
Abstract
Oncogenic fusions are major drivers in leukemogenesis and may serve as potent targets for treatment. DUX4/IGHs have been shown to trigger the abnormal expression of ERGalt through binding to DUX4-Responsive-Element (DRE), which leads to B-cell differentiation arrest and a full-fledged B-ALL. Here, we determined the crystal structures of Apo- and DNADRE-bound DUX4HD2 and revealed a clamp-like transactivation mechanism via the double homeobox domain. Biophysical characterization showed that mutations in the interacting interfaces significantly impaired the DNA binding affinity of DUX4 homeobox. These mutations, when introduced into DUX4/IGH, abrogated its transactivation activity in Reh cells. More importantly, the structure-based mutants significantly impaired the inhibitory effects of DUX4/IGH upon B-cell differentiation in mouse progenitor cells. All these results help to define a key DUX4/IGH-DRE recognition/step in B-ALL.
Similar content being viewed by others


Introduction
Oncogenic fusions are important causes/targets in leukemia and related treatment [1, 2]. Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy. B-ALL, which stems from the clonal proliferation of B lineage progenitors [3, 4], constitutes 80% of ALL cases [5]. Very recently, using next-generation sequencing technologies, a novel B-ALL oncogenic driver DUX4/IGH, derived from the insertion of chromosome fragments containing DUX4 gene into the IGH locus, has been reported in ~7% of B-ALL patients [3, 6, 7].
Double homeobox 4 (DUX4) is the core component of human subtelomeric macrostalelite D4Z4 [8]. It has been shown that 11 to >150 D4Z4 repeats can be identified in chromosome 4 and 10. In each of the D4Z4 repeat (~3.3 kb), it constitutes a single copy of the intronless DUX4 gene. At physiological condition, DUX4 is usually silenced [9]. In pathogenic condition known as facioscapulohumeral muscular dystrophy (FSHD) [10], the D4Z4 repeats are contracted to 1–10. The change of chromatin packing is thought to be a critical factor that leads to the abnormal expression of DUX4. Consistently, in B-cell ALL, the abnormal expression of DUX4/IGHs derived from chromosome translocation is also demonstrated to be a driver factor of leukemogenesis [3]. Furthermore, the deregulation of DUX4 is frequently associated with the biogenesis of ERGalt [4]. Very briefly, ERG is an ETS-family transcription factor playing important role in normal hematopoietic regulation [11] and is involved in the pathogenesis of T-ALL and acute myeloid leukemia [12,13,14]. ERGalt, derived from an abnormal transcription starting from the intron 6 of ERG, could induce lymphoid leukemia in Arf−/− mice [4]. Therefore, the ERGalt biogenesis is recognized as a reporter and a potent secondary hit in DUX4-driven leukemogenesis. However, due to the lack of structural information, it is not clear how DUX4/IGH recognizes, binds, and activates the transcription of its target genes via the double homeobox (HD1–HD2) domain.
In this report, we determined the crystal structures of Apo-DUX4HD2 and DNA-bound complex. The atomic structures, together with functional characterization, reveal a two-step clamp-like binding mechanism between double homeobox and TAATCTAAT DNA site, the latter of which is frequently observed in DUX4 target genes, and hence termed DUX4-Responsive-Element (DRE). Consistent with previous observation that a single HD domain is not sufficient for productive protein–DNA interaction, our data showed that both HD1 and HD2 are essential in DRE recognition. Single mutation in the major-groove-binding interfaces of HD1 and HD2 can significantly impair DUX4-DRE binding and DUX4-driven transactivation. When investigated in the context of DUX4/IGH and B-cell differentiation, structure-based mutations also abolish DUX4/IGH’s transcriptional activities and their aggressive/inhibitory roles upon B-cell differentiation. All these results have led to the novel proposal that the DUX4-DRE recognition and two-step clamp-like binding mechanism are critical for DUX4/IGH-driven transactivation and B-ALL leukemogenesis.
Materials and methods
Information concerning bacterial and eukaryotic cells, DNAs, and other materials used in this study is described in Supplementary Data. The experimental details of structure determination, functional/genetic characterizations including biolayer interferometry assay, transactivation assay, luciferase assay, B-cell differentiation assay, RNA-seq analysis etc. are also described in Supplementary Data. The statistics of the data collection and structure refinement are shown in Supplementary Table 1. Coordinates of Apo-DUX4HD2 and DUX4HD2-DNADRE have been deposited into the Protein Database Bank. The PDB codes are 5Z2S and 5Z2T, respectively.
Results
DUX4/IGH-driven transactivation in B-ALL
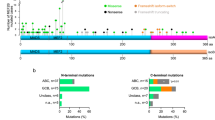
We have previously reported the genomic profiling of adult and pediatric B-cell ALL based on a cohort of Chinese patients [6]. Using RNA-seq technology, we observed that the abnormal expression of DUX4/IGHs often led to the overexpression of a subset of DUX4 target genes including PCDH17, STAP1, AGAP1, ERG alt , etc. (Fig. 1a). The expressions of DUX4 fusions were often associated with the genetic events of NRAS mutations (Supplementary Table 2). The breakpoints in DUX4 fusions were all located in the C-terminus of the protein (Fig. 1b). Consistent with previous observations [3, 4], human leukemia cell line Reh cells harboring the newly identified DUX4/IGHs showed the expression of ERGalt (Fig. 1c). All the DUX4 truncated mutants derived from Chinese patients harbor an intact N-terminal HD1–HD2 domain. This is also consistently observed in other B-ALL patients reported by the Japanese and American investigators [3, 4], suggesting that the double homeobox HD1–HD2 is critical for DUX4/IGH activity and B-ALL leukemogenesis. Supportively, it has been reported that wild-type (WT) DUX4 can selectively bind to DNA [4, 15,16,17]. Using published ChIP-seq data [10], we have investigated a number of DUX4/IGH target genes including ERGalt site TAATCTCAT (Supplementary Figure 1a,b). This has led to the discovery of a DUX4/IGH consensus binding site TAATCTAAT, and hence termed DRE (Supplementary Figure 1c).

DUX4/IGH-driven transactivation in Chinese B-ALL patients. a Heatmap of abnormal gene expression in DUX4-ALL patients (blue) and other B-ALL subgroups including MEF2D fusion (black), TCF3-PBX1 (red), ETV6-RUNX1 (green), ZNF384 (cyan) fusion, hyperdiploidy (purple), MLL fusion (brown), and BCR-ABL1 (gray). The DUX4-driven transactivation of genes in patient samples is highlighted with bracket symbol on the left. b Domain arrangement of WT DUX4 and its oncogenic fusions obtained from Chinese patients S1 to S5. DUX4/IGHJP3 was previously demonstrated to induce leukemia in mice [3]. c Transactivation activity of DUX4/IGHs. Western blot analysis of ERGalt in Reh cells overexpressing HA-tagged DUX4/IGH fusions. The bracket is used to highlight the expression of DUX4/IGHs
Crystal structures of Apo- and DNADRE-bound DUX4HD2
In order to investigate how DUX4/DUX4 fusions recognize DNA, we have determined the crystal structures of Apo-DUX4HD2 and DNADRE-bound complex at the resolutions of 1.5 and 2.6 Å, respectively (Fig. 2). The Apo structure is a classic homeobox fold with three α-helices, termed α1, α2, α3, respectively (Fig. 2a). The N-terminal α1, α2 helices are anti-parallel. The α3 helix is perpendicular to the axis of α1/α2 helices. Compatible with its DNA binding activity, the electrostatic surface of Apo-DUX4HD2 reveals a highly positively charged pocket delineated by residues Arg137, Trp141, Arg145, Arg148, and His149 in the α3 helix (Fig. 2b).

Crystal structures of Apo- and DNA-bound DUX4HD2. a Apo-form of HD2 domain. The homeobox subdomain is shown in cartoon representation. The N/C-termini and α1/α2/α3 are labeled. b Electrostatic surface of Apo-DUX4HD2. The highly positively charged patch in Apo-HD2 is delineated by residues Arg137, Trp141, Arg145, Arg148, and His149. The flexible N-terminal poly-Arg motif is shown with a dashed line. c Crystal structure of DUX4HD2-DNADRE complex. The HD2 molecules and TAATCTAAT sequences (DNADRE) are shown in cartoon representation and colored in cyan, green, and orange, respectively. d The electrostatic surfaces of the N-terminal loopRRKR and C-terminal α3 helix that engage with DNA binding. e The interface between the N-terminal RRKR motif and DNADRE minor groove. The poly-Arg residues are shown in stick representation. f The interface between the α3 helix and DNADRE major groove. Gln143 (yellow), Asn144 (red), and Arg148 (red) are shown in stick representation. The hydrogen bonds are highlighted with dashed lines. The TAAT nucleotides are labeled
The HD2-DNA complex structure reveals a clamp-like DNA-binding mechanism, in which the N-terminal R95RKR98 loop and the α3 helix interact with the TAAT site in the minor and major grooves, respectively (Fig. 2c, d). In the minor groove, the N-terminal R95RKR98 loop adopts an extended configuration making loosen interaction with the DNA backbone (Fig. 2e). In the asymmetry unit (ASU), two DUX4HD2 molecules were observed. The N-terminal R95RKR98 loop is highly flexible and might adopt different configurations for the binding of DNA minor groove. The two HD2 molecules in ASU are superimposed in the context of DNA binding. Upon DNA binding, Arg96/Lys97 or Arg95/Lys97 are utilized to make direct contract with the phosphate groups of DNA backbone in the minor groove (Supplementary Figure 2a). In compatible with the negatively charged nature of DNA molecules, the highly positively charged N-terminal RRKR loop could serve as important recruitment platform to initiate DNA binding via electrostatic interaction (see structure-based characterization below).
In the major groove, the α3 helix forms direct hydrogen bonds with DNA (Fig. 2f). The residues Arg137, Trp141, Arg145, and His149 are interacting with the DNA backbone (Supplementary Figure 2b). Compared with the Apo-DUX4HD2 structure, the guanidinium head group of Arg137 in the DNA-bound complex shifts 2.9 Å upward to form two direct hydrogen bonds with the phosphate groups in the DNA backbone. In parallel, His149, located in the opposite end of the α3 helix, moves 1.7 Å upward, flanking Arg145 via a side-chain π–π interaction. As a result, the side chains of Trp141, Arg145, and His149 all adopt extended configurations, with the alkyl side-chain in parallel to each other and the charged head groups pointing towards DNA. This has given rise to a pedestal-like base, on which the DNA backbone can rest. Inside the DNA major groove lie the residues Gln143, Asn144, and Arg148, termed QNR motif (Fig. 2f). The side-chains of these residues insert deep into the major groove reading the “barcode” of T1A2A3T4. Gln143 is located 4.6 Å away from the DRE nucleotides. Structural superimposition of Apo- and DNA-bound DUX4HD2 reveals interesting sidechain reshuffles of Asn144 and Arg148 upon the recognition of DRE. The carboxamide head group of Asn144 rotates 30° anti-clockwise to make two direct hydrogen bonds with the A2 nucleotide. In a concerted fashion, the guanidinium head group of Arg148 swings 5.3 Å into the major groove to form hydrogen bonds with the A3 site. The minor- and major-groove binding residues including RRRRHD1–RRKRHD2, QNRHD1–QNRHD2 motifs are highly conserved in DUX protein family (Supplementary Figure 2c), among which HD1 shares 54% sequence identity with HD2. In particular, Asn144 is strictly conserved in homeobox superfamily (Supplementary Figure 2d), highlighting the significance of DNA binding ability in DUX4/IGH-driven transactivation.
DUX4-DRE is important for DNA binding, transactivation, and B-cell differentiation
To characterize the crystal structure of DUX4HD2-DNADRE, the biolayer interferometry (BLI) was used. The recombinant DUX4HD2 and DUX4HD1–HD2 were shown to bind to a DRE sequence at K D values of 7.2 and 1.4 μM, respectively (Fig. 3a and Supplementary Figure 3a). By contrast, the mutations of R95A, R96A, K97A, and R98A in the poly-Arg motif significantly abolished the DNA binding activity of DUX4HD2 (Fig. 3b). Consistently, in the α3 helix region, Q143E, N144E, and R148E resulted in 4- to 9-fold weaker binding. The measured K D values of these mutants were 31, 55, and 67 μM, respectively (Fig. 3c). Supportively, similar structure-based mutations in DUX4HD1–HD2 completely abrogated DNA binding activity (Supplementary Figure 3b).

The BLI characterization of DNADRE binding by DUX4HD2 and mutants. a The interaction between DUX4HD2 and DNADRE. a WT DUX4HD2. b DUX4HD2 mutations in the minor groove binding residues, i.e., poly-Arg motif. c DUX4HD2 mutations in the major groove binding residues, i.e., QNR motif. A gradient concentration of 0.5–32 μM recombinant DUX4HD2 was used. Under these experiment conditions, the DNADRE was immobilized on the probe. Left, the association and dissociation curves. Right, the binding curves and K D values derived from Scatchard plots [32]
As previously reported [4], the ERGalt biogenesis could be used to monitor the transactivation activities of DUX4/IGH and mutants. The WT DUX4/IGH transactivated the expression of ERGalt (Fig. 4a and Supplementary Figure 4). Contrarily, the perturbation of DNA binding residues abrogated the biogenesis of ERGalt. The poly-Ala mutations of RRRRHD1–RRKRHD2 and QNRHD1–QNRHD2 motifs in either HD1 or HD2 completely abolished the transactivation activity of DUX4/IGH, suggesting that HD1 and HD2 domains are both required for DRE recognition.

DRE recognition is important for DUX4/IGH-driven transactivation and B-cell differentiation. a Transactivation activity of DUX4/IGH and mutants in Reh cells. Left column, the biogenesis of ERGalt in Reh cells that contain WT and mutant DUX4/IGHs was detected by western blot. Right column, the real-time PCR technique was used to confirm the DUX4-driven transactivation at mRNA level. Top row, mutations in HD2. Middle row, mutations in HD1. Bottom row, double mutations in HD1 and HD2. All experiments have been done at least with three independent replicates. b In vitro B cell differentiation assay of DUX4/IGH and mutants. FACS technique was used to analyze the effect of DUX4/IGH and mutants on the differentiating ability of mouse progenitor cells (Lin−/c-KitLow) into CD19 positive B cells. All experiments have been done at least with three independent replicates. Values are means ± SE
In order to investigate which DNA-binding motif (RRRRHD1–RRKRHD2 or QNRHD1–QNRHD2) could be more important, a series of single mutations were designed. The DUX4/IGH-driven transactivation activities (i.e., the abnormal biogenesis of ERGalt) of wild type and mutants were monitored in Reh cells. The single mutation in QNR motifs (i.e., N69A, R73A in HD1 and N144A, R148A in HD2) was sufficient to inhibit the DNADRE recognition and abolish subsequent ERGalt transactivation (Fig. 4a). Interestingly, the single mutations, Q68A and Q143A, failed to inhibit DUX4/IGH-driven transactivation. In marked contrast, the double Q68/143A mutation dramatically reduced ERGalt biogenesis, suggestive of rescue effect between HD1 and HD2 (Fig. 4a). Consistently, the complementary binding mode was also observed in the single mutations of RRRRHD1–RRKRHD2. Although single mutation in RRKRHD2 could preclude the formation of DUX4HD2-DNADRE (Fig. 3b), most single mutations in RRKRHD2 were less destructive, resulting in partial disruption of DUX4-driven transactivation (Fig. 4a), implying the DNA binding between DUX4/IGH and DNA might be rescued by the equivalent poly-Arg motif in HD1. Indeed, when an extra Arg/Lys to Ala mutation was engineered in the equivalent position (such as R20/95A, R21/96A, R22/K97A, R23/98A), the accumulative impacts were very clear in spite of the fact that the QNRHD1–QNRHD2 motif remained intact in these mutants (Fig. 4a). Supportively, similar results can be obtained using NALM6 cells (Supplementary Figure 4). All these results support a concerted binding clamp-like binding between HD1 and HD2. Furthermore, as shown in Fig. 4a, the single mutations in HD1 appear to be more destructive than those of HD2, prompting the hypothesis that two HDs might have different roles in DNA binding, in which HD1 might be more affinitive subunit to engage DRE site.
Next, we want to know whether the DRE recognition via HD domains could play a key role in disease mechanism. As demonstrated elsewhere [3], the leukemogenic activity of DUX4/IGH is constantly associated with the arrest of B-cell differentiation. We therefore addressed the phenotypic effect of distinct fusion proteins with WT or mutated DRE-binding motifs. In control experiment, ~85% primary murine progenitors transfected with vehicle (empty vector) could undergo normal B-cell differentiation as assessed by flow cytometry using antibodies against mouse CD19 (Fig. 4b). DUX4/IGH-transfected cells, by contrast, showed much less B-cell differentiation (~12%), in concordance with its transforming ability and leukemogenic effects [3]. The perturbations in the RRRRHD1–RRKRHD2 and QNRHD1–QNRHD2, which hampered the DNA binding and transactivation, also abrogated the inhibitory activity of DUX4/IGH on B-cell differentiation. As shown in Fig. 4b, most mutations rescued the lymphoid lineage differentiation at levels similar to that of the cells transfected with vehicle (i.e., 66 – 90% differentiation for R20/95A, R21/96A, R23/98A, N69/144A, R73/148A). Of note, cells transfected with R22/K97A and Q68/143A still bore certain levels of ERGalt expression (Fig. 4a) and showed only partially restored differentiation compartments in FACS analysis (44 and 27%, respectively) (Fig. 4b). These results taken together not only support our observation in the crystal structure of DUX4HD2-DNADRE, but also suggest DUX4HD1–HD2-DRE interacting motif as a potential therapeutic target for future treatment against B-ALL harboring DUX4/IGH fusion. Supportively, the structure-based mutations also inhibit the apoptotic function of WT DUX4 (Supplementary Figure 5a), and its transactivation ability on target gene as monitored by luciferase assay (Supplementary Figure 5b). Altogether, these have led to hypothesis that WT DUX4 and DUX4/IGH might share a similar DNA binding mechanism (see below).
However, until now, it is not clear how the C-terminal domain (CTD) of DUX4, whose molecular boundary is not yet clearly defined, might contribute to the transactivation activities of WT DUX4 and DUX4/IGHs. WT DUX4, overexpression of which often triggers apoptosis, is the main reason of FSHD disease [10]. In marked contrast, the loss/break-down of CTD has enabled DUX4/IGH to cause B-cell arrest and proliferation, and hence serves as an oncogenic driver in leukemia [3, 4]. In order to understand how CTD might influence the transactivation activity of DUX/IGH, a series of CTD truncated mutants were designed (Fig. 5a). Interestingly, when the CTD was restricted to residue 200, the DUX4/IGH activity was significantly impaired (Fig. 5b, c). Consistently, this correlates with the observation in B-ALL patients. The shortest version of DUX4/IGH derived from Patient S2 also displayed less ERGalt expression (Fig. 1c), suggestive of CTD contribution in DUX4/IGH and B-ALL. However, the size of CTD is not the only structural factor in DUX4/IGH’s transactivation activity. DUX41-412/IGH from Patient S5 displayed less ERGalt expression than those shorter fusion derivatives from Patients S3 and S4 (Fig. 1b, c). Similar results could be observed in CTD truncated mutants (Fig. 5b, c), suggesting that the overall structure/fold of CTD, which remains elusive, might play critical role in DUX4/IGH-driven transactivation.

The effect of C-terminal domain in DUX4/IGH-driven transactivation. a Scheme of CTD truncated mutants. The biogenesis of ERGalt in Reh cells by WT DUX4/IGH and C-terminal truncated mutants were monitored by western blot (b) and RT-PCR (c)
Abnormal transactivation controlled by DUX4HD1–HD2-DNADRE
The RNA-seq data mining of our 172 patients showed that the expression profile of DUX4/IGH positive ALL was significantly different from other B-ALL subtypes (Fig. 1a). Notably, a group of genes such as AGAP1, CHST2, CLEC12A, PCDH17, PTPRM, STAP1, etc., which all contain DREs, were found upregulated in patients harboring DUX4 fusions. This is further supported by the observation that knock-down (KD) of DUX4/IGH through shRNA in NALM6 cells [3], which contains an endogenous DUX4/IGH fusion, downregulated these target genes (Supplementary Figure 6a). We next investigated the function of these DUX4/IGH-targeted genes by FACS profiling and apoptotic assay. Firstly, FACS profiling showed that the over-expression of AGAP1, but none of other target genes, led to a partial arrest of B-cell differentiation in murine progenitor cells (Supplementary Figure 6b). Secondly, the genetic KD of AGAP1 in NALM6 cells exerted a significant impact on apoptosis, whereas KD of other DUX4/IGH target genes (except for CHST2) did not show obvious apoptotic effects (Supplementary Figure 6c). More importantly, as monitored by luciferase assays, the structure-based mutations significantly abolished the DUX4/IGH-driven transactivation of AGAP1 (Supplementary Figure 6d). Supportively, abnormal expression of AGAP1 is frequently observed in B-ALL patients with DUX4 fusions reported elsewhere [4, 18]. These results highlight a critical DUX4HD1–HD2-DNADRE recognition in B-ALL pathogenesis (Fig. 6). However, although AGAP1 displays potential in B-cell differentiation, further investigation such as in vivo characterization involving the use of knock-out mice model is required to establish the leukemogenic role of DUX4/IGH target gene.

Oncogenic network controlled by DUX4HD1–HD2-DNADRE recognition in B-ALL pathogenesis. A two-step cooperative binding/transactivation mechanism via HD1 and HD2 domains. In the first step (top-left panel), poly-Arg motifs lead the initial DNA binding via a non-specific eletrostatic attraction. This in turn primes the second step (top-right panel) for the intimate/specific clamp-like association between DUX4-DNADRE via QNRHD1–QNRHD2 motifs. The two-step DUX4/IGH is a critical step in B-ALL leukemogenesis (low panel). Based on 54% sequence identity, the HD1 model in this figure is built by homology modeling using DUX4HD2 coordinates. The covalent linkage between HD1 and HD2 is shown in dashed line. CTD is indicated with octagon. The symbols “?” are used to highlight/prompt further investigations to understand (1) what role CTD might play in DUX4- and DUX4/IGH-driven transactivations; (2) whether DUX4/IGH target genes such as AGAP1 could accelerate/or is required in DUX4/IGH-mediated leukemogenesis
Discussion
Protein diversity in DUX4
DUX4, a member of homeobox protein family, is exclusively found in placental mammals [19, 20]. Compared to other homeobox proteins, DUX4 contains two adjacent HD1 and HD2 domain, connected by a short linker of 15 residues. Thus far, no structural information is available for the double homeobox. In this section, we will focus our discussion on the protein diversity in DUX4 double homeobox.
It has been proposed that DUX4 belongs to the PAIRED (PRD) homeobox subclass because their HD domains share the most sequence identity (Supplementary Figure 2d) [21]. In addition, the tandem arrangement of double homeobox is in striking similarity with the domain architecture observed in PRD proteins. The PAI sub-domain is a homeobox, and the RED sub-domain can fold into a HD-like structure with three α-helices [20, 22]. Hence PAI-RED appears to be a double-homeobox-like structure. However, it has been shown that the RED sub-domain is not really a HD domain in term of DNA binding and recognition. As demonstrated in the structural and functional study of PAXPAIRED-DNA complex, the RED sub-domain is not essential in DNA binding [23]. In line with this observation, many studies have shown that a single homeobox domain is not sufficient to mediate DNA binding/recognition for productive transactivation. Thus far, it has been reported that homeobox proteins often require: (1) the protein–protein interaction involving a second transcription factor; (2) inter-molecular dimerization; and (3) an extra intra-molecule DNA binding domain to bind DNA [24,25,26,27].
In DUX4, the presence of double homeobox appears to be sufficient for DNA recognition and transactivation. The gain of an extra HD domain in DUX4, possibly derived from gene reshuffle and infusion [19], gives rise to a powerful transcription factor for TAATCTAAT (DRE) site (Supplementary Figure 1). In B-ALL patients, nearly all the DUX4/IGHs contain a structural and functional intact double homeobox, leading to the hypothesis that the DUX4 double homeobox might play the central role in DNA binding and transactivation. Indeed, when the Reh cells were transfected with DUX4 fusion with different IGH tails, ERGalt biogenesis were consistently observed (Fig. 1b, c). When the endogenous expression of DUX4/IGH was suppressed by shRNA, the target genes containing DRE sites were consistently downregulated (Supplementary Figure 6a). More importantly, when the DNA binding interface in either DUX4HD1 or DUX4HD2 was mutated, the binding affinity and transactivation function were severely impaired (Figs. 3 and 4). Altogether, these results suggest that the evolutionary tinkering of double homeobox design is an important mean to endow DUX4 with transactivation activity.
Two-step clamp-like binding mechanism
In this report, the crystal structure of DUX4HD2-DRE reveals a novel two-step clamp-like DNA binding mechanism for double homeobox. In the asymmetric unit (ASU), it contains two HD2 molecules and a DNA duplex of TAATCTAAT (DRE) sequence. Based on homology modeling, a HD1 molecule can be mapped onto DRE site. Supportively, the C-termini of HD1 and the N-termini of HD2 are both correctly positioned, giving ample space to envisage the missing linker in between. More importantly, the QNRHD1 motif is predicted to insert into the DNA major groove engaging the direct hydrogen bonds with TAAT. In parallel, the RRRR motif of HD1 wraps around the DNA minor groove (Fig. 6). All these data suggest that: (1) HD1 can bind TAAT sequence in the same way as HD2 and (2) the presence of an extra HD domain might enhance its overall binding. Indeed, the BLI characterization showed that the double homeobox binds to a DRE sequence at a K D value of 1.4 μM, >5 fold stronger than HD2 alone (Fig. 3a and Supplementary Figure 3a).
The two-step binding mode is further supported by structure-based mutagenesis. The single mutation of QNRHD1–QNRHD2 (i.e., N69A, N144A, R73A, R148A) could ~100% inhibit transactivation, suggesting both HD1 and HD2 are essential. In marked contrast, single mutations of RRRRHD1–RRKRHD2 were less dramatic, implying that (1) poly-Arg motif is less critical when compared to the QNR motif; (2) the existence of cooperative binding between HD1 and HD2. This is further supported by the observation that double mutations of RRRRHD1–RRKRHD2 (i.e., R20/95A, R21/96A, R22/K97A, R23/98A) resulted a complete loss of transactivation even in the presence of QNRHD1–QNRHD2 motifs (Fig. 4). This is also the case in BLI characterization, the poly-Glu mutation of RRRR–RRKR resulted in no binding between double homeobox and DNA (Supplementary Figure 3b). These data appear to suggest that the DNA binding of poly-Arg motifs might precede the interaction of TAAT-QNR. Based on these observations, we proposed a two-step clamp-like DRE recognition mechanism by DUX4 double homeobox (Fig. 6). In the first step, the highly positively charged RRRRHD1–RRKRHD2 is important to direct DUX4 towards its DNA substrate (via electrostatic interaction). In this step, the HD1 subdomain might take the leading role to make the first kiss with target DNA. The non-specific association between RRRRHD1–RRKRHD2 with DNA in turn enables the double homeobox to recognize the DRE site via QNRHD1–QNRHD2. The double kiss between TAATs and QNR motifs might be critical to “clamp” DUX4-DRE into an intimate association that enables productive transactivation of its target genes (Fig. 6). Similar DNA binding mode should also present in WT DUX4 (Supplementary Figure 5). In addition, the CTD domain, which clear contributes to DUX4/IGH-driven transactivation (Fig. 5), should remain an interesting subject for future investigation in the context of WT DUX4 and its oncogenic derivatives.
Oncogenic potential of DUX4-DRE
It has been reported that homeobox-containing protein including PRD-type subclass is involved in hematopoiesis and leukemogenesis [28]. The abnormal expression of PAX5, a member of the PRD-type subclass, derived from chromosome translocation with the IGH locus can deregulate the lymphoid cell gene expression program and contribute to B and T-lineage neoplasms [29,30,31]. Recent studies showed that, the homeobox-containing DUX4 can also translocate into the IGH locus, and the aberrant expression of DUX4 can cause B-cell leukemia in immunocompromised mice [3]. A follow-up study by Zhang and co-workers showed that the aberrant expression of DUX4/IGHs were constantly associated with the production of a novel ERGalt isoform that can also contribute to leukemogenesis in vivo [4]. These observations have led to the proposal of DUX4/IGH-ERGalt axis in B-ALL development. However, not all patients in DUX4/IGH subgroup were ERGalt positive (Supplementary Table 2), suggestive of other genetic lesion in B-ALL. The RNA-seq data mining of our 172 patients showed that the expression profile of DUX4/IGH positive ALL was significantly different from other B-ALL subtypes (Fig. 1 and Supplementary Figure 1). Using FACS and apoptotic assays, we have found that AGAP1, like DUX4/IGH and ERGalt, could contribute to B cells proliferation and differentiation. However, it should be noted, cytoplasm-bound AGAP1 is not a transcription factor like ERG or ERGalt, and its oncogenic pathway to B-cell arrest is not yet clear. Further investigations such as in vivo study in the background of AGAP1 knock-out mice should be carried out to characterize its leukemogenic cross-talk with DUX4/IGH and ERGalt.
References
de The H, Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer. 2010;10:775–83.
Hehlmann R, Hochhaus A, Baccarani M, European LeukemiaNet. Chronic myeloid leukaemia. Lancet. 2007;370:342–50.
Yasuda T, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48:569–74.
Zhang J, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48:1481–1489.
Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373:1541–52.
Liu YF, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–83.
Lilljebjorn H, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7:11790.
Gatica LV, Rosa AL. A complex interplay of genetic and epigenetic events leads to abnormal expression of the DUX4 gene in facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2016;26:844–52.
Lemmers RJ, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–4.
Geng LN, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 2012;22:38–51.
Loughran SJ, et al. The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat Immunol. 2008;9:810–9.
Tursky ML, et al. Overexpression of ERG in cord blood progenitors promotes expansion and recapitulates molecular signatures of high ERG leukemias. Leukemia. 2015;29:819–27.
Tsuzuki S, Taguchi O, Seto M. Promotion and maintenance of leukemia by ERG. Blood. 2011;117:3858–68.
Thoms JA, et al. ERG promotes T-acute lymphoblastic leukemia and is transcriptionally regulated in leukemic cells by a stem cell enhancer. Blood. 2011;117:7079–89.
Krom YD, et al. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet. 2013;9:e1003415.
De Iaco A, et al. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat Genet. 2017;49:941–5.
Hendrickson PG, et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet. 2017;49:925–34.
Harvey RC, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–84.
Leidenroth A, Hewitt JE. A family history of DUX4: phylogenetic analysis of DUXA, B, C and Duxbl reveals the ancestral DUX gene. BMC Evol Biol. 2010;10:364.
Burglin TR, Affolter M. Homeodomain proteins: an update. Chromosoma. 2016;125:497–521.
Holland PW. Evolution of homeobox genes. Wiley Interdiscip Rev Dev Biol. 2013;2:31–45.
Gehring WJ, Affolter M, Burglin T. Homeodomain proteins. Annu Rev Biochem. 1994;63:487–526.
Xu W, Rould MA, Jun S, Desplan C, Pabo CO. Crystal structure of a paired domain-DNA complex at 2.5 A resolution reveals structural basis for Pax developmental mutations. Cell. 1995;80:639–50.
Verrijzer CP, et al. The DNA binding specificity of the bipartite POU domain and its subdomains. EMBO J. 1992;11:4993–5003.
Grueneberg DA, Natesan S, Alexandre C, Gilman MZ. Human and Drosophila homeodomain proteins that enhance the DNA-binding activity of serum response factor. Science. 1992;257:1089–95.
Wilson DS, Guenther B, Desplan C, Kuriyan J. High resolution crystal structure of a paired (Pax) class cooperative homeodomain dimer on DNA. Cell. 1995;82:709–19.
Wilson D, Sheng G, Lecuit T, Dostatni N, Desplan C. Cooperative dimerization of paired class homeo domains on DNA. Genes Dev. 1993;7:2120–34.
Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64.
Iida S, et al. The t(9;14)(p13; q32) chromosomal translocation associated with lymphoplasmacytoid lymphoma involves the PAX-5 gene. Blood. 1996;88:4110–7.
Busslinger M, Klix N, Pfeffer P, Graninger PG, Kozmik Z. Deregulation of PAX-5 by translocation of the Emu enhancer of the IgH locus adjacent to two alternative PAX-5 promoters in a diffuse large-cell lymphoma. Proc Natl Acad Sci USA. 1996;93:6129–34.
Souabni A, Jochum W, Busslinger M. Oncogenic role of Pax5 in the T-lymphoid lineage upon ectopic expression from the immunoglobulin heavy-chain locus. Blood. 2007;109:281–9.
Abdiche Y, Malashock D, Pinkerton A, Pons J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal Biochem. 2008;377:209–17.
Acknowledgements
This work was supported by research grants 81770142, 81370620, 81570120, 31070645 from National Natural Scientific Foundation of China (to GM), a research grant 20152504 from “Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant Support” (to GM), “The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institute of Higher Learning” (to GM), a research grant 11JC1407200 from SMSTC (to GM), a research grant 12ZZ109 from SME (to GM), We thank the personnel of beamlines BL17U/18U1/19U1 (SSRF/NFPS, Shanghai, China) for help during data collection.
Author contributions
Conceived and designed the experiments: ZC, S-JC, GM. Performed the experiments: XD, WZ, HW, JH, MZ, PW, HZ. Analyzed the data: XD, WZ, HW, JH, MZ, PW, HZ, ZC, S-JC, GM. Preparation of the figures: XD, WZ, HW, JH, ZC, S-JC, GM. Wrote the paper: GM.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, X., Zhang, W., Wu, H. et al. Structural basis of DUX4/IGH-driven transactivation. Leukemia 32, 1466–1476 (2018). https://doi.org/10.1038/s41375-018-0093-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0093-1
This article is cited by
-
Acute lymphoblastic leukaemia
Nature Reviews Disease Primers (2024)
-
Emerging molecular subtypes and therapeutic targets in B-cell precursor acute lymphoblastic leukemia
Frontiers of Medicine (2021)
-
Long-read sequencing unveils IGH-DUX4 translocation into the silenced IGH allele in B-cell acute lymphoblastic leukemia
Nature Communications (2019)
-
DUX4HD2-DNAERG structure reveals new insight into DUX4-Responsive-Element
Leukemia (2019)
