White D, Hughes T, Valent P, Burchert A, et al. Reduced CD62L expression on T cells and increased soluble CD62L levels predict molecular response to tyrosine kinase inhibitor therapy in early chronic-phase chronic myelogenous leukemia. J Clin Oncol. 2016;35:175–84.
Ilander M, Olsson-Strömberg U, Schlums H, Guilhot J, Brück O, Lähteenmäki H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia. 2017;31:1108–16.
Schütz C, Inselmann S, Sausslele S, Dietz CT, Müller MC, Eigendorff E, et al. Expression of the CTLA-4 ligand CD86 on plasmacytoid dendritic cells (pDC) predicts risk of disease recurrence after treatment discontinuation in CML. Leukemia. 2017;32:1–8.
Imagawa J, Tanaka H, Okada M, Nakamae H, Hino M, Murai K, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol. 2015;2:e528–e535.
Koskenvesa P, Kreutzman A, Rohon P, Pihlman M, Vakkila E, Räsänen A, et al. Imatinib and pegylated IFN-α2b discontinuation in first-line chronic myeloid leukemia patients following a major molecular response. Eur J Haematol. 2014;92:413–20.
Rea D, Henry G, Khaznadar Z, Etienne G, Guilhot F, Nicolini F et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. 2017.
Caocci G, Martino B, Greco M, Abruzzese E, Trawinska MM, Lai S, et al. Killer immunoglobulin-like receptors can predict TKI treatment-free remission in chronic myeloid leukemia patients. Exp Hematol. 2015;43:1015–8.
Mizoguchi I, Yoshimoto T, Katagiri S, Mizuguchi J, Tauchi T, Kimura Y, et al. Sustained upregulation of effector natural killer cells in chronic myeloid leukemia after discontinuation of imatinib. Cancer Sci. 2013;104:1146–53.
Branford S, Yeung DT, Parker WT, Roberts ND, Purins L, Braley JA, et al. Prognosis for patients with CML and 10% BCR-ABL1 after 3 months of imatinib depends on the rate of BCR-ABL1 decline. Blood. 2014;124:511–8.
Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Leukemia. 2006;108:1809–20.
Cross NCP, White HE, Colomer D, Ehrencrona H, Foroni L, Gottardi E, et al. Laboratory recommendations for scoring deep molecular responses following treatment for chronic myeloid leukemia. Leukemia. 2015;29:999–1003.
Blom S, Paavolainen L, Bychkov D, Turkki R, Mäki-Teeri P, Hemmes A, et al. Systems pathology by multiplexed immunohistochemistry and whole-slide digital image analysis. Sci Rep. 2017;7:15580.
Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100.
Xing F, Member S, Yang L. Robust nucleus/cell detection and segmentation in digital pathology and microscopy images: a comprehensive review. IEEE Rev Biomed Eng. 2016;9:234–63.
Caicedo JC, Cooper S, Heigwer F, Warchal S, Qiu P, Molnar C, et al. Data-analysis strategies for image-based cell profiling. Nat Methods. 2017;14:849–63.
Yosef YB, H. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 2017;57:289–300.
Tibshirani R. Regression selection and shrinkage via the lasso. J R Stat Soc B. 1996;58:267–88.
Guinney J, Wang T, Laajala TD, Winner KK, Bare JC, Neto EC, et al. Prediction of overall survival for patients with metastatic castration-resistant prostate cancer: development of a prognostic model through a crowdsourced challenge with open clinical trial data. Lancet Oncol. 2017;18:132–42.
Hung H, Chin‐Tsang C. Estimation methods for time‐dependent AUC models with survival data. Can J Stat. 2009;38:8–26.
RStudio Team. RStudio: Integrated Development for R. Boston, MA: RStudio, Inc.; 2015.
Pfirrmann M, Baccarani M, Saussele S, Guilhot J, Cervantes F, Ossenkoppele G, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia. 2015;30:48–56.
Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:1–10.
Saglio G, Kim D-W, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–9.
Kantarjian HM, Hochhaus A, Saglio G, De Souza C, Flinn IW, Stenke L, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011;12:841–51.
Cortes JE, Saglio G, Kantarjian HM, Baccarani M, Mayer J, Boqué C, et al. Final 5-year study results of DASISION: the dasatinib versus imatinib study in treatment-Naïve chronic myeloid leukemia patients trial. J Clin Oncol. 2016;34:2333–40.
Mustjoki S, Richter J, Barbany G, Ehrencrona H, Fioretos T, Gedde-Dahl T, et al. Impact of malignant stem cell burden on therapy outcome in newly diagnosed chronic myeloid leukemia patients. Leukemia. 2013;27:1520–6.
Costantini A, Mancini S, Giuliodoro S, Butini L, Regnery CM, Silvestri G, et al. Effects of cryopreservation on lymphocyte immunophenotype and function. J Immunol Methods. 2003;278:145–55.
Christiansson L, Söderlund S, Svensson E, Mustjoki S, Bengtsson M, Simonsson B, et al. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in sokal high risk chronic myeloid leukemia. PLoS ONE. 2013;8:1–12.
Song J-X, Dian Z-J, Wen Y, Mei F, Li R-W, Sa Y-L. Assessment of the number and phenotype of macrophages in the human BMB samples of CML. Biomed Res Int. 2016;2016:1–9.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860.
Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22.
Mumprecht S, Schürch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia–specific T cells results in T-cell exhaustion and disease progression. Blood. 2009;114:1528 LP–1536.
Held SAE, Heine A, Kesper AR, Schönberg K, Beckers A, Wolf D, et al. Interferon gamma modulates sensitivity of CML cells to tyrosine kinase inhibitors. Oncoimmunology. 2016;5:e1065368.
Schürch C, Riether C, Amrein MA, Ochsenbein AF. Cytotoxic T cells induce proliferation of chronic myeloid leukemia stem cells by secreting interferon-γ. J Exp Med. 2013;210:605–21.
Riether C, Gschwend T, Huguenin A-L, Schürch CM, Ochsenbein AF. Blocking programmed cell death 1 in combination with adoptive cytotoxic T-cell transfer eradicates chronic myelogenous leukemia stem cells. Leukemia. 2015;29:1781–5.
Larmonier N, Janikashvili N, LaCasse CJ, Larmonier CB, Cantrell J, Situ E, et al. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol. 2008;181:6955–63.
Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17:1094–1100.
Hekim C, Ilander M, Yan J, Michaud E, Smykla R, Vähä-Koskela M, et al. Dasatinib changes immune cell profiles concomitant with reduced tumor growth in several murine solid tumor models. Cancer Immunol Res. 2017;5:157–69.
Mustjoki S, Auvinen K, Kreutzman A, Rousselot P, Hernesniemi S, Melo T, et al. Rapid mobilization of cytotoxic lymphocytes induced by dasatinib therapy. Leukemia. 2013;27:914–24.
Kreutzman A, Ilander M, Porkka K, Vakkila J, Mustjoki S. Dasatinib promotes Th1-type responses in granzyme B expressing T-cells. Oncoimmunology. 2014;3:e28925.
Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306.
Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, Lugli A, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med. 2012;10:205.
Acknowledgements
This study was supported by the Doctoral Program in Biomedicine (DPBM) in the University of Helsinki and personal grants (O.B.) from Emil Aaltonen Foundation, Hematology Research Foundation, Paulo Foundation, Duodecim Foundation, research grants (S.M.) from the Finnish Cancer Institute, Finnish Cancer Organizations, Sigrid Juselius Foundation, Signe and Ane Gyllenberg Foundation, State funding for university-level health research in Finland, the EUTOS project for CML 2016, and investigator-initiated research grant from Novartis. We thank Tiina Kasanen (Hematology Research Unit Helsinki), and the Department of Clinical Chemistry, Laboratory of Hematology at the Helsinki University Hospital for technical support with flow cytometry. We are grateful to the members of the Hematology Research Unit Helsinki for discussions and technical help.
Author contributions
Conception and design: O.B., O.D., O.K., S.B., T.P., and S.M. Collection and assembly of data: A.R., O.B., O.D., K.P., P.Kov., P.Kos., and S.M. Data analysis and interpretation: H.C., O.B., O.D., R.T., S.B., S.M., T.A., and T.P. Manuscript writing: all authors. Final approval of manuscript: All authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
K.P. has received honoraria and research funding from Pfizer, Novartis and Bristol-Myers Squibb. S.M. has received honoraria and research funding from Pfizer, Novartis, and Bristol-Myers Squibb. S.B. has received consultant fees from Fimmic. T.A. has received research funding from Novartis. P.Kos. has received honoraria from Novartis, Pfizer, and Incyte.
Electronic Fementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brück, O., Blom, S., Dufva, O. et al. Immune cell contexture in the bone marrow tumor microenvironment impacts therapy response in CML. Leukemia 32, 1643–1656 (2018). https://doi.org/10.1038/s41375-018-0175-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0175-0
This article is cited by
-
Prognostic Significance of Regulatory T-Cells and PD-1 + CD8 T-Cells in Chronic Myeloid Leukemia Patients Treated with Generic Imatinib
Indian Journal of Hematology and Blood Transfusion (2024)
-
Blockade of PD-1 and TIM-3 Ameliorates CD8+ T Cell Exhaustion in a Mouse Model of Chronic Myeloid Leukemia
Cell Biochemistry and Biophysics (2024)
-
Single-cell analysis of immune recognition in chronic myeloid leukemia patients following tyrosine kinase inhibitor discontinuation
Leukemia (2024)
-
Leukaemia exposure alters the transcriptional profile and function of BCR::ABL1 negative macrophages in the bone marrow niche
Nature Communications (2024)
-
Functional biomaterials for biomimetic 3D in vitro tumor microenvironment modeling
In vitro models (2023)
