
Abstract
Ulcerative Colitis (UC) has been reported to be related to Porphyromonas gingivalis (P. gingivalis). Porphyromonas gingivalis peptidylarginine deiminase (PPAD), a virulence factor released by P. gingivalis, is known to induce inflammatory responses. To explore the pathological relationships between PPAD and UC, we used homologous recombination technology to construct a P. gingivalis strain in which the PPAD gene was deleted (Δppad) and a Δppad strain in which the PPAD gene was restored (comΔppad). C57BL/6 mice were orally gavaged with saline, P. gingivalis, Δppad, or comΔppad twice a week for the entire 40 days (days 0−40), and then, UC was induced by dextran sodium sulfate (DSS) solution for 10 days (days 31−40). P. gingivalis and comΔppad exacerbated DDS-induced colitis, which was determined by assessing the parameters of colon length, disease activity index, and histological activity index, but Δppad failed to exacerbate DDS-induced colitis. Flow cytometry and ELISA revealed that compared with Δppad, P. gingivalis, and comΔppad increased T helper 17 (Th17) cell numbers and interleukin (IL)-17 production but decreased regulatory T cells (Tregs) numbers and IL-10 production in the spleens of mice with UC. We also cocultured P. gingivalis, Δppad, or comΔppad with T lymphocytes in vitro and found that P. gingivalis and comΔppad significantly increased Th17 cell numbers and decreased Treg cell numbers. Immunofluorescence staining of colon tissue paraffin sections also confirmed these results. The results suggested that P. gingivalis exacerbated the severity of UC in part via PPAD.
Similar content being viewed by others


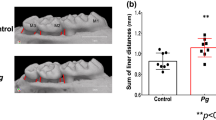
Introduction
Periodontitis, the most common oral infectious disease caused by bacterial biofilms, results in the loss of periodontal attachment and alveolar bone, and it is a major etiology of tooth loss in adults1. Increasing evidence suggests that periodontitis is a risk factor for many systemic diseases, for instance, intestinal diseases2,3. Inflammatory bowel disease (IBD) is an idiopathic relapsing and remitting intestinal inflammatory disease, including ulcerative colitis (UC) and Crohn’s disease (CD). The etiology and pathogenesis of UC are the result of complex interactions among environmental factors, intestinal microorganisms, genetic susceptibility, and immune factors, among which immunomodulatory disorder is the key factor. The activation of intestinal mucosal immune responses is the immediate cause of the occurrence and development of UC and affects its prognosis4. Studies have demonstrated that UC patients possess a significantly higher incidence of periodontitis, deeper periodontal pocket depth, and more tooth loss than healthy controls5,6. Furthermore, oral bacteria derived from periodontitis may translocate to the intestine, and together with the intestinal microbiome, cause intestinal epithelial cell barrier dysfunction and amplify intestinal inflammation7. Kitamoto et al. found that periodontitis contributes to the progression of colitis via a dual microbiome and immune mechanism. After reaching a certain level, oral microbiota pathogens in saliva will colonize the intestine. In addition, the excessive oral bacteria can induce the abnormal immune responses by migratory T helper 17 (Th17) cells8. Alterations in the structure of the small intestine, including epithelial stratification, altered villous height, and neutrophil infiltration, are observed in rats with ligature-induced periodontitis9. Supplementation with intestinal probiotics may alleviate these defects10. Oral infection with Porphyromonas gingivalis impairs colonic motor functions, suggesting that periodontal pathogens can be defined as regulators of the host response in IBD11,12. Proinflammatory cytokines produced and activated locally in the oral cavity might enter the bloodstream and circulate to the intestines, thus impacting IBD13,14. This evidence indicates that periodontitis may be closely related to colitis through a substantially altered microbiome.
P. gingivalis is an anaerobic bacterium and a main periodontal pathogen that resides in periodontitis lesions15. P. gingivalis expresses many virulence factors, including proteases, endotoxins, organic acids, and others that directly attack gingival tissues1,16. These virulence factors also allow P. gingivalis to induce abnormal immune responses in the host and lead to an ecological imbalance between the host and microorganisms17,18,19. After the initial bacterial insult, the host’s immune/inflammatory responses, partially driven by T lymphocytes, play a great role in the breakdown of periodontal tissues20,21. Increased Th17 cell proportions and interleukin (IL)-17 levels were observed in local gingival tissues from patients with periodontitis compared with those from healthy controls22,23, and these changes were reflected in the systemic circulation in serum24,25. Regulatory T cells (Tregs) are strongly associated with disease progression and reduced immune-inflammatory responses and function by suppressing the proliferation and proinflammatory cytokine production of Th1 and Th17 cells21. Increasing the numbers of Tregs have been used to regulate the imbalance of Th17/Tregs and successfully reduce P. gingivalis-induced alveolar bone loss26,27. Th17 cells and Tregs also play crucially important roles during the advancement of DSS-induced colitis28,29. Th17 cells, through an IL-17-dependent mechanism, stimulate the activation of infiltrating inflammatory cells and induce diffuse hemorrhage of the colonic mucosa, accelerating crypt abscess formation and UC exacerbation30. Because they promote the production of the anti-inflammatory cytokines TGF-β and IL-10 or limit the amplification of proinflammatory Th17 cells to suppress ongoing colitis, Tregs are regarded as the primary immunosuppressive cells in UC30.
Peptidylarginine deiminase (PAD) is a protein-modifying enzyme and a critical virulence factor of P. gingivalis, and it was first described in 198131. PAD participates in many vital physiological processes, including cellular differentiation, immune responses, and gene transcription32,33. PAD can modify and transform the host proteins by catalyzing and converting peptidyl-arginine to peptidyl-citrulline—a procedure known as citrullination34. Protein citrullination can elicit an abnormal autoimmune response by changing the immunological activity of chemokines and altering the structure and function of proteins35,36. Disruptions of normal PAD activities lead to abnormal citrullination, resulting in multiple inflammatory diseases, such as rheumatoid arthritis, Alzheimer’s disease, and UC37,38. Citrullination stimulates the activation of neutrophils and macrophages in the initial stages of innate immune responses and promotes the progression of intestinal inflammation39. The role of citrullination in IBD was first reported in 2006, and the levels of citrullinated peptides in colonic biopsies from IBD patients were higher than those from the control subjects40. Immunohistochemistry showed that PAD staining was strong in the intestinal lamina propria cells of mice with DSS-induced acute colitis and patients with UC, confirming that citrullination can promote inflammation28. Porphyromonas gingivalis peptidylarginine deiminase (PPAD) is synthesized and released by P. gingivalis via membrane vesicles41,42, and can accelerate protein citrullination in gingival tissues. Whether the PAD secreted by P. gingivalis contributes to UC has not been reported.
In this study, we explored the significant effect of P. gingivalis on UC, which resulted in the exacerbation of inflammation in a mouse model. This exacerbation was due to PPAD, which induced an abnormal immune response and elevated the Th17/Treg ratio. Our findings offer further insights into the pathogenic effects of PPAD on systemic diseases.
Results
P. gingivalis exacerbated the intestinal inflammation of mice treated with DSS
After 19 days of pretreatment, mice were inoculated with P. gingivalis strains on day 0 (Fig. 1a). In the first 30 days, no abnormalities were detected after each gavage of bacteria, and the body weight gain was consistent with the normal growth rate in all the mice. On the 32nd day, one day after the administration of DSS, the mice in the DSS groups exhibited the following changes: furs became dry, the luster disappeared, activity decreased, and water intake was reduced. On the 34th day, blood stains appeared in the perianal area. The appearance of occult blood and blood in the stool consistently increased, which was accompanied by body weight loss over time. As shown in Fig. 1b, the mice in the DSS group exhibited significantly greater body weight loss (P < 0.01) than the mice in the NC group, while the mice in the P. gingivalis + DSS group exhibited even greater weight loss (P < 0.05) than the mice in the DSS group.

Clinical assessment indicates that P. gingivalis exacerbates UC. Schematic of the animal experimental design for the three groups (n = 6 in each group). b Mice with colitis showed significant body weight loss from 35th to 40th day after DSS induction. P. gingivalis suspension exacerbated body weight loss compared with DSS treatment (P < 0.01). c DAI scores calculated on 35th, 38th, and 40th day. The bar chart showing the typical presentation of inflammation in mice on 35th day after drinking the DSS solution. The DAI scores of the P. gingivalis + DSS group were significantly higher than those of the NC group and DSS group (P < 0.01). d The colons of the DSS group became shorter and exhibited more hyperemia and edema than those of the NC group (P < 0.01). The P. gingivalis + DSS group presented the shortest colons, the most severe inflammation, and the bloodiest stool (P < 0.05). e Representative H&E staining of colons from the three groups. The green and yellow arrows in the digital photographs of mice with colitis show the lymphocytic infiltration and partial exfoliation of the intestinal epithelium. The pathological changes observed after feeding a P. gingivalis suspension to mice with colitis by gavage were worse than those observed in the mice with colitis; these changes included large areas of deep ulcers, destruction of epithelial cells, exfoliation of intestinal epithelium and intestinal gland, exfoliation of basement membrane, and infiltration of neutrophils and lymphocytes. f The bar chart of the HAI scores indicated that damage in the P. gingivalis + DSS group was much worse than that in the DSS group (P < 0.01) and NC group (P < 0.01). The data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.000 1 by independent 2-tailed Student’s t test and one-way ANOVA combined with Mann-Whitney U test
In these experiments, the disease activity index (DAI) score increased with the time, indicating that the colitis model had been successfully established. The mice in the P. gingivalis + DSS group possessed higher DAI scores than those in the NC group (P < 0.000 1) and DSS group (P < 0.05) (Fig. 1c). Colon specimens of the mice in each group after 10 days of DSS exposure are presented in Fig. 1d and show that the colon length of the mice in the P. gingivalis + DSS group was shorter than that of the mice in the other two groups (NC group, P < 0.000 1; DSS group, P < 0.05), suggesting more severe inflammation.
The DSS-induced colitis model we used in the experiment was characterized by severely damaged intestinal epithelium. H&E colonic staining (Fig. 1e) showed that DSS-induced colitis caused multifocal small ulcers, disrupted epithelial cells and crypts, reduced goblet cell numbers and lymphocyte infiltration, and incomplete mucosal structure. However, the mice treated with both DSS and P. gingivalis exhibited exacerbated histological damage, as shown by the exfoliation of the intestinal epithelium and gland, fragmentation of the basement membrane, and serious infiltrations of the mucosa by inflammatory cells. The crypt defects were obvious, and diffuse mucosal hemorrhage and extended erosion were observed. The histological activity index (HAI) scores further indicated that the intestinal epithelium lesions of the P. gingivalis + DSS group were the worst among all the three groups (NC group, P < 0.000 1; DSS group, P < 0.05) (Fig. 1f).
P. gingivalis increased Th17 cell number and decreased Treg numbers in colitis mice
The ratio of Th17 cells to Tregs can reflect the systemic inflammatory balance in UC. Proinflammatory and anti-inflammatory cytokines play crucial roles in the progression of colitis by DSS-induced43. As shown in Fig. 2, we investigated the ratio of Th17 cells to Tregs in the spleen and the expression levels of the related cytokines in the serum to determine whether P. gingivalis could exacerbate colitis by altering the Th17/Treg balance.

P. gingivalis-exacerbated UC is associated with an imbalance in Th17/Tregs. a, b Flow cytometric analysis and bar chart showing that compared with the NC group and the DSS group, P. gingivalis significantly induced the transformation of CD4+ T cells into proinflammatory Th17 cells (P < 0.01) and inhibited the generation of Tregs (P < 0.05). c ELISA showing that compared with the other two groups, P. gingivalis administration to mice with colitis significantly increased the expression level of IL-17 (P < 0.01) and decreased expression of IL-10 in serum (P < 0.05). All the indicators were measured in six replicates. The data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.000 1 by independent 2-tailed Student’s t test and one-way ANOVA combined with Mann-Whitney U test
Compared with the NC group, the DSS and P. gingivalis + DSS groups exhibited significantly increased numbers of CD4+ IL-17A+ cells (Th17 cell population) (P < 0.000 1) and protein expression level of IL-17 (P < 0.000 1) (Fig. 2a, b). In contrast, CD4+ CD25+ Foxp3+ cell numbers (Treg population) and IL-10 expression were significantly decreased in the DSS mice and were further decreased in the P. gingivalis + DSS mice (Tregs, P < 0.000 1; IL-10, P < 0.000 1) (Fig. 2c, d). Moreover, increases in the CD4+ IL-17A+ cell numbers and IL-17 expression and decreases in the CD4+ CD25+ Foxp3+ cell numbers and IL-10 expression were observed in the P. gingivalis + DSS groups compared to the DSS groups (Th17, P < 0.01; IL-17, P < 0.01; Tregs, P < 0.05; IL-10, P < 0.05).
P. gingivalis and the PPAD gene complemented strain (comΔppad) increased Th17 cell numbers and decreased Treg cell numbers compared with the P. gingivalis strain in which the PPAD gene was deleted (Δppad)
The identification results of the P. gingivalis W83 wild-type strain, Δppad, and comΔppad by PCR are shown in the Supplementary Figure. Lanes 1−3 show that the P. gingivalis 16S rRNA PCR products of the three strains are all positive, with about 200 bp. As shown in Lanes 4−6, the expressions of erythromycin resistance gene in P. gingivalis W83 wild-type strain is negative, while positive in Δppad and comΔppad. Lanes 7−9 demonstrate that the expressions of PPAD gene in P. gingivalis W83 wild-type strain and comΔppad are positive, but negative in Δppad. We used the supernatants of the cultures of the three strains as conditioned media to coculture T lymphocytes extracted from untreated C57BL/6 mice spleens. The proportion of IL-17A+ CD4+ cells and the protein expression level of IL-17A appeared to be significantly increased by P. gingivalis-conditioned growth medium (Th17, P < 0.000 1; IL-17, P < 0.000 1) and comΔppad-conditioned growth medium (Th17, P < 0.000 1; IL-17, P < 0.000 1) compared with Δppad-conditioned growth medium. In addition, the proportion of Tregs (P. gingivalis, P < 0.01; comΔppad, P < 0.01) and the expression level of IL-10 (P. gingivalis, P < 0.01; comΔppad, P < 0.01) were significantly decreased (Fig. 3).

The proportions of T cell subsets and the expression of cytokines in vitro. a, b Flow cytometric analysis and bar chart showing that the P. gingivalis- and comΔppad-conditioned culture media significantly stimulated activated T lymphocytes to undergo a transformation into Th17 cells and decreased the generation of Tregs compared to Δppad-conditioned culture medium (P < 0.05). c ELISA showing that compared to Δppad-conditioned culture medium, P. gingivalis- and comΔppad-conditioned culture medium significantly increased the expression levels of IL-17 and decreased the expression levels of IL-10 in the cell supernatants (P < 0.05). Five replicates were analyzed per sample. The data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.000 1 by independent 2-tailed Student’s t test and one-way ANOVA combined with Mann-Whitney U test
Deleting PPAD reduced the severity of UC
Compared with the DSS group, the clinical indicators of the Δppad + DSS group revealed that Δppad did not exacerbate the colitis phenotype (body weight loss, P > 0.05; DAI, P > 0.05; colon length, P > 0.05) (Fig. 4b–d). The mice in the Δppad + DSS group exhibited histopathological characteristics similar to those in the DSS group (HAI, P > 0.05) (Fig. 4e, f). The intestinal inflammation of the Δppad + DSS group was significantly milder than that of the P. gingivalis + DSS group (body weight loss, P < 0.05; DAI, P < 0.05; colon length, P < 0.05; HAI, P < 0.05) and comΔppad + DSS group (body weight loss, P < 0.05; DAI, P < 0.05; colon length, P < 0.05; HAI, P < 0.05) (Fig. 4b–f).

P. gingivalis and comΔppad exacerbate UC compared to Δppad in vivo. a Schematic of the animal experimental design in the five groups (n = 6 in each group). b The body weight loss of the DSS group and Δppad + DSS group was less than that of the P. gingivalis + DSS group and comΔppad + DSS group (P < 0.05). c The bar chart shows that the severity of colitis in the P. gingivalis + DSS group and comΔppad + DSS group was most serious among the five groups as determined assessed by the DAI scores (P < 0.05). The scores of the Δppad + DSS group were like those of the DSS group (P > 0.05). d Representative colons of the five groups. The specimens of the P. gingivalis + DSS group and comΔppad + DSS group were shorter than those in the other three groups (P < 0.05). The Δppad + DSS group presented a length that was approximately similar to that of the DSS group (P > 0.05). e Representative H&E staining of the colons from the five groups. The comΔppad + DSS group showed pathological lesions similar to those of the P. gingivalis + DSS group, with large areas of deep ulcers, destruction of epithelial cells, exfoliation of intestinal epithelium, and intestinal gland, fragmentation of basement membrane, and infiltration of neutrophils and lymphocytes. Δppad administration to mice with colitis led to pathological changes similar to those of mice with colitis, including lymphocytic infiltration and partial exfoliation of the intestinal epithelium. f Bar chart showing that the HAI score of the P. gingivalis + DSS group was equal to that of the comΔppad + DSS group (P > 0.05) and worse than that of the Δppad + DSS group and DSS group (P < 0.05). The data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.000 1 by independent 2-tailed Student’s t test and one-way ANOVA combined with Mann-Whitney U test
As shown in Fig. 5, the flow cytometry and ELISA results demonstrated that the administration of Δppad by gavage to mice with colitis mice did not promote abnormal immune responses of splenic T lymphocytes. The proportions of T cell subsets and the expression levels of cytokines were similar between the Δppad + DSS group and DSS group (Th17, P > 0.05; IL-17, P > 0.05; Tregs, P > 0.05; IL-10, P > 0.05). Compared with the Δppad + DSS groups, the P. gingivalis + DSS group and comΔppad + DSS group exhibited significantly increased percentages of Th17 cells (P. gingivalis, P < 0.01; comΔppad, P < 0.01) (Fig. 5a, b) and the expression levels of proinflammatory cytokine of IL-17 (P. gingivalis, P < 0.01; comΔppad, P < 0.01) (Fig. 5c), and decreased percentages of the suppressor T cells of Tregs (P. gingivalis, P < 0.05; comΔppad, P < 0.05) (Fig. 5a, b) and expression levels of IL-10 (P. gingivalis, P < 0.05; comΔppad, P < 0.05) (Fig. 5c).

P. gingivalis- and comΔppad-exacerbated UC is associated with an imbalance in Th17/Tregs. a, b Flow cytometric analysis and bar chart showing that compared with Δppad, P. gingivalis and comΔppad induced the transformation of CD4+ T cells into proinflammatory Th17 cells and simultaneously inhibited the generation of Tregs (P < 0.05), but no difference was observed between mice administered Δppad and DSS and mice with colitis administered physiological saline (P > 0.05). c ELISA showing that compared with Δppad, P. gingivalis and comΔppad administration caused the increased expression of IL-17 and decreased expression of IL-10 in serum (P < 0.05). The expression levels of the two cytokines were similar in the mice administered Δppad and DSS mice and the mice with colitis (P > 0.05). All indicators were measured with six replicates. The data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.000 1 by independent 2-tailed Student’s t test and one-way ANOVA combined with Mann-Whitney U test
PPAD induced an abnormal proinflammatory immune response in the colon
We harvested mouse colons at the end of the experiment on the 41st day. Immunostaining indicated a dramatic increase in the expression of IL-17 and a decrease in the expression of IL-10 in the colons of the P. gingivalis + DSS group and comΔppad + DSS group compared with those of the control group (IL-17, P < 0.000 1; IL-10, P < 0.000 1). The expression of IL-17 was significantly increased and the expression of IL-10 was decreased in the CD4+ T cells of the colon samples of the P. gingivalis + DSS group and comΔppad + DSS group compared with those of the DSS group and Δppad + DSS group (IL-17, P < 0.05; IL-10, P < 0.05) (Fig. 6), indicating that bacteria that secrete PPAD can cause proinflammatory immune responses of CD4+ T cells in colon tissue.

P. gingivalis and comΔppad exacerbate UC by upregulating IL-17 compared to Δppad as shown by immunofluorescence staining analysis. a Colon sections of mice from the NC group, DSS group, P. gingivalis + DSS group, Δppad + DSS group, and comΔppad + DSS group were stained with anti-CD4 and anti-IL-17 antibodies. Scale bar: 50 μm. b The IL-17 intensity in the CD4+ T cells of mice was quantified (n = 6 in each group). The data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.000 1 by independent 2-tailed Student’s t test and one-way ANOVA combined with Mann-Whitney U test
Discussion
Periodontitis and UC are both chronic inflammatory diseases mediated by complex interactions between immune responses and the microbes. P. gingivalis, as the major pathogenic bacterium that causes periodontitis, is considered a risk factor for many systemic diseases, such as diabetes mellitus, rheumatoid arthritis, lupus erythematosus, Alzheimer’s disease, and UC44. In our in vivo study, the mice treated with P. gingivalis by gavage exhibited a more severe clinical presentation than the mice treated with DSS alone, indicating that P. gingivalis can exacerbate the inflammation of UC. Kitamoto et al. found that symbiotic oral pathogens and effector memory T cells did not produce IFN-γ during periodontitis8. However, after reaching the intestine, these bacteria-cell symbionts produced IFN-γ and caused colitis. Intestinal inflammation disrupts the homeostasis of normal intestinal flora, allowing the oral bacteria to defeat and displace the resident bacteria in the intestine. Furthermore, oral inflammation, such as periodontitis, can increase the number of oral pathogens. After reaching a certain threshold, this number will increase the probability of the successful passage of these oral pathogens through the acidic environment of the stomach. The synergistic effect of these two conditions can promote the ectopic oral bacterial colonization of the intestine. In this regard, oral bacteria will exacerbate existing intestinal inflammation, but do not affect healthy intestines8.
Despite the fact that inflammation is a distinguishing characteristic of periodontitis, P. gingivalis not only causes inflammation but also acts as a potent inducer of abnormal immune responses45. P. gingivalis can induce abnormal cellular immunological responses46,47. In patients with periodontitis, Th17 cell numbers and related cytokine production are increased and positively correlated with periodontal destruction due to P. gingivalis, whereas Treg cell numbers and related cytokine production are decreased and negatively correlated with periodontal destruction22. The imbalance of Th17/Tregs has recently been proven to be a cause of the pathogenesis of periodontitis and many autoimmune diseases48,49. An emerging viewpoint is that UC is associated with immune-inflammatory responses, which are the result of the abnormal immune responses of T lymphocytes to specific microbes in genetically susceptible populations50. We also investigated the proportions of Th17 and Tregs among splenic T lymphocytes and the expression levels of IL-17 and IL-10 in the serum. According to the results, we concluded that P. gingivalis significantly increased the proportion of Th17 cells and the level of IL-17 and inversely decreased the proportion of Tregs and the level of IL-10. Bacteria-reactive CD4+ T cells in the gut have been proposed to play an influential role in the development of IBD51,52. Under certain circumstances, Th17 cells in the intestine undergo pathogenic transformation to produce IFN-γ and cause intestinal inflammation53,54. These cells can also trigger many autoimmune diseases, such as UC, by enhancing the permeability of cells and promoting the recruitment and activation of inflammatory cells, which consequently leads to inflammatory changes in the body55. A number of studies have discovered that the number of Th17 cells in the peripheral blood of UC patients is significantly increased and positively correlated with disease activity56,57. Li et al58. further confirmed that the inhibition of Th17 cell function in intestinal tissues significantly reduces the degree of colitis in mice with DSS-induced. High levels of IL-17 in the peripheral blood and colonic mucosa can lead to a highly inflammatory state in the host, which is closely related to the pathogenesis of UC59.
Tregs are a subset of T cells that are identified by CD4 and CD25 expression, are mainly responsible for the negative regulation of the inflammatory responses, and perform the functions of preventing, weakening, or terminating various inflammatory reactions21. Tregs can inhibit the activities of dendritic cells and macrophages that contribute to innate immunity60. Tregs also inhibit the development of various inflammatory diseases by secreting the critical cytokine of IL-10, which is characterized by its anti-inflammatory properties. IL-10 can downregulate the transcription and secretion of IL-6, TNF-α, and some other proinflammatory factors in the intestinal tract to suppress T cells and maintain tolerance functions, and maintaining mucosal homeostasis61. The metabolites of intestinal microorganisms can alleviate intestinal inflammatory symptoms in mice by increasing the number of Tregs and the expression level of IL-10 62. It has been noted that the balance of Th17 cells and Tregs is very important for controlling inflammation and maintaining intestinal immune homeostasis63.
P. gingivalis expresses dozens of virulence factors, such as gingipains, fimbrillin peptides, lipopolysaccharides, outer membrane vesicles, and various enzymes. By effectively manipulating the immunosuppression through the various virulence factors, P. gingivalis may play a role in periodontitis and related systemic diseases. Much of the pathogenicity of P. gingivalis is owing to its ability to damage the host’s immune defenses. PPAD is an enzyme secreted by P. gingivalis in the oral cavity and can induce the abnormal inflammatory responses and autoimmune reactions in the host by increasing protein citrullination35,36,64. To confirm the proinflammatory effect of PPAD, we constructed Δppad by silencing the expression function of PPAD, and we restored the PPAD gene by homologous recombination to construct comΔppad. Murine splenic T lymphocytes were stimulated with P. gingivalis-, Δppad-, and comΔppad-conditioned media. It is clear that P. gingivalis and comΔppad can significantly promote the transformation of CD4+ T cells into proinflammatory Th17 cells while inhibiting the generation of Tregs. By investigating cocultured cell supernatants, it was observed that P. gingivalis and comΔppad induced significant increases in the expression of IL-17 and decreases in the expression of IL-10. In the last part of this study, we verified the impact of the PPAD gene by orally gavaging live P. gingivalis, Δppad, and comΔppad separately into UC mice. The oral gavage of P. gingivalis and comΔppad to UC mice resulted in more severe clinical symptoms and colonic damage than the oral gavage of Δppad and the control to UC mice. The immunological indicator results were similar to the results of the in vitro cytological experiments. Compared with Δppad and the control, wild-type P. gingivalis strain and the complemented strain stimulated increased proportions of Th17 cells and increased expression of IL-17 but decreased proportions of Tregs and expression of IL-10 in mice with UC. PAD is activated extracellularly or in the cytoplasm by calcium ions, and activated PAD can promote protein citrullination, disrupt the host immune balance, and cause excessive inflammatory responses65. Citrulline levels are biomarkers of human intestinal function. Citrullination is a posttranslational modification that occurs by the conversion of arginine residues, and it can lead to pathogenesis by regulating the transcription of cytokines and the production of proinflammatory proteins66. PAD-mediated hypercitrullination is able to inhibit the expression of Th2 cytokines and amplifying the production of Th17 cytokines, resulting in an imbalance in the Th17/Tregs ratio67. Our immunofluorescence staining results showed that the wild-type strain expressing PPAD caused an increased expression of the proinflammatory cytokine of IL-17 and decreased expression of the protective cytokine of IL-10 in colon tissues. The same result was also observed with the complemented strain. These results indicate that the PPAD gene can result in an abnormal immune response in the intestinal tissue Fig. 7.

P. gingivalis and comΔppad exacerbate UC by downregulating IL-10 compared to Δppad as shown by immunofluorescence staining analysis. a Colon sections of mice from NC group, DSS group, P. gingivalis + DSS group, Δppad + DSS group, and comΔppad + DSS group were stained with anti-CD4 and anti-IL-10 antibodies. Scale bar: 50 μm. b The IL-10 intensity in the CD4+ T cells of mice was quantified (n = 6 in each group). The data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.000 1 by independent 2-tailed Student’s t test and one-way ANOVA combined with Mann-Whitney U test
In this study, we can conclude that P. gingivalis exacerbates intestinal inflammation in UC via PPAD. The possible mechanism is that PPAD induces the transformation of naive CD4+ T cells into proinflammatory Th17 cells and inhibits the generation of anti-inflammatory Tregs. The interaction between PPAD and citrullinated proteins and the mechanism by which colitis is exacerbated is still unclear. We will explore the mechanism by which PPAD exacerbates UC in future studies. The in-depth study of PPAD has broadened our horizon and provides a new research direction for exploring the related virulence factors expressed by periodontal pathogens and the abnormal immune responses of intestinal diseases. In the future, we can target PPAD to prevent the progression and exacerbation of UC by increasing the quantity and function of Tregs and maintaining the balance of Th17/Tregs in intestinal homeostasis.
Materials and methods
Preparation of bacterial strains and growth conditions
The P. gingivalis W83 wild-type strain was cultured for 5 days at 37 °C in Brain Heart Infusion (BHI) broth supplemented with vitamin K (1 μg·mL−1), hemin (5 μg·mL−1) and 5% defibrinated sheep blood in an anaerobic chamber as previously described68. The Escherichia coli (E. coli) DH5α strain was used for DNA cloning and cultured in a broth and on agar plates containing the appropriate antibiotics.
Construction and identification of P. gingivalis mutant strain (Δppad) and complemented strain (comΔppad)
The PPAD gene is an outer membrane protein, which is located at the position of 1509227–1510897 bp (GenBank accession number 2552184; locus tag PG1424) in the genome of the P. gingivalis W83 strain, and the total length of the gene is 1671 bp. The upstream region of the PPAD gene and the erythromycin resistance cassette ermF/ermAM were amplified by PCR and inserted into the XbaI and SphI sites and the SmaI and XbaI sites, respectively, of the pUC19 plasmid (Genscript Biotech Co., Nanjing, China). Then, the downstream region of the PPAD gene was inserted into the SacI and SmaI sites to construct the deletional inactivation plasmid. Alternatively, instead of the downstream region of the PPAD gene was inserted into the SacI and SmaI sites to construct the complemented plasmid. The correct placement and orientation of the inserted DNA segments were verified by sequencing. These two modified plasmids were integrated into the P. gingivalis W83 genome by electroporation to generate Δppad and comΔppad. The Δppad and comΔppad were subcultured on erythromycin-containing selection plates3.
To identify P. gingivalis W83, Δppad and comΔppad, total DNA of these three strains were extracted by Genomic DNA Mini Preparation Kit (#D0063, Beyotime Biotechnology, Shanghai, China), PCR was performed with primers targeting P. gingivalis 16S rRNA-, Erm-, or PPAD-specific regions, which were designed by Primer Premier 6.0 (supplementary table). The three strains were centrifuged at 10 000 × g for 10 min and then adjusted to a final concentration of 1 × 108 colony forming units (CFUs)/mL based on ultraviolet spectrophotometry colorimetry at 450 nm for further use.
Animals and dextran sodium sulfate (DSS)-induced UC
Six-week-old, healthy, specific pathogen-free (SPF) female C57BL/6 mice weighing approximately (20.0 ± 1.0) g were obtained from Beijing Vital River Laboratory Animal Technology Co. We used the female sex only, as female C57BL/6 mice have been reported to be more susceptible to UC modeling in the literature69. All the mice were acclimatized under SPF conditions, exposed to 12 h cycles of light and dark, and fed with standard rodent chow and water ad libitum in clear cages for one week. Then, all the mice were administered azithromycin in their drinking water (10 mg per 500 mL) for 5 days to eliminate the original intestinal flora, followed by a 7-day antibiotic-free period before the initiation of DSS administration at eleven weeks of age70. DSS (#160110, molecular weight: 36 000–50 000; MP Biomedicals, Santa Ana, USA) dissolved in drinking water (3% (wt/vol)) was provided ad libitum to those mice for 10 days to induce an acute colitis71.
Grouping and experimental design
All the mice were randomly divided into five groups (n = 6 per group): the normal control group (NC), DSS group (DSS), P. gingivalis + DSS group (P. gingivalis + DSS), Δppad + DSS group (Δppad + DSS), and comΔppad + DSS group (comΔppad + DSS). The mice in the P. gingivalis + DSS group, Δppad + DSS group, and comΔppad + DSS group were administered a total of 108 CFUs of live P. gingivalis W83, Δppad, or comΔppad suspended in 100 μL sterile saline twice per week by oral gavage for the entire 40 days (days 0−40) and an additional 3% DSS for the last 10 days (days 31–40)71,72,73. In the DSS groups, the mice were orally gavaged with saline twice per week (days 0−40) and 3% DSS for 10 days (days 31–40). The mice in the NC group were orally gavaged with saline twice per week for the entire 40 days and offered normal drinking water.
The daily observation was conducted to record the weight, water/food consumption, piloerection, stool consistency, occult blood, and hematochezia throughout the experiment. At the end of the experiment (day 41, Fig. 1a), peripheral blood was collected from the inner canthus for cytokine detection. All the mice were humanely sacrificed, and colon segments and spleens were harvested for follow-up analyses.
Assessment of the severity of colitis by measuring the DAI and HAI
In the colitis model, disease severity is usually related to shortened colon length caused by intestinal inflammation74. Oral administration of DSS leads to loss of body weight, changes in stool consistency, and bloody diarrhea that mimic the parameters of the clinical presentation of humans with UC and can be used to evaluate the DAI score71. Colon segments were harvested, fixed in 4% paraformaldehyde, dehydrated with ethanol, and embedded in paraffin. Paraffin sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E) for histological analyses and calculating the HAI score75 (supplementary table).
Culture of T cells with P. gingivalis-, Δppad-, or comΔppad-conditioned medium
P. gingivalis, Δppad, and comΔppad were maintained in BHI broth at 37 °C for 24 h in an anaerobic chamber. A total of 1 × 108 CFU bacteria were quantified, and the supernatants of three strains were collected by centrifugation at 10 000 × g and filtered through a 0.22 μm filter. Twelve-well plates were coated with anti-mouse CD3 (2 µg·mL−1 in PBS, 1 mL per well) (#100340, Biolegend, California, USA) per well at 4 °C overnight before T cell isolation.
Spleens were collected from healthy C57BL/6 mice and passed through a 70 μm cell strainer. Further clearing of the noncellular debris was achieved by centrifugation at 500 × g for 5 min. The resulting debris-free single-cell suspension was washed and filtered through a 40 μm cell strainer and centrifuged again. After the final resuspension, the total numbers of T lymphocytes were counted under a hemocytometer. The T lymphocytes were washed and resuspended in RPMI-1640 medium (#21870084, Gibco, Carlsbad, USA) supplemented with 10% fetal bovine serum (#10100139C, Gibco, Carlsbad, USA), penicillin-streptomycin solution (100 U·mL−1 penicillin/100 μg·mL−1 streptomycin) (#SV30010, HyClone, South Logan, USA), anti-mouse CD28 antibody (2 μg·mL−1) (#102116, Biolegend, California, USA), and TGF-β (2 ng·mL−1) (#100-21-2, Peprotech, Rocky Hill, USA). IL-6 (50 ng·mL−1) (#216-16-2, Peprotech, Rocky Hill, USA) was added to the RPMI-1640 medium to differentiate Th17 cells, while IL-2 (2 ng·mL−1) (#212-12-5, Peprotech, Rocky Hill, USA) was added to the medium to differentiate Tregs. T lymphocytes were cocultured with the bacterial medium in 12-well plates without the coating buffer in humidified air with 5% CO2 at 37°C for 3 days. Then, the cells and supernatants were harvested for analyses.
Determination of the percentages of T cell subsets by flow cytometric analysis
A total of 1 × 106 splenic T lymphocytes in vivo and three kinds of in vitro cocultured T cells described above were separately washed and resuspended in FACS buffer containing phosphate-buffered saline (PBS) and 0.5% bovine serum albumin (BSA). The suspension was incubated with fluorescently labeled monoclonal antibodies for 1 h at 4 °C in the dark. Anti-mouse PerCP-CD4 (#46-0041-82, Thermo Fisher Scientific, Waltham, USA) and anti-mouse PE-IL-17 (#12-7177-81, Thermo Fisher Scientific, Waltham, USA) monoclonal antibodies were prepared to identify Th17 cells, while anti-mouse PerCP-CD4, anti-mouse APC-CD25 (#17-0251-82, Thermo Fisher Scientific, Waltham, USA), and anti-mouse PE-Foxp3 (#12-5773-82, Thermo Fisher Scientific, Waltham, USA) monoclonal antibodies were prepared to identify Tregs.
After washing three times, the cells were resuspended again in PBS containing 0.1% BSA for flow cytometry (#Canto II, BD Biosciences, New Jersey, US). The data were analyzed with FlowJo (#V10.5, BD Biosciences, New Jersey, US).
Detection of the cytokines IL-17 and IL-10 in sera and cell supernatants
Peripheral blood was harvested from the inner canthus and then centrifuged at 3,000 rpm for 10 min to isolate the serum. Mouse cytokine ELISA kits (anti-mouse IL-17A: #88-7371-88, Thermo Fisher Scientific, Waltham, USA; anti-mouse IL-10: #88-7105-88, Thermo Fisher Scientific, Waltham, USA) were used to analyze the levels of the cytokines IL-17 and IL-10 in the sera and cell supernatants. The samples were analyzed in triplicate, and the concentrations were calculated according to standard curves.
Immunofluorescence staining analysis
Paraffin sections (6 μm) were deparaffinized, rehydrated, prepared, and double-stained with the following antibodies: (i) rat anti-CD4 (1:500; BD Pharmingen, California, USA) with rabbit anti-IL-17 (1:500; Novus Biologicals, Littleton, Colorado, USA) and (ii) rat anti-CD4 (1:500; BD Pharmingen, California, USA) with goat anti IL-10 (1:200; R&D, California, USA). The sections were incubated with the antibodies in a humidified chamber overnight at 4 °C. After being washed with PBS, the sections were incubated with a mixture of Alexa 488-conjugated donkey anti-goat (1:500; Jackson ImmunoResearch, Pennsylvania, USA) and Cy3-conjugated donkey anti-rat (1:500; Jackson ImmunoResearch, Pennsylvania, USA) secondary antibodies for 2 h at room temperature. All of the sections were counterstained with DAPI (1:10 000; #PP0131, Beyotime Biotechnology, Shanghai, China). Images were obtained using a FluoView confocal microscope (#FV3000, Olympus, Tokyo, Japan). Immunostaining quantification was completed using ImageJ software.
Statistical analysis
GraphPad Prism 8.0 (GraphPad software Corp., California, USA) was used for the statistical analysis. Comparisons between two groups were performed using an independent two-tailed Student’s t test, and one-way analysis of variance (ANOVA) combined with the Mann−Whitney U test was used to determine the significance of the differences among multiple groups. The data are shown as the means ± SDs. P < 0.05 was considered statistically significant.
References
Pihlstrom, B. L., Michalowicz, B. S. & Johnson, N. W. Periodontal diseases. Lancet 366, 1809–1820 (2005).
Loos, B. G. Systemic markers of inflammation in periodontitis. J. Periodontol. 76, 2106–2115 (2005).
Wegner, N. et al. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 62, 2662–2672 (2010).
Kaushal, S., Singh, A. K., Lal, N., Das, S. K. & Mahdi, A. A. Effect of periodontal therapy on disease activity in patients of rheumatoid arthritis with chronic periodontitis. J. Oral. Biol. Craniofac. Res. 9, 128–132 (2019).
Habashneh, R. A., Khader, Y. S., Alhumouz, M. K., Jadallah, K. & Ajlouni, Y. The association between inflammatory bowel disease and periodontitis among Jordanians: a case-control study. J. Periodontal Res. 47, 293–298 (2012).
Tan, C. X., Brand, H. S., de Boer, N. K. & Forouzanfar, T. Gastrointestinal diseases and their oro-dental manifestations: Part 2: Ulcerative colitis. Br. Dent. J. 222, 53–57 (2017).
Nakajima, M. et al. Oral administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS One 10, e0134234 (2015).
Kitamoto, S. et al. The intermucosal connection between the mouth and gut in commensal pathobiont-driven colitis. Cell 182, 447–462 (2020). e414.
Messora, M. R. et al. Probiotic therapy reduces periodontal tissue destruction and improves the intestinal morphology in rats with ligature-induced periodontitis. J. Periodontol. 84, 1818–1826 (2013).
Foureaux Rde, C. et al. Effects of probiotic therapy on metabolic and inflammatory parameters of rats with ligature-induced periodontitis associated with restraint stress. J. Periodontol. 85, 975–983 (2014).
Van Dyke, T. E., Dowell, V. R. Jr., Offenbacher, S., Snyder, W. & Hersh, T. Potential role of microorganisms isolated from periodontal lesions in the pathogenesis of inflammatory bowel disease. Infect. Immun. 53, 671–677 (1986).
Gangula, P. et al. Polybacterial periodontal pathogens alter vascular and gut BH4/nNOS/NRF2-Phase II enzyme expression. PLoS One 10, e0129885 (2015).
Lira-Junior, R. & Figueredo, C. M. Periodontal and inflammatory bowel diseases: is there evidence of complex pathogenic interactions? World J. Gastroenterol. 22, 7963–7972 (2016).
Nguyen, C. M., Kim, J. W., Quan, V. H., Nguyen, B. H. & Tran, S. D. Periodontal associations in cardiovascular diseases: the latest evidence and understanding. J. Oral. Biol. Craniofac. Res. 5, 203–206 (2015).
How, K. Y., Song, K. P. & Chan, K. G. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front. Microbiol. 7, 53 (2016).
Fiorillo, L. et al. Porphyromonas gingivalis, periodontal and systemic implications: a systematic review. Dent J. 7, 114 (2019).
Olsen, I., Lambris, J. D. & Hajishengallis, G. Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J. Oral. Microbiol. 9, 1340085 (2017).
Olsen, I. & Hajishengallis, G. Major neutrophil functions subverted by Porphyromonas gingivalis. J. Oral. Microbiol. 8, 30936 (2016).
Hajishengallis, G., Darveau, R. P. & Curtis, M. A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10, 717–725 (2012).
Diaz-Zuniga, J. et al. Differential human Th22-lymphocyte response triggered by Aggregatibacter actinomycetemcomitans serotypes. Arch. Oral. Biol. 78, 26–33 (2017).
Gonzales, J. R. T- and B-cell subsets in periodontitis. Periodontol 2000 69, 181–200 (2015).
Dutzan, N. et al. Interleukin-21 expression and its association with proinflammatory cytokines in untreated chronic periodontitis patients. J. Periodontol. 83, 948–954 (2012).
Adibrad, M. et al. Signs of the presence of Th17 cells in chronic periodontal disease. J. Periodontal Res. 47, 525–531 (2012).
Qi, Y. et al. Role of serum IL-23/IL-17 axis in the relationship between periodontitis and coronary heart disease. Int J. Periodontics Restor. Dent. 33, 185–191 (2013).
Wang, H. et al. Interaction between oral lichen planus and chronic periodontitis with Th17-associated cytokines in serum. Inflammation 36, 696–704 (2013).
Wang, L., Wang, J., Jin, Y., Gao, H. & Lin, X. Oral administration of all-trans retinoic acid suppresses experimental periodontitis by modulating the Th17/Treg imbalance. J. Periodontol. 85, 740–750 (2014).
Jin, Y., Wang, L., Liu, D. & Lin, X. Tamibarotene modulates the local immune response in experimental periodontitis. Int. Immunopharmacol. 23, 537–545 (2014).
Chumanevich, A. A. et al. Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G929–G938 (2011).
Volarevic, V. et al. Galectin-3 regulates indoleamine-2,3-dioxygenase-dependent cross-talk between colon-infiltrating dendritic cells and T regulatory cells and may represent a valuable biomarker for monitoring the progression of ulcerative colitis. Cells 8, 709 (2019).
Matricon, J., Barnich, N. & Ardid, D. Immunopathogenesis of inflammatory bowel disease. Self Nonself. 1, 299–309 (2010).
Fujisaki, M. & Sugawara, K. Properties of peptidylarginine deiminase from the epidermis of newborn rats. J. Biochem. 89, 257–263 (1981).
Witalison, E. E., Thompson, P. R. & Hofseth, L. J. Protein arginine deiminases and associated citrullination: physiological functions and diseases associated with dysregulation. Curr. Drug Targets 16, 700–710 (2015).
Vermilyea, D. M., Ottenberg, G. K. & Davey, M. E. Citrullination mediated by PPAD constrains biofilm formation in P. gingivalis strain 381. NPJ Biofilms Microbiomes 5, 7 (2019).
Machold, K. P. et al. Very recent onset rheumatoid arthritis: clinical and serological patient characteristics associated with radiographic progression over the first years of disease. Rheumatology 46, 342–349 (2007).
Li, S., Yu, Y., Yue, Y., Zhang, Z. & Su, K. Microbial infection and rheumatoid arthritis. J. Clin. Cell Immunol. 4, 174 (2013).
Koziel, J., Mydel, P. & Potempa, J. The link between periodontal disease and rheumatoid arthritis: an updated review. Curr. Rheumatol. Rep. 16, 408 (2014).
Dreyton, C. J. et al. Optimization and characterization of a pan protein arginine deiminase (PAD) inhibitor. In Probe Reports from the NIH Molecular Libraries Program (Bethesda (MD), 2010).
Cau, L., Mechin, M. C. & Simon, M. Peptidylarginine deiminases and deiminated proteins at the epidermal barrier. Exp. Dermatol. 27, 852–858 (2018).
Dragoni, G., De Hertogh, G. & Vermeire, S. The role of citrullination in inflammatory bowel disease: a neglected player in triggering inflammation and fibrosis? Inflamm. Bowel Dis. 27, 134–144 (2021).
Makrygiannakis, D. et al. Citrullination is an inflammation-dependent process. Ann. Rheum. Dis. 65, 1219–1222 (2006).
McGraw, W. T., Potempa, J., Farley, D. & Travis, J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect. Immun. 67, 3248–3256 (1999).
McInnes, I. B. & Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 365, 2205–2219 (2011).
Zhang, M. et al. Butyrate inhibits interleukin-17 and generates Tregs to ameliorate colorectal colitis in rats. BMC Gastroenterol. 16, 84 (2016).
Hajishengallis, G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 15, 30–44 (2015).
Darveau, R. P., Hajishengallis, G. & Curtis, M. A. Porphyromonas gingivalis as a potential community activist for disease. J. Dent. Res. 91, 816–820 (2012).
Moutsopoulos, N. M. et al. Porphyromonas gingivalis promotes Th17 inducing pathways in chronic periodontitis. J. Autoimmun. 39, 294–303 (2012).
Olsen, I., Taubman, M. A. & Singhrao, S. K. Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and Alzheimer’s disease. J. Oral. Microbiol. 8, 33029 (2016).
Eskan, M. A. et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol. 13, 465–473 (2012).
Garlet, G. P. et al. Regulatory T cells attenuate experimental periodontitis progression in mice. J. Clin. Periodontol. 37, 591–600 (2010).
Indriolo, A., Greco, S., Ravelli, P. & Fagiuoli, S. What can we learn about biofilm/host interactions from the study of inflammatory bowel disease. J. Clin. Periodontol. 38, 36–43 (2011).
Calderon-Gomez, E. et al. Commensal-specific CD4(+) cells from patients with Crohn’s disease have a T-helper 17 inflammatory profile. Gastroenterology 151, 489–500e483 (2016).
Shen, C., Landers, C. J., Derkowski, C., Elson, C. O. & Targan, S. R. Enhanced CBir1-specific innate and adaptive immune responses in Crohn’s disease. Inflamm. Bowel Dis. 14, 1641–1651 (2008).
Ahern, P. P. et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 33, 279–288 (2010).
Harbour, S. N., Maynard, C. L., Zindl, C. L., Schoeb, T. R. & Weaver, C. T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl Acad. Sci. USA 112, 7061–7066 (2015).
Kotake, S. et al. Elevated ratio of Th17 cell-derived Th1 cells (CD161(+)Th1 cells) to CD161(+)Th17 cells in peripheral blood of early-onset rheumatoid arthritis patients. Biomed. Res. Int. 2016, 4186027 (2016).
Slebioda, T. J. et al. TL1A as a potential local inducer of IL17A expression in colon mucosa of inflammatory bowel disease patients. Scand. J. Immunol. 82, 352–360 (2015).
Yamada, A. et al. Role of regulatory T cell in the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 22, 2195–2205 (2016).
Li, Y. H. et al. Berberine ameliorates chronic relapsing dextran sulfate sodium-induced colitis in C57BL/6 mice by suppressing Th17 responses. Pharm. Res. 110, 227–239 (2016).
Li, Q. et al. Total glycosides of peony protects against inflammatory bowel disease by regulating IL-23/IL-17 axis and Th17/Treg balance. Am. J. Chin. Med. 47, 177–201 (2019).
Anderson, K. M. et al. Dual destructive and protective roles of adaptive immunity in neurodegenerative disorders. Transl. Neurodegener. 3, 25 (2014).
Roers, A. et al. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J. Exp. Med. 200, 1289–1297 (2004).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013).
Ueno, A. et al. Increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm. Bowel Dis. 19, 2522–2534 (2013).
Scher, J. U., Bretz, W. A. & Abramson, S. B. Periodontal disease and subgingival microbiota as contributors for rheumatoid arthritis pathogenesis: modifiable risk factors? Curr. Opin. Rheumatol. 26, 424–429 (2014).
Jones, J. E., Causey, C. P., Knuckley, B., Slack-Noyes, J. L. & Thompson, P. R. Protein arginine deiminase 4 (PAD4): current understanding and future therapeutic potential. Curr. Opin. Drug Discov. Devel. 12, 616–627 (2009).
Wegner, N. et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol. Rev. 233, 34–54 (2010).
Chang, H. H. et al. A molecular signature of preclinical rheumatoid arthritis triggered by dysregulated PTPN22. JCI Insight 1, e90045 (2016).
Fletcher, H. M., Schenkein, H. A. & Macrina, F. L. Cloning and characterization of a new protease gene (prtH) from Porphyromonas gingivalis. Infect. Immun. 62, 4279–4286 (1994).
Jang, J. et al. Daucosterol suppresses dextran sulfate sodium (DSS)-induced colitis in mice. Int. Immunopharmacol. 72, 124–130 (2019).
Velsko, I. M. et al. Fusobacterium nucleatum alters atherosclerosis risk factors and enhances inflammatory markers with an atheroprotective immune response in ApoE(null) mice. PLoS One 10, e0129795 (2015).
Alex, P. et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 15, 341–352 (2009).
Boyer, E. et al. Oral dysbiosis induced by Porphyromonas gingivalis is strain-dependent in mice. J. Oral. Microbiol. 12, 1832837 (2020).
Sato, K. et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci. Rep. 7, 6955 (2017).
Wirtz, S. et al. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 12, 1295–1309 (2017).
Obermeier, F. et al. Interferon-gamma (IFN-gamma)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin. Exp. Immunol. 116, 238–245 (1999).
Acknowledgements
We are grateful to Chen Li and Yanshu Li for assistance in electroporation during the construction of Δppad and comΔppad, Xinwen Zhang for immunofluorescence staining, and Yanan Ma for statistical analysis. This study was supported by grants from the National Natural Science Foundation of China (81870771), and the plan of the talents for Liaoning development (XLYC1802129).
Author information
Authors and Affiliations
Contributions
X.Z., J.L., and Y.P. conceived and designed the study; X.Z., Y.L., N.Y., and C.Z. have contributed for data analysis; X.Z., Z.L., S.Z.hang, Y.L., Q.L., J.L., and D.L. performed the experiments; X.Z. wrote the paper with comments from other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
All the procedures involving the animals, which were given humane care following the concept of replacement, refinement, and reduction (the 3Rs), were approved by the ethics committee of China Medical University (approval number 2018155). The use of mice in this study complied with Animal Research Reporting In Vivo Experiments (ARRIVE) guidelines.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, X., Liu, J., Zhang, C. et al. Porphyromonas gingivalis exacerbates ulcerative colitis via Porphyromonas gingivalis peptidylarginine deiminase. Int J Oral Sci 13, 31 (2021). https://doi.org/10.1038/s41368-021-00136-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41368-021-00136-2
This article is cited by
-
The oral microbiome in autoimmune diseases: friend or foe?
Journal of Translational Medicine (2023)

{kind=link}