
Abstract
Tens-of-thousands of chemicals are registered in the U.S. for use in countless processes and products. Recent evidence suggests that many of these chemicals are measureable in environmental and/or biological systems, indicating the potential for widespread exposures. Traditional public health research tools, including in vivo studies and targeted analytical chemistry methods, have been unable to meet the needs of screening programs designed to evaluate chemical safety. As such, new tools have been developed to enable rapid assessment of potentially harmful chemical exposures and their attendant biological responses. One group of tools, known as “non-targeted analysis” (NTA) methods, allows the rapid characterization of thousands of never-before-studied compounds in a wide variety of environmental, residential, and biological media. This article discusses current applications of NTA methods, challenges to their effective use in chemical screening studies, and ways in which shared resources (e.g., chemical standards, databases, model predictions, and media measurements) can advance their use in risk-based chemical prioritization. A brief review is provided of resources and projects within EPA’s Office of Research and Development (ORD) that provide benefit to, and receive benefits from, NTA research endeavors. A summary of EPA’s Non-Targeted Analysis Collaborative Trial (ENTACT) is also given, which makes direct use of ORD resources to benefit the global NTA research community. Finally, a research framework is described that shows how NTA methods will bridge chemical prioritization efforts within ORD. This framework exists as a guide for institutions seeking to understand the complexity of chemical exposures, and the impact of these exposures on living systems.
Similar content being viewed by others
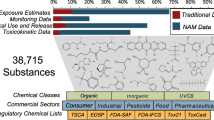
Introduction
The last decade has witnessed pronounced transformations in approaches for linking chemical exposures to human and ecological health. Toxicity testing methods that support chemical safety evaluations have evolved rapidly, ushering in an era defined by high-throughput screening (HTS) and chemical prioritization [1, 2]. Two US-based testing programs—the Toxicity Testing in the 21st Century (Tox21) Federal Consortium and the EPA Toxicity Forecaster (ToxCast) project—have together evaluated over 8000 chemical substances across hundreds of bioassays [3,4,5]. Efforts are underway to map the derived bioactivity data to key events along adverse outcome pathways (AOPs) in support of 21st century risk assessments and regulatory decisions [6, 7]. Risk-based decisions, however, are weakened without quantitative knowledge of exposure, processes that link exposure and target dose, and the impact of target dose on AOPs [8,9,10,11]. Noting this challenge, the exposure science community has mirrored recent advances in toxicity testing, developing both predictive and empirical methods for rapid acquisition of chemical exposure data [8, 9, 12, 13]. Many measurement-based methods are borne out of successes in the metabolomics field. For example, high-resolution mass spectrometry (HRMS), a common metabolomics tool, now allows rapid characterization of hundreds to thousands of compounds in a given environmental (e.g., surface water), residential (e.g., house dust), or biological (e.g., serum) sample. Whereas metabolomics has mostly eyed endogenous compounds, the emerging field of “exposomics” has broadened the analytical focus to include xenobiotic compounds [14, 15]. Popular open metabolomics databases, in fact, are expanding to include large lists of man-made compounds, as well as known and predicted metabolites of xenobiotics and naturally-occurring compounds [16,17,18,19]. Furthermore, software developers are adapting existing tools, and developing new tools, to better meet the needs of the growing exposomics community [20, 21]. In time, these adaptations will enable fully integrated research workflows that seamlessly bridge empirical knowledge of stressors and biological adaptations to those stressors [10, 18].
The concept of the “exposome” was introduced in 2005 by Dr. Christopher Wild as a way to represent all life-course environmental exposures from the prenatal period onwards [22]. Since that time, exposomics, like any nascent field, has evolved in concept, definition, and practice. While multiple definitions now exist, it is generally agreed upon that the exposome represents the totality of exposures experienced by an individual (human or other), and that these exposures reflect exogenous and endogenous stressors originating from chemical and non-chemical sources [23, 24]. By definition, chemical components of the exposome are measureable in media with which a receptor comes into contact. For humans, these media include—but are not limited to—food, air, water, consumer products (e.g., lotions), articles (e.g., clothing), house dust, and building materials. Biological media further offer a window into the exposome, and have been a focus of many analytical efforts [25,26,27,28].
In most instances, analytical chemistry-based exposome research has moved away from “targeted” methods and towards suspect screening analysis (SSA) and non-targeted analysis (NTA) methods. Suspect screening studies are those in which observed but unknown features (generally defined in HRMS experiments by an accurate mass, retention time [RT], and mass spectrum) are compared against a database of chemical suspects to identify plausible hits [21, 29]. True NTA (also called “untargeted”) studies are those in which chemical structures of unknown compounds are postulated without the aid of suspect lists [21, 29]. While clear differences exist in the methods used for SSA and NTA, the term “non-targeted analysis” is commonly used to describe both SSA and NTA experiments. As such, the abbreviation “NTA” is used here in a general sense to describe this entire genre of research. Within this NTA realm, emphasis is generally placed on characterizing compounds that are unknown or poorly studied, and, more importantly, on examining compounds that are significantly related to an exposure source (environmental forensics), health status, or some other measure of interest. NTA studies are gaining in popularity [30], but the rapid and accurate characterization of large suites of chemical unknowns remains challenging. Appropriate resources and efficient methods must therefore be identified to propel NTA methods away from a niche field and into mainstream public health laboratories.
EPA’s Office of Research and Development (ORD) has pioneered many HTS strategies for toxicity testing, exposure forecasting, and risk-based prioritization over the past decade. In support of these efforts, EPA’s ToxCast project, administered within the National Center for Computational Toxicology (NCCT), has procured and manages a rich library of individual chemicals [5]. NCCT further develops, curates, and manages databases and dashboards that house information on these and many other compounds of relevance to environmental health. Whereas these collective tools are the basis for EPA’s HTS activities (designed to potentially inform regulatory decisions), they have seldom been considered as resources for the exposomics research community, and remain underutilized in NTA experiments. In a recent article [31], we demonstrated the power of ORD resources for guiding novel NTA workflows. Our pilot-scale study showed that ORD tools can be effectively used to identify, prioritize, and confirm novel compounds in samples of house dust. It further indicated that certain novel compounds (i.e., those never before measured in house dust) are ubiquitous environmental contaminants and likely to activate specific biological pathways. Additional studies have reported similar findings based on analyses of house dust and other media [32,33,34]. Together, these studies underscore a limited understanding of the compounds present in our environments. Yet, they also highlight a need for, and clear advantage of, integrating NTA research efforts with those already established to support risk-based chemical prioritization.
The purpose of this article is to provide a clear road map for integrating NTA research with current chemical screening initiatives. The article first discusses NTA methods as tools for discovering the exposome. It then provides a brief history and synopsis of current activities within ORD, with specific emphasis on activities that relate to NTA research. A summary of an EPA-led collaborative trial is then presented, which exploits ORD resources to advance NTA research efforts. A multi-step framework is finally offered, which is being used by EPA scientists to maximize data used in, and knowledge gained from, NTA experiments. The information provided herein will enable NTA practitioners to make greater use of valuable resources that service 21st century chemical testing programs. It will further allow scientists and decision makers to make direct use of NTA data when performing risk-based chemical prioritizations. Together these actions will enable more efficient, comprehensive, and relevant evaluations of chemical safety.
Methods, results, and discussion
NTA as a tool for exposome research
The concept of the exposome has been in existence for more than a decade. During this period, a number of modified definitions have been proposed to place emphasis on: 1) external vs. internal exposure sources (e.g., the “eco-exposome” [8] and the “endogenous exposome” [35]); 2) research applications for specific media (e.g., the “blood exposome” [24, 36] and the “tooth exposome” [37]); and 3) general analytical strategies (e.g., “top-down exposomics” vs. “bottom-up exposomics” [23, 38]). Regardless of the definition and application, it is generally agreed that NTA methods are a key to discovering the breadth of all exposures, and more importantly, which exposures are associated with disease. Different portions of the exposome have now been characterized using suites of analytical tools, which range from low resolution gas chromatography mass spectrometry (GC/MS) platforms, to ultra-high resolution Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR/MS) platforms. Many recent investigations have focused on polar organic compounds, which are often readily detected using liquid chromatography (LC) coupled with high resolution Orbitrap or time-of-flight mass spectrometry (TOF/MS) [28]. Hybrid systems, such as quadrupole-Orbitrap and quadrupole-TOF mass spectrometers (Q-TOF/MS), further enable compound identification using both precursor ion detection in full-scan MS mode, and product ion detection in MS/MS mode. These HRMS hybrid systems are quickly becoming the most commonly used tools in NTA laboratories [28].
High-resolution MS instruments generate data on thousands of molecular features, which represent unknown compounds generally described in terms of their monoisotopic masses, retention times, and isotope distributions. In some cases, these data are accompanied by fragmentation spectra (via MS/MS analysis) and predicted molecular formulae. The job of the analyst is to proffer chemical structures that are consistent with these observed features. Current guidance recommends binning structures based upon the certainty of assignment [39]. “Tentative candidates” are proposed structures that are consistent with experimental data, but not necessarily unequivocal top hits. “Probable structures” are those not confirmed with standards but named as top candidates using library spectrum matches and other diagnostic evidence (e.g., RTs associated with a specific method). Finally, “confirmed structures” are those that have been verified using a reference standard.
Multiple tentative candidates can exist for a given molecular feature. As such, it is expected that, for a given NTA experiment, the number of compounds within each bin will be ordered as follows: tentative candidates > probable structures > confirmed structures. Exact ratios across bins vary from lab-to-lab and medium-to-medium based on available resources (e.g., authentic standards), tools (e.g., MS/MS-enabled platforms), and experience/expertise. Yet, it is clear that the number of unknowns will continue to outweigh the number of knowns for the foreseeable future. The goal, then, is to enable knowledge-based ascension, for any feature of interest (e.g., those associated with measures of biological perturbation), from labeling as a tentative candidate, to probable structure, to confirmed compound.
The rise from tentative candidate to probable structure is conditional upon the availability of sufficient diagnostic evidence. Probable structures are generally those that have high-scoring library spectrum matches, relatively large numbers of sources or references in public databases, and predicted retention behavior that is consistent with observations about the unknowns [39]. A number of open access tools exist for ranking tentative candidates and naming probable structures (e.g., MetFrag [http://c-ruttkies.github.io/MetFrag/] and STOFF-IDENT [https://www.lfu.bayern.de/stoffident/#!home]. These tools, as well as those available from instrument vendors, often rely on large public databases (e.g., ChemSpider [http://www.chemspider.com/] and PubChem [https://pubchem.ncbi.nlm.nih.gov/]) for the initial identification of tentative candidates, and subsequent ranking based on data sources/references. Some tools predict and evaluate retention behavior using logP-based or logD-based models that vary in sophistication [40]. Finally, to enable spectral matching, most tools utilize existing reference spectra, which are available via vendors and open databases (e.g., mzCloud™ [https://www.mzcloud.org/], MassBank [http://www.massbank.jp/index.html?lang=en], and MoNA [http://mona.fiehnlab.ucdavis.edu/]), or theoretical spectra, which are generated from fragmentation prediction tools such as CFM-ID (http://cfmid.wishartlab.com/), MetFrag (http://c-ruttkies.github.io/MetFrag/), and MAGMa [41].
The combination of these approaches has proven successful in the characterization of unknowns in a variety of media. Yet, opportunities exist to further enhance these tools for future investigations. For example, there is a growing need extend screening libraries to include not just known parent chemicals, but predicted metabolites and environmental degradants—compounds which are believed to comprise a substantial portion of the exposome [16]. Indeed, as stated in a recent review by Escher and colleagues “…a very small number of the thousands of compounds detectable in a sample can actually be identified, leaving the largest fraction of chemicals at the level of a known accurate mass (or molecular formula) and retention time. Any improvements here rely strongly on a better assignment of likely structures… based on a prediction of fragmentation, ionization, or chromatographic retention times supported by more comprehensive mass spectra databases” [10]. From these statements it is clear that significant improvements to NTA workflows are needed, as are appropriate resources (e.g., chemicals on which to build reference databases and model training sets) that can enable these improvements. The following section details projects and resources within ORD that are now being used, by EPA scientists and the broader scientific community, to enhance NTA methods and workflows.
Highlights from EPA’s Office of Research and Development
High throughput bioactivity screening and the ToxCast project
In 2007, the National Research Council (NRC) of the National Academies of Science (NAS) published “Toxicity Testing in the 21st Century”, a report calling for greater focus on mechanistic (i.e., pathway-based) understanding of toxicity [2]. At that time, the advent of HTS had enabled the pharmaceutical industry to: 1) rapidly screen many hundreds or thousands of chemicals; 2) screen against targets having greater relevance to humans; and 3) make specific inferences pertaining to the biological pathways involved with toxicity [42]. In many cases, the potential for bioactivity within human or specific ecological species could be targeted using in vitro methods, along with proteins and cells derived from tissues of the species in question. Noting these advancements, and the recommendations of the NRC, the National Institutes of Health (NIH) National Toxicology Program (NTP), the NIH National Center for Advancing Translational Sciences (NCATS), and EPA formed the Federal Tox21 consortium, which was soon joined by the US Food and Drug Administration (FDA). The goal of this consortium was to use modern HTS approaches to better assess chemical toxicity, especially for many thousands of chemicals for which little or no toxicity data were available [1]. To date, over 8000 chemical substances (including pharmaceuticals, plasticizers, pesticides, fragrances, and food additives) have been tested, robotically and uniformly at the NCATS intramural testing facility, across over 100 HTS assays (consisting of nuclear receptor target assays and cell-based viability assays) [3].
The EPA-contributed portion of Tox21 includes more than 3800 unique compounds. Many of these compounds have undergone additional HTS across more than 800 assay endpoints as part of a separate EPA testing program, known as the ToxCast project [4]. This EPA testing program has expanded in tandem with the Tox21 program, enlisting a number of contract-administered, commercially available assay systems, many of which were originally developed to service the pharmaceutical industry’s drug discovery programs. ToxCast assay technologies span a broad suite of high and medium-throughput screening targets and cell-based systems, and provide for more extensive biological screening of EPA’s ToxCast library, effectively complementing the available Tox21 assays. ToxCast testing has been conducted in phases. The Phase I library included 310 compounds, which were primarily pesticides that have been well characterized by animal toxicity studies, along with small sets of high-priority environmental chemicals (e.g., bisphenol A [BPA]) and toxicologically active metabolites (e.g., mono(2-ethylhexyl)phthalate [MEHP]). Phase II testing examined Phase I chemicals across new assays. It further broadened the chemical library to include more than 700 industrial chemicals, known toxicants and carcinogens, alternative “green” chemicals, food-additives, and failed pharmaceuticals. Phase II testing also included ~800 additional chemicals that underwent limited testing in endocrine-relevant assays only. A rolling “Phase III” is ongoing with the goals of: 1) broadening assay endpoint coverage across the nearly 1800 compounds in the Phase II library, 2) expanding upon the Phase II library with newly added priority chemicals, and 3) applying strategic testing to the larger EPA Tox21 library [5].
ToxCast HTS is typically conducted in concentration-response format, with statistical analysis used to estimate the concentration of chemical needed to cause bioactivities in any given assay [4]. Many chemical-assay combinations are inactive at even the highest tested concentration [43, 44]. Those assays that show systematic response with concentration are referred to as “hits”, with a portion of assay hits occurring at concentrations below ranges of cytotoxicity. A series of statistically-derived and biologically-derived models for predicting in vivo effects have been developed using ToxCast HTS hits as predictors, and archival in vivo animal studies as evaluation data. To date, some pathways are better covered than others due to available technologies and EPA priorities (e.g., there are 18 assays that indicate activity related to estrogen receptor alpha (ERα) activation [45]). ToxCast assay results have been made publically available by multiple means at the conclusion of each testing phase, and at regular intervals [4].
Most ToxCast and Tox21 assays have focused on parent compound effects [46]. There are two primary reasons for a lack of testing on metabolites, degradants, and transformation products. First, some of these compounds are highly reactive and cannot be effectively assessed until metabolically competent systems are created. Second, sufficient quantities of these compounds are needed to provide to multiple testing facilities–many metabolites, degradants, and transformation products are not available on the market, and have therefore not been tested [5]. An extensive library of thousands of ToxCast chemicals does exist, however, allowing independent laboratories to perform experiments on matched chemical samples [5]. Nominations for new test chemicals are welcomed, with the biggest limitation being the ability to acquire sufficient quantities of the compounds of interest. We note that while most of the in vitro assays do not have metabolic competency, some assays using primary human hepatocytes or pluripotent liver cells (e.g., HepaRG) do allow the assessment of metabolic effects on the liver [47]. Additional research is ongoing to apply structure-based metabolism prediction methods and to augment other important assays with metabolic competency.
High throughput exposure screening and the ExpoCast project
While thousands of chemicals have been profiled for bioactivity using HTS, many of these chemicals are lacking data on exposure [48], which hinders risk-based evaluation. Many more chemicals exist without exposure or bioactivity data, and are in need of “exposure-based prioritization” prior to HTS and risk assessment [49]. The EPA’s exposure forecaster (ExpoCast) project was therefore developed to generate the data, tools, and evaluation methods required to produce rapid and scientifically-defensible estimates of exposure [11], and to confidently relate these estimates to concentrations that exhibit bioactivity (identified via HTS) [50,51,52]. Since the inception of ExpoCast, EPA has organized and analyzed extant data; collected new data on chemical properties, uses, and occurrence [53, 54]; and evaluated/developed mathematical models for predicting exposures across thousands of compounds [55]. With regards to mathematical modeling, a meet-in-the-middle approach has proven valuable. Using this approach, forward modeling predictions (e.g., those from mechanistic exposure models) have been compared against exposure estimates inferred from down-stream monitoring data (e.g., human biomarker measures, which cover only a small fraction of the overall chemicals of interest). Statistical comparisons of forward model predictions vs. biomarker-based estimates allows global examination of model performance and the impact of specific modeling assumptions on final exposure predictions [56]. The concepts and strategies for this meet-in-the-middle approach have been described elsewhere and implemented at EPA as part of a Systematic Empirical Evaluation of Models (SEEM) framework [57].
The SEEM framework allows for crude extrapolation from chemicals with monitoring data to chemicals without such data. To date, this approach has relied upon exposures inferred from urinary biomarker data as reported in the Centers for Disease Control and Prevention’s (CDC) National Health and Nutrition Examination Survey (NHANES). Notable findings of SEEM work include: 1) fate and transport models–that can predict exposure for thousands of chemicals following industrial releases (i.e., “far-field” sources) and migration through the environment [58, 59]–are limited in their ability to describe urinary biomarker data [57]; 2) chemicals present in urine often reflect “near-field” sources in the home, such as consumer products and articles of commerce (e.g., furniture and flooring) [57]; and 3) five factors (production volume, use in consumer products, use in industrial processes, use as a pesticidal active, and use as a pesticidal inert) are able to explain roughly half of the chemical-to-chemical variance in median exposure rates inferred from NHANES urine data [60].
Consistent with these findings, new mechanistic models have been developed with a focus on near-field exposure pathways [61, 62]; the incorporation of predictions from these new models into the SEEM framework has the potential to refine consensus exposure predictions for data-poor chemicals. In order to parameterize these models, however, information is needed on product formulation—that is, the concentration of chemicals in a product. Goldsmith and colleagues addressed this need by cataloging thousands of Material Safety Data Sheets (MSDS) for products sold by a major U.S. retailer, allowing searches for chemical presence in reviewed products [53]. Dozens of similar product ingredient databases now exist from other sources and were recently aggregated into EPA’s Chemical and Product Categories (CPCat) database [54]. Listings within this aggregated database include chemicals declared by the manufacturer or observed through laboratory analysis. It is noteworthy that certain formulated products (e.g., personal care products) have specific labeling guidelines that make ingredient information more prevalent, whereas other products (e.g., household cleaning products, and “durable goods” such as apparel or furniture) are governed by narrow (or non-existent) chemical reporting requirements, and therefore have limited formulation data [49].
A challenge in using product ingredient databases for mechanistic exposure modeling is the qualitative nature of the formulation data. Even when chemicals are listed as being present in a product, concentration values are often not provided. National production volume data are available for many chemicals, but typically binned into category ranges that can span an order of magnitude, and not directly linked to specific releases or intended use. Further, many chemicals determined to be present in urine by NHANES (generally as metabolites) do not even appear on lists of highly produced chemicals, indicating that they are produced at low levels (less than 25,000 lb/year) or do not emanate from monitored production processes. Finally, while some data exist for chemicals deliberately added to objects, many chemicals are introduced to products through packaging, and are therefore present despite not being explicitly labeled [54]. Noting these limitations, machine learning models have been developed at EPA to fill knowledge gaps related to product chemistries. These models utilize physico-chemical properties [63] and/or chemical structure information [64] to predict functional uses for individual compounds. Functional use estimates are then combined with consumer product ingredient databases (described above) to develop screening-level concentration estimates (“generic formulations”) for select products. These screening-level estimates are appropriate for some applications (e.g., chemical prioritization), but may not be well-suited for rigorous quantitative analyses. Additional product composition data are therefore needed to expand coverage across additional products and non-intentional ingredients, and to support the development of exposure predictions fit for higher-tier safety assessments.
Based on existing product/product-use information, along with environmental and biological monitoring data, it’s clear that chemical exposures often co-occur, leading to the potential for mixture effects on biological systems. To date, limited bioactivity-based HTS has been performed on chemical mixtures, owing, in part, to the vast number of mixtures that could conceivably be tested. Exposure information, however, is now being used to address this limitation. In particular, knowledge of chemical co-occurrence in media [65] and formulations [54] are being used to reduce the number of permutations considered for HTS. As an example, in a chemical library of 1000 unique compounds, there are more than 10300 combinations of compounds that could be evaluated using HTS assays. The role of exposure-based priority setting is to identify known or possible (i.e., those that are likely to occur) chemical mixtures that first require screening, and to set aside mixtures that may never occur. A recent ExpoCast analysis demonstrated the value of this approach using existing measures from CDC’s NHANES. Specifically, Kapraun and colleagues considered chemical co-occurrence using urine and blood measures, and ultimately identified a tractable number of chemical combinations that occurred in greater than 30% of the U.S. population [66]. The techniques utilized by Kapraun and colleagues now make it possible to readily evaluate chemicals for potentially hazardous synergies. Yet, analyses to date are beholden to limited datasets of target analytes. As such, broad measurement-based datasets are now required to further examine the extent to which chemical exposures co-occur in a consistent, predictable, and biologically-relevant manner.
The Distributed Structure-Searchable Toxicity (DSSTox) database
Data generated from EPA’s ToxCast and ExpoCast programs are now stored within EPA’s DSSTox database. The original DSSTox web site was launched in 2004, providing a common access point for several thousand environmental chemicals associated with four publicly available toxicity databases pertaining to carcinogenicity, aquatic toxicity, water disinfection by-products, and estrogen-receptor binding activity. This collection of DSSTox data files offered a highly-curated, standardized set of chemical structures that was well-suited for structure-activity modeling [67, 68]. The quality of mappings between chemical identifiers (names, registry numbers, etc.) and their corresponding structures provided the community with a comprehensive set of mappings to a unified DSSTox structure index. This structure index became the underpinning of the current DSSTox chemical database.
DSSTox continued to expand over the next decade with additional chemical structure files of interest to the toxicology and environmental science communities, including lists of high-production volume (HPV) chemicals, indexed lists of public microarray experiment databases, FDA drugs, and risk assessment lists (e.g., EPA’s Integrated Risk Information System [https://cfpub.epa.gov/ncea/iris2/atoz.cfm]). From 2007 onward, the database was enlisted to serve as the cheminformatics backbone of the ToxCast and Tox21 programs, with DSSTox curators registering all chemicals entering both screening libraries [5]. This enabled the mapping of in vitro and in vivo data to chemical structures, the latter through indexing of the NTP bioassay database and EPA’s Toxicity Reference Database (ToxRefDB) [69]. By mid-2014, the manually curated DSSTox database had grown to over 20,000 chemical substances (spanning more than a dozen inventories) of high priority to EPA research programs (archived DSSTox content available for download at ftp://ftp.epa.gov/dsstoxftp).
Despite the growth of DSSTox from 2007–2014, coverage did not extend to larger EPA inventories (e.g., the Toxic Substances Control Act [TSCA] inventory, https://www.epa.gov/tsca-inventory and the Endocrine Disruption Screening Program universe, https://www.epa.gov/endocrine-disruption), which were beginning to define a putative “chemical exposure landscape” [48, 70]. The focused nature of DSSTox stemmed from rate-limiting manual curation efforts, which ensured high quality structure-identifier mappings, but limited opportunities for DSSTox to more broadly support EPA research and regulatory efforts. A number of large chemically-indexed databases (such as PubChem, ChemSpider, ChEMBL, ChemIDPlus, and ACToR) eventually provided access points for additional chemical structures and identifiers. Curation efforts, however, demonstrated high rates of inaccuracies and mis-mapped chemical identifiers in these public domain chemical databases (e.g., a name or registry number incorrectly mapped to one or more structures), a common situation that has previously been reported [71, 72]. As such, the decision was ultimately made to expand DSSTox using publicly available resources, while also recognizing the limitations of those resources, and preserving the aspects of quality curation upon which DSSTox was built.
The product of database expansion efforts, known as DSSTox version 2 (V2), was developed using algorithmic curation techniques, both alone and in support of focused, ongoing manual curation efforts. A key constraint applied to the construction of DSSTox_V2 was the requirement for a 1:1:1 mapping among the preferred name for a chemical (chosen to be unique), the active (or current) Chemical Abstracts Services Registration Number (CAS-RN), and the chemical structure, as could be uniquely rendered in mol file format. Subject to these constraints (i.e., disallowing conflicts) chemical structures and uniquely mapped identifiers were sequentially loaded into DSSTox_V2 from the following public databases: the EPA Substance Registry Services (SRS) database (containing the public TSCA chemical inventory, accessed at https://iaspub.epa.gov/sor_internet/registry/substreg/); the National Library of Medicine’s (NLM) ChemIDPlus (part of the TOXNET suite of databases, accessed at https://chem.nlm.nih.gov/chemidplus/); and the National Center for Biotechnology Information’s (NCBI) PubChem database (the portion containing registry number identifiers along with other chemical identifiers, accessed at https://pubchem.ncbi.nlm.nih.gov/). Based on the number of sources that agreed on mappings of identifiers to structures, these public data were loaded with a quality control annotation (qc_level) ranging from low to high. Publicly indexed substances containing structures and identifiers that conflicted with existing DSSTox information were not registered; they were either queued for manual curation if considered important to EPA research programs, or were set aside to be loaded at a later date with appropriate documentation of the conflict.
In addition to the programmatic incorporation of non-conflicting portions of SRS, ChemIDPlus and PubChem into DSSTox_V2, both manual and programmatically assisted curation has continued to address critical gaps in coverage of high-interest environmental lists, including pesticides, food additives, chemicals of potential concern for endocrine disruption [73], chemicals with known functional use in products [54], and substances on the public EPA hydraulic fracturing chemicals list (https://cfpub.epa.gov/ncea/hfstudy/recordisplay.cfm?deid=332990). With these latest additions, the DSSTox database now has over 750,000 records, with more than 60,000 manually curated or having consistent identifier assignments in three or more public databases constituting the highest qc_level content. The clean mapping of structural identifiers (names, CAS-RN) to chemical structures provides an essential underpinning to robust and accurate cheminformatics workflows. Elements of such workflows, designed to support quantitative structure-activity relationship (QSAR) modeling as part of EPA’s ToxCast and ExpoCast programs, are now being surfaced through EPA’s CompTox Chemistry Dashboard.
The CompTox Chemistry Dashboard
The CompTox Chemistry Dashboard (hereafter, referred to as the “Dashboard”), developed at NCCT, is a freely accessible web-based application and data hub. Chemical substances surfaced via the Dashboard are hosted in the DSSTox database with associated identifiers (e.g., CAS-RN, systematic and trivial names). The Dashboard is used to search DSSTox using a simple alphanumeric text entry box (Fig. 1a). A successful search will result in a chemical page header (Fig. 1b) that provides:

The CompTox Chemistry Dashboard home page (a) and an example chemical page header (b)
-
1.
a chemical structure image (with ability to download in mol file format);
-
2.
intrinsic properties (e.g., molecular formula and monoisotopic mass);
-
3.
chemical identifiers (e.g., systematic name, SMILES string, InChI string, and InChIKey);
-
4.
related compounds (based on molecular skeleton search, molecular similarity search, and chemical presence in various mixtures and salt forms);
-
5.
a listing of databases in which the chemical is present (e.g., ToxCast and Tox21); and
-
6.
a record citation including a unique DSSTox substance identifier (DTXSID).
Below the header is a series of individual data tabs (Fig. 1b). The “Chemical Properties” and “Environmental Fate and Transport” tabs contain experimental properties assembled from various sources; presented values reflect recent efforts of NCCT to curate specific datasets in support of prediction algorithms [74, 75]. The “Synonyms” tab lists all associated systematic and trivial names, and various types of CAS-RN (i.e., active, deleted, and alternate, with the associated flags). The “External Links” tab lists a series of external resources associated with the chemical in question. The “Exposure” tab includes information regarding chemical weight fractions in consumer products, product use and functional use categories, NHANES monitoring data, and predicted exposure using the ExpoCast models. The “Bioassays” tab provides access to details of the ToxCast data and bioassay data available in PubChem. The “Toxicity” values tab includes data gathered from multiple EPA databases and documents, and various online open data sources. The “Literature” tab allows a user to choose from a series of queries, and perform searches against Google Scholar and Pubmed. It further integrates PubChem widgets for articles and patents. In general, all tabular data surfaced on the Dashboard can be downloaded as either tab-separated value files or Excel files, or included into an SDF file with the chemical structure.
An advanced search on the Dashboard (Fig. 2a) allows for mass searching, molecular formula searching, and molecular formula generation (based on a mass input). A batch search (Fig. 2b) further allows users to input lists of chemical names, CAS numbers, InChI Keys and other identifiers, and to retrieve formulae, masses, DTXSIDs, and other data related to chemical bioactivity and exposure. Various slices of data associated with the Dashboard are available as open data and can be obtained via the downloads page (https://comptox.epa.gov/dashboard/downloads). A detailed help file regarding how to use the Dashboard is also available online (https://comptox.epa.gov/dashboard/help).

The CompTox Chemistry Dashboard advanced search menu (a) and batch search menu (b)
Summary of EPA’s NTA workshop and collaborative trial
In August 2015, ORD’s National Exposure Research Laboratory (NERL) and Chemical Safety for Sustainability (CSS) research program jointly hosted an NTA-focused workshop in Research Triangle Park, North Carolina. The purpose of the workshop was to bring together world experts in exposure science, toxicology, cheminformatics, and analytical chemistry to discuss opportunities for collaboration and research integration. Invited presentations focused on research and regulatory drivers; existing data, tools, and resources that are being used to support HTS programs (as described in the previous sections); and NTA methods that are being developed and applied to characterize the exposome. Presentations from EPA science leaders called for engagement among research communities and highlighted how individual groups stand to benefit from shared knowledge and resources. Needs of the exposure scientists (representing the ExpoCast project), toxicologists (representing the ToxCast project), and analytical chemists (representing NTA projects) were articulated during the workshop as follows:
Needs of exposure scientists (ExpoCast) to support HT exposure screening:
-
Measurements of chemicals in consumer products and articles of commerce
-
Measurements of chemicals in environmental/residential media
-
Measurements of chemicals in biological media
Needs of toxicologists (ToxCast) to support HT bioactivity screening:
-
Prioritized lists of candidate parent (registered) chemicals
-
Prioritized lists of candidate degradants/metabolites
-
Prioritized lists of candidate chemical mixtures
Needs of analytical chemists (NTA) to support exposome research:
-
Large, relevant, curated, and open chemical databases for compound identification
-
Informatics tools for candidate selection and prioritization
-
Large inventories of chemical standards and reference spectra for candidate confirmation
-
Laboratory networks to support comprehensive analyses and standardized methods
The needs of the exposure scientists reflect the general lack of measurement data that are required to parameterize and ultimately evaluate exposure models. The needs of the toxicology community reflect the challenge of utilizing HTS methods to characterize bioactivity across tens-of-thousands of known compounds, and many more possible degradants, metabolites, and mixtures. Finally, the needs of the analytical chemistry community reflect the resources that are required for a holistic examination of the exposome.
Two days of discussion on these needs led to the planning and development of a research collaboration that will benefit all invested parties. A primary goal of the research collaboration is to answer the following questions:
-
1.
How can resources procured for HTS research in support of chemical safety evaluations be used to advance NTA methods?
-
2.
How can measurement data generated from NTA methods be used to direct HTS research and strengthen chemical safety evaluations?
EPA’s Non-Targeted Analysis Collaborative Trial (ENTACT) was developed in direct response to these questions. ENTACT makes full use of EPA’s ToxCast library of approximately 4000 compounds, is designed to be conducted in three parts, and involves international participants spanning more than 25 government, academic, and private/vendor laboratories. For part I of ENTACT, approximately 1200 compounds from the ToxCast library were combined into a series of synthetic mixtures, with ~100 to ~400 compounds included in each mixture. Laboratories participating in ENTACT will perform blinded analyses of these mixtures using their state-of-the art NTA methods. Individual methods will span a variety of separation and detection techniques, instruments, software, databases, and workflows. Results will be compiled by EPA and used to determine which NTA tools are best suited for the detection of specific compounds or groups of compounds. They will further indicate the extent to which sample complexity affects NTA method performance. Finally, they will serve as the basis for future QSAR models that predict the likelihood of a given compound being detected by a selected analytical method.
Part I of ENTACT evaluates NTA method performance using samples of fully synthetic mixtures. Part II, on the other hand, evaluates NTA method performance using extracts of true environmental and biological samples. Here, extracts of reference material house dust (National Institute of Standards and Technology [NIST] Standard Reference Material [SRM] 2585), human serum (NIST SRM 1957), and silicone passive air samplers were shared across laboratories to determine the region of chemical space that can be characterized using specific NTA methods, by sample type. To explore the extent to which the matrices affect extraction and other method performance parameters, each sample has also been fortified with a mixture of ToxCast chemicals prior to extraction. As such, laboratories participating in ENTACT have received two extracts of each medium—one based on a fortified reference sample and one based on an unaltered reference sample. Results of part II analyses will identify the most suitable methods for characterizing specific chemicals within a given medium. Perhaps more importantly, they will indicate how comprehensively a concerted effort of top laboratories can characterize compounds within house dust, human serum, and passive air samplers.
Parts I and II of ENTACT have been open to all interested laboratories, resources permitting. Part III, however, has been open only to instrument vendors, and select institutions that manage large open databases/software in support of NTA workflows. For part III, the full ToxCast chemical library, totaling ~4000 unique substances, is being shared for the purpose of generating reference mass spectra across a variety of instruments and analytical conditions (e.g., ionization source, ionization mode, collision energy, MS level). Institutions receiving these compounds will generate individual spectral records and make them available to EPA for further public use. Institutions may also make spectral records available to the public, or their customers, via addition to existing databases or development of compound libraries. Collectively, these efforts will enable users of many MS and HRMS platforms to rapidly screen for the presence of ToxCast chemicals in samples of their choosing. Results from these screening-level analyses will then provide provisional measurement data (e.g., presence/absence in a given medium) across thousands of compounds for which exposure data are currently lacking. These data will ultimately allow an improved understanding of aggregate exposures (i.e., one compound, multiple exposure pathways), cumulative exposures (i.e., multiple compounds, multiple exposure pathways, one biological target), and the contribution of ToxCast chemicals to the exposome.
Framework for research integration
A formalized framework is needed to ensure maximum benefit of ENTACT to both exposome and chemical screening research programs. The primary function of the framework, as shown in Fig. 3, is to highlight how and where existing chemical screening tools (i.e., ToxCast, ExpoCast, DSSTox, and the CompTox Chemistry Dashboard) can be leveraged to enhance NTA efforts, and ways in which NTA data can allow for more informed chemical screening.

A framework for integrating NTA methods and data with HTS tools (ToxCast, ExpoCast, and the CompTox Chemistry Dashboard [with the underlying DSSTox database]) available from EPA’s Office of Research and Development
Sample analysis
The first step within the framework is the physical analysis of products/articles, environmental samples, and/or biological samples using NTA methods (Fig. 3). Irrespective of the medium in question, no single analysis method, no matter how refined, is able to characterize the full chemical contents of a given sample. The use of multiple methods and analytical platforms, however, can greatly extend surveillance capabilities. A goal of ENTACT is to determine the chemical space applicability domain for a given method. Trial results will inform the breadth of approaches required to adequately characterize a given medium, or to address a given research, public health, or regulatory need. For example, trial results will indicate the number and types of methods required to screen for all ToxCast chemicals in a suite of consumer products. Trial results will also identify compounds that have yet to be considered as part of ToxCast/Tox21 but are present in select environmental and biological media. As described in detail below, latter steps of the framework determine which of these compounds, if any, should be prioritized for bioactivity screening.
Candidate identification and evaluation
Within ORD, the CompTox Chemistry Dashboard, and the underlying DSSTox database, are primary NTA tools for candidate identification and evaluation (Fig. 3). Initial work has determined that the Dashboard can effectively identify “known unknowns” in samples using data source ranking techniques as developed by Little et al [76]. Here, the Dashboard is used to search unidentified features from HRMS experiments within a mass range, or by an exact formula, and the most likely candidate chemicals are those with the highest data source counts [77]. Data source ranking alone, however, does not provide sufficient evidence for a “probable” compound classification [39]. The Dashboard is therefore incorporating additional data streams, models, and functionality to increase certainty when assigning structures to unknown compounds. For example, chemical functional use data from EPA’s CPCat database are now available through the Dashboard and can be incorporated into workflows to filter lists of tentative structures. A new and enhanced version of CPCat, the Consumer Products Database (CPDat), has been developed, made available as a beta release in the March, 2017 update to the Dashboard, and further provides predicted functional uses for chemicals with no known use data. This information can help determine the likelihood that a given compound would be present in a given sample (e.g., a textile dye is more likely than a drug to be found in house dust) [77]. In addition, physicochemical properties of candidate chemicals are available within the Dashboard, and can be used to predict the likelihood of environmental media occurrence, and the suitability of a selected laboratory method (e.g., extraction solvent, separation technique, ionization mode) for detection.
The utilization of relevant data streams within the Dashboard can improve the confidence in structural assignments, but a true one-pass analysis requires the ability to search large lists of unidentified features exported from an HRMS instrument. Batch search capability within the Dashboard (Fig. 2b) now enables users to search thousands of instrument generated molecular formulae at once and receive back the top ten most likely candidate chemicals with associated chemical data (e.g., identifiers, properties, structures, etc.). A further enhancement to this search capability is the inclusion of “MS-ready” structures, whereby all chemicals within the database have been desalted, desolvated, and had stereochemistry removed to represent the forms of chemicals observed via HRMS. In addition to this feature, and the aforementioned features for data source ranking and functional use filtering, spectral matching capabilities will eventually provide supporting evidence for compound identification. Specifically, linking Dashboard records to those from open spectral libraries (e.g., MassBank and MoNa) and fragmentation prediction resources (e.g., MetFrag and CFM-ID) will allow for further confidence in probable identifications. Finally, incorporation of empirical reference spectra from vendors participating in ENTACT will allow rapid screening for a large suite of ToxCast compounds.
Candidate prioritization
Once probable structures have been proposed, chemical standards are used for feature confirmation, and in some cases, quantitation. As additional standards become available, incremental advances are to be expected in the percentages of probable and confirmed structures relative to tentative candidates. By definition, however, the ability to confirm compounds will always be limited by the availability of chemical standards. This limitation is likely to persist given the cost and time associated with standard synthesis. As such, focus must be given to tools for prioritizing tentative candidates that require further study. In other words, methods should be employed that help determine which tentative compounds require further study, and which are potentially of little health consequence.
In a previous pilot study [31], we identified molecular features in house dust samples using LC-TOF HRMS, proposed tentative candidates by screening observed molecular features against the DSSTox database (which included, at the time, ~33 K compounds), and prioritized tentative candidates for further analysis using data from ToxCast and ExpoCast [31]. Priority candidates - those predicted to have high bioactivity, exposure potential, or both - were examined to identify which candidates could be further classified as probable structures. ToxCast standards were ultimately used to confirm a manageable list of compounds. About half of the confirmed chemicals, according to a review of the published literature, had never before been measured in house dust. This pilot study paved the way for a number of NTA studies now being conducted by EPA/ORD, and serves as the basis for the framework proposed here. It was further featured in the recent NRC report “Using 21st Century Science to Improve Risk-Related Evaluations” as an example of an “…innovative approach for identifying and setting priorities among chemicals for additional exposure assessment, hazard testing, and risk assessment that complements the current hazard-oriented paradigm” [9]
ToxCast and ExpoCast data exist for thousands of DSSTox chemicals, and are freely available to the public via the Dashboard. The Dashboard can therefore be used to identify tentative candidates (via formula or mass-based searching), and then sort these candidates based on potential for human (or ecological) contact and biological response. Figure 3 depicts how ToxCast and ExpoCast data were used in our previous dust analysis, and are now integrated into the research framework. As shown in Fig. 3, exposure and bioactivity estimates for tentative candidates are combined into a prioritization algorithm, along with estimates of feature abundance (i.e., average peak intensity across samples) and detection frequency. EPA’s Toxicological Prioritization Index (ToxPi) software is then used to generate graphical displays for each tentative candidate [78]. Here, each pie wedge represents a weighted and normalized value for the selected variable. The scoring algorithm and ToxPi graphical representation are completely customizable—new variables and different weighting schemes can be easily applied. To date, our internal analyses have given more weight to candidates with elevated detection frequency and evidence of bioactivity.
Exposure and bioactivity evaluation
While exposure and bioactivity data are available for thousands of chemicals, the majority of DSSTox compounds (~ 99%) are without these data. With regards to priority scoring, compounds with data are considered separately from those without data. A bifurcation of the research workflow is shown in Fig. 3 to depict this differentiation. Here, compounds with data are shown to undergo a series of steps to enable exposure evaluation, whereas compounds without data are further considered as part of a bioactivity evaluation.
EPA’s HT exposure models and ExpoCast framework make use of and predict environmental and biological concentrations of known compounds. Often, limited data are available as model inputs and for model parameterization, which can lead to large uncertainties in media concentration or final exposure estimates. Chemical measurements are therefore needed to help parameterize, evaluate, and refine existing models. A major goal of the proposed research framework is to enable NTA data to meet these needs. Here, the initial focus is on compounds classified as probable structures, and ranked as high-priority using the ToxPi approach. As a first step, to the extent that resources allow, these compounds are confirmed using existing standards - provisional concentrations may then be estimated using a variety of techniques [79]. These concentration estimates are then compared to predictions from HT exposure models. Agreement between predicted and estimated concentrations provide confidence in model performance. Sizable disparities between model predicted and laboratory estimated values, however, may prompt re-evaluation of model structures and parameters, and/or follow-up laboratory analyses. Specifically, targeted methods may be developed and applied in instances where NTA-estimated concentrations significantly exceed model predicted values and encroach on exposure thresholds that are consistent with predicted biological activity. The final product of these steps is strengthened assessments of potential risk for confirmed high-priority compounds.
As DSSTox increases in size, so does the number of probable structures for which exposure and bioactivity data are unavailable. For a given experiment, it is not uncommon to have ten times as many probable structures without exposure and bioactivity data than probable structures with this data. It is critical that these compounds are not disregarded from further analysis based on existing data limitations. Rather, these compounds must pass a cursory evaluation for bioactivity before being exempted from further consideration. QSAR modeling has been applied to determine which compounds are most likely to be bioactive, and therefore higher priority. For example, the Collaborative Estrogen Receptor Activity Prediction Project recently predicted ER activity across a set over 32,000 chemical structures [73]. Using these predictions, candidate compounds can be prioritized, and attempts made at confirmation using standards and/or additional targeted analysis procedures. Confirmed high-priority compounds are eventually nominated for in vitro screening through the ToxCast program. Results of the ToxCast assays, as well as any new ExpoCast predictions, are ultimately collated within the DSSTox database and Chemistry Dashboard, and used to support Agency prioritization efforts and eventual decisions.
Conclusions and outlook
Studies at EPA are now being planned and executed with this integrated research framework in mind. Analyses as part of ENTACT are underway (as of January 2017) and will be a source of measurement data for thousands of ToxCast compounds, and chemicals, degradants, metabolites, and mixtures not currently considered by ToxCast/Tox21. The content of DSSTox and the functionality of the Dashboard are constantly expanding, including the addition of chemical datasets provided by other parties interested in NTA, thereby allowing better access to chemistry data and tools for supporting cheminformatics applications and NTA workflows. Also expanding are the exposure and bioactivity data being generated by the ExpoCast and ToxCast projects, respectively. Semi-quantitative NTA measures across a variety of media will soon enable evaluation and refinement of ExpoCast predictions. When examined using bioactivity prediction models, these NTA measures will further yield prioritized lists of compounds that can be considered for ToxCast screening.
It is worth noting that measurement data from NTA studies will not parallel those from targeted studies in terms of accuracy and precision. The NTA community will surely face challenges when comparing semi-quantitative data over time, and across analytical platforms and labs. Standardized approaches will therefore be needed to ensure the appropriate generation, communication, and use of NTA measurement data. Numeric results from ENTACT are expected to shed light on the severity of this issue (i.e., the amount of variability in semi-quantitative measures from one experiment to the next) and act as a large training set for future concentration prediction models. Ultimately, NTA data are intended to be fit-for-purpose—that is, to support screening-level activities. Targeted measures will always be the benchmark for risk-based decisions and actions, and therefore must be generated in tandem, as needed, with NTA measures (Fig. 3). Such a combined measurement scheme will provide a solid foundation for 21st century chemical safety evaluations, and an improved understanding of the chemical composition of the exposome.
References
Collins FS, Gray GM, Bucher JR. Toxicology. Transforming environmental health protection. Science. 2008;319:906–7.
NRC. Toxicity testing in the 21st century: a vision and a strategy. Washington, DC: National Academies Press; 2007.
Tice RR, Austin CP, Kavlock RJ, Bucher JR. Improving the human hazard characterization of chemicals: a Tox21 update. Environ Health Perspect. 2013;121:756–65.
Kavlock R, Chandler K, Houck K, Hunter S, Judson R, Kleinstreuer N, et al. Update on EPA’s ToxCast program: providing high throughput decision support tools for chemical risk management. Chem Res Toxicol. 2012;25:1287–302.
Richard AM, Judson RS, Houck KA, Grulke CM, Volarath P, Thillainadarajah I, et al. ToxCast chemical landscape: paving the road to 21st century toxicology. Chem Res Toxicol. 2016;29:1225–51.
Edwards SW, Tan YM, Villeneuve DL, Meek ME, McQueen CA. Adverse outcome pathways-organizing toxicological information to improve decision making. J Pharmacol Exp Ther. 2016;356:170–81.
Kleinstreuer NC, Sullivan K, Allen D, Edwards S, Mendrick DL, Embry M, et al. Adverse outcome pathways: From research to regulation scientific workshop report. Regul Toxicol Pharmacol: RTP. 2016;76:39–50.
NRC. Exposure science in the 21st century: a vision and a strategy. Washington, DC: National Academies Press; 2012.
NRC. Using 21st century science to improve risk-related evaluations. Washington, DC: National Academies Press; 2017.
Escher BI, Hackermuller J, Polte T, Scholz S, Aigner A, Altenburger R, et al. From the exposome to mechanistic understanding of chemical-induced adverse effects. Environ Int. 2017;99:97–106.
Hubal EA. Biologically relevant exposure science for 21st century toxicity testing. Toxicol Sci. 2009;111:226–32.
Cohen Hubal EA, Richard AM, Shah I, Gallagher J, Kavlock R, Blancato J, et al. Exposure science and the U.S. EPA National Center for Computational Toxicology. J Expo Sci Environ Epidemiol. 2010;20:231–6.
Egeghy PP, Sheldon LS, Isaacs KK, Ozkaynak H, Goldsmith MR, Wambaugh JF, et al. Computational exposure science: an emerging discipline to support 21st-century risk assessment. Environ Health Perspect. 2016;124:697–702.
Wishart D, Arndt D, Pon A, Sajed T, Guo AC, Djoumbou Y, et al. T3DB: the toxic exposome database. Nucleic Acids Res. 2015;43:D928–34.
Neveu V, Moussy A, Rouaix H, Wedekind R, Pon A, Knox C, et al. Exposome-Explorer: a manually-curated database on biomarkers of exposure to dietary and environmental factors. Nucleic Acids Res. 2017;45:D979–84
Menikarachchi LC, Hill DW, Hamdalla MA, Mandoiu II, Grant DF. In silico enzymatic synthesis of a 400,000 compound biochemical database for nontargeted metabolomics. J Chem Inf Model. 2013;53:2483–92.
Rothwell JA, Urpi-Sarda M, Boto-Ordonez M, Llorach R, Farran-Codina A, Barupal DK, et al. Systematic analysis of the polyphenol metabolome using the Phenol-Explorer database. Mol Nutr Food Res. 2016;60:203–11.
Warth B, Spangler S, Fang M, Johnson C, Forsberg E, Granados A, et al. Exposome-scale investigations guided by global metabolomics, pathway analysis, and cognitive computing. Anal Chem. 2017;89:11505–13.
Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, et al. HMDB 3.0–The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–7.
Edmands WM, Petrick L, Barupal DK, Scalbert A, Wilson MJ, Wickliffe JK, et al. compMS2Miner: an automatable metabolite identification, visualization, and data-sharing R package for high-resolution LC-MS data sets. Anal Chem. 2017;89:3919–28.
Schymanski EL, Singer HP, Longree P, Loos M, Ruff M, Stravs MA, et al. Strategies to characterize polar organic contamination in wastewater: exploring the capability of high resolution mass spectrometry. Environ Sci Technol. 2014;48:1811–8.
Wild CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomark Prev. 2005;14:1847–50.
Rappaport SM. Implications of the exposome for exposure science. J Expo Sci Env Epid. 2011;21:5–9.
Rappaport SM, Smith MT. Epidemiology. Environment and disease risks. Science. 2010;330:460–1.
Rappaport SM. Biomarkers intersect with the exposome. Biomarkers. 2012;17:483–9.
Miller GW, Jones DP. The nature of nurture: refining the definition of the exposome. Toxicol Sci. 2014;137:1–2.
Pleil JD, Stiegel MA. Evolution of environmental exposure science: using breath-borne biomarkers for “discovery” of the human exposome. Anal Chem. 2013;85:9984–90.
Andra SS, Austin C, Patel D, Dolios G, Awawda M, Arora M. Trends in the application of high-resolution mass spectrometry for human biomonitoring: An analytical primer to studying the environmental chemical space of the human exposome. Environ Int. 2017;100:32–61.
Krauss M, Singer H, Hollender J. LC-high resolution MS in environmental analysis: from target screening to the identification of unknowns. Anal Bioanal Chem. 2010;397:943–51.
Enders JR, Phillips MB, Clewell HJ, Clewell RA, Strynar MJ, Ulrich EM, et al. Application of non-targeted exposure analysis in assessment: opportunities and challenges. In preparation.
Rager JE, Strynar MJ, Liang S, McMahen RL, Richard AM, Grulke CM, et al. Linking high resolution mass spectrometry data with exposure and toxicity forecasts to advance high-throughput environmental monitoring. Environ Int. 2016;88:269–80.
Brack W, Ait-Aissa S, Burgess RM, Busch W, Creusot N, Di Paolo C, et al. Effect-directed analysis supporting monitoring of aquatic environments–An in-depth overview. Sci Total Environ. 2016;544:1073–118.
Fang M, Webster TF, Stapleton HM. Activation of human peroxisome proliferator-activated nuclear receptors (PPARgamma1) by semi-volatile compounds (SVOCs) and chemical mixtures in indoor dust. Environ Sci Technol. 2015;49:10057–64.
Phillips KA, Yau A, Favela KA, Isaacs K, Grulke CM, Richard AM, et al. Suspect screening analysis of chemicals in consumer products. Submitted.
Nakamura J, Mutlu E, Sharma V, Collins L, Bodnar W, Yu R, et al. The endogenous exposome. DNA Repair (Amst). 2014;19:3–13.
Rappaport SM, Barupal DK, Wishart D, Vineis P, Scalbert A. The blood exposome and its role in discovering causes of disease. Environ Health Persp. 2014;122:769–74.
Andra SS, Austin C, Arora M. The tooth exposome in children’s health research. Curr Opin Pediatr. 2016;28:221–7.
Lioy PJ, Rappaport SM. Exposure science and the exposome: an opportunity for coherence in the environmental health sciences. Environ Health Perspect. 2011;119:A466–7.
Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, et al. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ Sci Technol. 2014;48:2097–8.
McEachran AD, Mansouri K, Newton SR, Beverly B, Sobus JR, Williams AJ. A comparison of three chromatographic retention time prediction models. Submitted.
Ridder L, van der Hooft JJ, Verhoeven S. Automatic compound annotation from mass spectrometry data using MAGMa. Mass Spectrom. 2014;3:S0033.
Judson R, Richard A, Dix D, Houck K, Elloumi F, Martin M, et al. ACToR–aggregated computational toxicology resource. Toxicol Appl Pharmacol. 2008;233:7–13.
Judson R, Houck K, Martin M, Richard AM, Knudsen TB, Shah I, et al. Analysis of the effects of cell stress and cytotoxicity on in vitro assay activity across a diverse chemical and assay space. Toxicol Sci. 2016;153:409.
Judson RS, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, Mortensen HM, et al. In vitro screening of environmental chemicals for targeted testing prioritization: the ToxCast project. Environ Health Perspect. 2010;118:485–92.
Browne P, Judson RS, Casey WM, Kleinstreuer NC, Thomas RS. Screening chemicals for estrogen receptor bioactivity using a computational model. Environ Sci Technol. 2015;49:8804–14.
Pinto CL, Mansouri K, Judson R, Browne P. Prediction of estrogenic bioactivity of environmental chemical metabolites. Chem Res Toxicol. 2016;29:1410–27.
Rotroff DM, Beam AL, Dix DJ, Farmer A, Freeman KM, Houck KA, et al. Xenobiotic-metabolizing enzyme and transporter gene expression in primary cultures of human hepatocytes modulated by ToxCast chemicals. J Toxicol Environ Health B Crit Rev. 2010;13:329–46.
Egeghy PP, Judson R, Gangwal S, Mosher S, Smith D, Vail J, et al. The exposure data landscape for manufactured chemicals. Sci Total Environ. 2012;414:159–66.
Egeghy PP, Vallero DA, Hubal EAC. Exposure-based prioritization of chemicals for risk assessment. Environ Sci Policy. 2011;14:950–64.
Wambaugh JF, Wetmore BA, Pearce R, Strope C, Goldsmith R, Sluka JP, et al. Toxicokinetic triage for environmental chemicals. Toxicol Sci. 2015;147:55–67.
Wetmore BA, Wambaugh JF, Allen B, Ferguson SS, Sochaski MA, Setzer RW, et al. Incorporating high-throughput exposure predictions with dosimetry-adjusted in vitro bioactivity to inform chemical toxicity testing. Toxicol Sci. 2015;148:121–36.
Wetmore BA, Wambaugh JF, Ferguson SS, Sochaski MA, Rotroff DM, Freeman K, et al. Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol Sci. 2012;125:157–74.
Goldsmith MR, Grulke CM, Brooks RD, Transue TR, Tan YM, Frame A, et al. Development of a consumer product ingredient database for chemical exposure screening and prioritization. Food Chem Toxicol. 2014;65:269–79.
Dionisio KL, Frame AM, Goldsmith M-R, Wambaugh JF, Liddell A, Cathey T, et al. Exploring consumer exposure pathways and patterns of use for chemicals in the environment. Toxicol Rep. 2015;2:228–37.
Mitchell J, Arnot JA, Jolliet O, Georgopoulos PG, Isukapalli S, Dasgupta S, et al. Comparison of modeling approaches to prioritize chemicals based on estimates of exposure and exposure potential. Sci Total Environ. 2013;458-460:555–67.
Chadeau-Hyam M, Athersuch TJ, Keun HC, De Iorio M, Ebbels TM, Jenab M, et al. Meeting-in-the-middle using metabolic profiling–a strategy for the identification of intermediate biomarkers in cohort studies. Biomarkers. 2011;16:83–8.
Wambaugh JF, Setzer RW, Reif DM, Gangwal S, Mitchell-Blackwood J, Arnot JA, et al. High-throughput models for exposure-based chemical prioritization in the ExpoCast project. Environ Sci Technol. 2013;47:8479–88.
Arnot JA, Brown TN, Wania F, Breivik K, McLachlan MS. Prioritizing chemicals and data requirements for screening-level exposure and risk assessment. Environ Health Perspect. 2012;120:1565–70.
Arnot JA, Mackay D, Webster E, Southwood JM. Screening level risk assessment model for chemical fate and effects in the environment. Environ Sci Technol. 2006;40:2316–23.
Wambaugh JF, Wang A, Dionisio KL, Frame A, Egeghy P, Judson R, et al. High throughput heuristics for prioritizing human exposure to environmental chemicals. Environ Sci Technol. 2014;48:12760–7.
Csiszar SA, Ernstoff AS, Fantke P, Meyer DE, Jolliet O. High-throughput exposure modeling to support prioritization of chemicals in personal care products. Chemosphere. 2016;163:490–8.
Isaacs KK, Glen WG, Egeghy P, Goldsmith MR, Smith L, Vallero D, et al. SHEDS-HT: an integrated probabilistic exposure model for prioritizing exposures to chemicals with near-field and dietary sources. Environ Sci Technol. 2014;48:12750–9.
Isaacs KK, Goldsmith MR, Egeghy P, Phillips K, Brooks R, Hong T, et al. Characterization and prediction of chemical functions and weight fractions in consumer products. Toxicol Rep. 2016;3:723–32.
Phillips KA, Wambaugh JF, Grulke CM, Dionisio KL, Isaacs KK. High-throughput screening of chemicals as functional substitutes using structure-based classification models. Green Chem. 2017;19:1063–74.
Tornero-Velez R, Egeghy PP, Cohen Hubal EA. Biogeographical analysis of chemical co-occurrence data to identify priorities for mixtures research. Risk Anal. 2012;32:224–36.
Kapraun DF, Wambaugh JF, Ring CL, Tornero-Velez R, Setzer RW. A method for identifying prevalent chemical combinations in the US population. Environ Health Perspect. 2017;125:087017.
Richard AM, Yang C, Judson RS. Toxicity data informatics: supporting a new paradigm for toxicity prediction. Toxicol Mech Methods. 2008;18:103–18.
Richard AM. DSSTox Website launch: Improving public access to databases for building structure-toxicity prediction models. Preclinica. 2004;2:103–8.
Martin MT, Judson RS, Reif DM, Kavlock RJ, Dix DJ. Profiling chemicals based on chronic toxicity results from the U.S. EPA ToxRef Database. Environ Health Perspect. 2009;117:392–9.
Judson R, Richard A, Dix DJ, Houck K, Martin M, Kavlock R, et al. The toxicity data landscape for environmental chemicals. Environ Health Perspect. 2009;117:685–95.
Williams AJ, Ekins S. A quality alert and call for improved curation of public chemistry databases. Drug Discov Today. 2011;16:747–50.
Williams AJ, Ekins S, Tkachenko V. Towards a gold standard: regarding quality in public domain chemistry databases and approaches to improving the situation. Drug Discov Today. 2012;17:685–701.
Mansouri K, Abdelaziz A, Rybacka A, Roncaglioni A, Tropsha A, Varnek A, et al. CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ Health Perspect. 2016;124:1023–33.
Mansouri K, Grulke CM, Richard AM, Judson RS, Williams AJ. An automated curation procedure for addressing chemical errors and inconsistencies in public datasets used in QSAR modelling. SAR QSAR Environ Res. 2016;27:939–65.
Zang Q, Mansouri K, Williams AJ, Judson RS, Allen DG, Casey WM, et al. In silico prediction of physicochemical properties of environmental chemicals using molecular fingerprints and machine learning. J Chem Inf Model. 2017;57:36–49.
Little JL, Williams AJ, Pshenichnov A, Tkachenko V. Identification of “known unknowns” utilizing accurate mass data and ChemSpider. J Am Soc Mass Spectrom. 2012;23:179–85.
McEachran AD, Sobus JR, Williams AJ. Identifying known unknowns using the US EPA’s CompTox chemistry dashboard. Anal Bioanal Chem. 2017;409:1729–35.
Reif DM, Sypa M, Lock EF, Wright FA, Wilson A, Cathey T, et al. ToxPi GUI: an interactive visualization tool for transparent integration of data from diverse sources of evidence. Bioinformatics. 2013;29:402–3.
Go YM, Walker DI, Liang Y, Uppal K, Soltow QA, Tran V, et al. Reference Standardization for mass spectrometry and high-resolution metabolomics applications to exposome research. Toxicol Sci. 2015;148:531–43.
Acknowledgements
The authors thank Adam Biales, Myriam Medina-Vera, John Kenneke, Sania Tong-Argao, Timothy Buckley, Annette Guiseppi-Elie, Jennifer Orme-Zavaleta, Tina Bahadori, Russell Thomas, Robert Kavlock, and Thomas Burke from EPA for their guidance and support. The authors also thank Barbara Wetmore, Peter Egeghy, and Jeffre Johnson from EPA for their thoughtful reviews of this manuscript. The United States Environmental Protection Agency through its Office of Research and Development funded and managed the research described here. It has been subjected to Agency administrative review and approved for publication. Julia Rager and Andrew McEachran were supported by an appointment to the Internship/Research Participation Program at the Office of Research and Development, U.S. Environmental Protection Agency, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and EPA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sobus, J.R., Wambaugh, J.F., Isaacs, K.K. et al. Integrating tools for non-targeted analysis research and chemical safety evaluations at the US EPA. J Expo Sci Environ Epidemiol 28, 411–426 (2018). https://doi.org/10.1038/s41370-017-0012-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41370-017-0012-y
Keywords
This article is cited by
-
Free and open-source QSAR-ready workflow for automated standardization of chemical structures in support of QSAR modeling
Journal of Cheminformatics (2024)
-
Screening for drinking water contaminants of concern using an automated exposure-focused workflow
Journal of Exposure Science & Environmental Epidemiology (2024)
-
Improving predictions of compound amenability for liquid chromatography–mass spectrometry to enhance non-targeted analysis
Analytical and Bioanalytical Chemistry (2024)
-
Predicting RP-LC retention indices of structurally unknown chemicals from mass spectrometry data
Journal of Cheminformatics (2023)
-
A precision environmental health approach to prevention of human disease
Nature Communications (2023)
