
Abstract
Osteoarthritis (OA) is the most common form of arthritis. It is characterized by progressive destruction of articular cartilage and the development of chronic pain and constitutes a considerable socioeconomic burden. Currently, pharmacological treatments mostly aim to relieve the OA symptoms associated with inflammation and pain. However, with increasing understanding of OA pathology, several potential therapeutic targets have been identified, enabling the development of disease-modifying OA drugs (DMOADs). By targeting inflammatory cytokines, matrix-degrading enzymes, the Wnt pathway, and OA-associated pain, DMOADs successfully modulate the degenerative changes in osteoarthritic cartilage. Moreover, regenerative approaches aim to counterbalance the loss of cartilage matrix by stimulating chondrogenesis in endogenous stem cells and matrix anabolism in chondrocytes. Emerging strategies include the development of senolytic drugs or RNA therapeutics to eliminate the cellular or molecular sources of factors driving OA. This review describes the current developmental status of DMOADs and the corresponding results from preclinical and clinical trials and discusses the potential of emerging therapeutic approaches to treat OA.
Similar content being viewed by others


Introduction
The key feature of osteoarthritis (OA) is the gradual loss of articular cartilage. Other OA-related manifestations include osteophyte formation at joint margins and bone remodeling that accompanies bone marrow lesions and subchondral bone sclerosis1,2,3,4. Synovial inflammation and meniscal damage are common features of OA. All of these OA manifestations collectively lead to the impairment of joint function and the development of chronic pain, and OA is widely considered a whole-joint disease5.
OA treatment has been largely limited to steroidal or nonsteroidal anti‐inflammatory drugs that provide symptomatic relief from pain and inflammation6. Next-generation OA treatments, often referred to as disease-modifying OA drugs (DMOADs), are under development and aim to modify the underlying OA pathophysiology and alleviate the associated structural damage to prevent long-term disability. Although DMOADs are not yet available in the pharmaceutical market, several clinical trials are ongoing7. One group of promising DMOADs delays cartilage degeneration by targeting pro-inflammatory cytokines, the proteolytic activities of catabolic enzymes, and the Wnt pathway. Another group of drugs stimulates the regenerative potential of cartilage to counteract matrix loss in osteoarthritic cartilage. The emerging DMOAD therapies under active investigation aim to eliminate senescent chondrocytes or use RNA-based approaches to modulate OA-inducing mechanisms.
DMOADs based on the molecular mechanisms underlying OA pathogenesis
Based on recent advances in our understanding of the mechanisms underlying OA pathogenesis, various DMOADs have been developed. In particular, an imbalance between matrix anabolism and catabolism contributes to osteoarthritic cartilage degeneration4,8. The DMOADs that are currently in clinical trials aim to restore the homeostasis of matrix metabolism.
Pro-inflammatory cytokines and matrix-degrading enzymes
Therapeutic strategies targeting pro-inflammatory cytokines, matrix-degrading enzymes, or Wnt signaling have been developed to delay the catabolism of cartilage matrix in OA patients.
Targeting pro-inflammatory cytokines
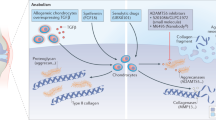
Interleukin (IL)-1 and tumor necrosis factor (TNF) are the most well-characterized pro-inflammatory cytokines and stimulate the production of inflammatory mediators, such as prostaglandin E, nitric oxide synthase, chemokines, and other cytokines, in the joint microenvironment9,10,11,12,13. Furthermore, IL-1 and TNF directly promote the expression of matrix metalloproteinases (MMPs) and other matrix-degrading enzymes involved in cartilage degeneration9,10. Therefore, there have been rigorous attempts to treat OA by inhibiting the IL-1 and TNF pathways (Fig. 1). However, the results of clinical trials of therapeutic candidates that block these pro-inflammatory cytokines have been rather unsatisfactory despite the fact that these candidates effectively suppress the inflammatory phenotypes in chondrocytes in vitro14. Intra-articular injection of anakinra, an IL-1 receptor antagonist that obstructs the receptor binding of both IL-1α and IL-1β, into 160 individuals with knee OA did not reduce OA-associated pain or cartilage turnover during weeks 4–12 of administration in a random controlled trial (NCT00110916, phase II clinical trial)15. Likewise, a randomized double-blind controlled trial (NCT00110942, phase II clinical trial) of AMG108, which is a monoclonal antibody against IL-1 receptor type I that blocks the receptor binding of both IL-1α and IL-1β, did not provide sufficient clinical benefits16. ABT-981 (a dual neutralizing antibody against IL-1α and IL-1β) was tested in patients with hand17 or knee18 OA. Neither two phase II clinical trial (NCT02384538 and NCT02087904) showed substantially improved outcomes, indicating that ABT-981 is ineffective in treating OA. In a clinical trial involving 43 hand OA patients with random allocation to groups administered adalimumab (TNF antibody) or placebo for 12 weeks, no significant difference in hand pain was noted between the two groups19. Similarly, in a trial of 90 patients with hand OA, etanercept (a decoy receptor that binds to TNF) did not differ from placebo in alleviating pain after 24 weeks of administration20.

IL-1 and TNF are the major pro-inflammatory cytokines that stimulate the production of matrix-degrading enzymes and inflammatory mediators in joint tissues. MMP and ADAMTS family members degrade the extracellular matrix components of cartilage, promoting osteoarthritic cartilage destruction.
Targeting matrix-degrading enzymes
MMPs are a family of zinc-dependent proteolytic enzymes that degrade the components of the extracellular matrix21. Various MMPs are upregulated in the degenerating cartilage of OA patients22,23. Although some of the developed MMP inhibitors have shown notable effects on preclinical OA models24,25,26,27,28, only a few have entered clinical trials for patients with mild-to-moderate knee OA (Fig. 1).
The clinical efficacy of PG-116800, a small-molecule inhibitor with a high affinity for MMP-2, -3, -8, -9, -13, and -14, was tested in 401 patients with knee OA with random allocation to treatment groups that also included a placebo group29. No statistically significant difference in knee-joint space width or the Western Ontario and McMaster Universities Osteoarthritis Index WOMAC score was observed between the test and placebo groups. Furthermore, some side effects, such as restricted joint motion and arthralgia, were observed in the test group29. Although the cause of these adverse effects remains unclear, MMP inhibitors may broadly affect the matrix turnover in musculoskeletal tissues other than cartilage30. Accordingly, these studies provide evidence that broad-spectrum MMP inhibitors are unlikely to be suitable for OA treatment due to their side effects.
MMP-13 has attracted the most attention as a promising therapeutic target because it has the highest substrate specificity against type II collagen, the most abundantly present collagen in cartilage. Wang et al. examined the effect of CL82198, a specific MMP-13 inhibitor, on inhibiting MMP-13 activity in a preclinical model of OA24. In mice with surgically induced OA, different doses of CL82198 or control saline was intraperitoneally injected daily beginning 1 day after the surgery. OA progression was significantly alleviated after CL82198 administration. No follow-up clinical studies on this compound have been performed yet. Recently, Baragi et al. developed the MMP-13 inhibitor ALS 1-0635 and evaluated its efficacy in an OA rat model31. The researchers orally administered ALS 1-0635 to rats twice a day for 3 weeks and found that ALS 1-0635 protected the cartilage from osteoarthritic destruction. Of note, frequent administration of the relatively high dose of 60 mg/kg ALS 1-0635 was effective, suggesting potential shortcomings associated with the low substrate specificity of ALS 1-0635.
ADAMTS-4 and -5 are principal enzymes responsible for cleaving aggrecan, the major proteoglycan in articular cartilage. Knockout of Adamts5 but not Adamts4 in mice alleviated OA-induced proteoglycan loss in cartilage, suggesting that ADAMTS-5 is the primary enzyme responsible for aggrecan cleavage32,33. GLPG1972 is a highly selective, orally bioavailable small molecule that inhibits ADAMTS-534 (Fig. 1). Two phase I studies (NCT02851485 and NCT03311009) showed that GLPG1972 was safe and well tolerated without any evident adverse events35,36. The drug caused a decrease up to 53% decrease in the serum levels of the aggrecan neo-epitope generated by ADAMTS-5 catalytic activities. Unfortunately, a recent phase II study (NCT03595618) with 938 patients did not meet the primary endpoint, pending detailed results to be reported37. Another novel ADAMTS-5 inhibitor under development is nanobody M6495. Nanobodies are single-domain monoclonal antibodies whose antigen-binding sites are composed of one heavy chain; thus nanobodies are markedly smaller in size than conventional monoclonal antibodies38. M6495 is a bifunctional nanobody that can bind to both ADAMTS-5 metalloproteinase/disintegrin domains and human serum albumin (Fig. 1). The binding of M6495 with albumin extends its half-life in vivo39. In a phase I clinical trial (NCT03224702), M6495 was subcutaneously injected into healthy male subjects and demonstrated an acceptable safety and tolerability profile40. Another phase I study (NCT03583346) was conducted to validate the safety and efficacy profile in OA patients. The results are expected to be announced in the near future.
The cysteine cathepsin family is composed of eleven members41,42. Cathepsins B, H, K, L, and S are the best-known members of the cathepsin family and can degrade native collagens and other components of the ECM43,44,45. In particular, increased expression of cathepsin K has been observed in the degenerative cartilage of human OA46,47. Multiple selective cathepsin K inhibitors have been shown to be effective in treating OA in animal models, ameliorating cartilage degeneration48,49 or joint pain50,51. MIV-711, an orally administered small-molecule cathepsin K inhibitor, is in clinical development for OA treatment (Fig. 1). In a phase I trial (NCT03443453) evaluating bioavailability, MIV-711 was found to be safe and well tolerated in healthy subjects52. MIV-711 did not meet the primary endpoint for the Numeric Rating Scale (NRS) knee pain score in the phase II clinical trial and its extension substudy (NCT02705625 and NCT03037489). Nevertheless, MIV-711 has shown some beneficial effects in terms of cartilage thickness, as assessed by radiological analysis, and OA-associated pain measured according to WOMAC53,54,55, leaving room to further improve the clinical efficacy of cathepsin K inhibitors.
The Wnt pathway
The Wnt signaling pathway is transduced through a large family of Wnt glycoproteins (19 genes in mammals)56. β-Catenin is one of the important protein in canonical Wnt signaling, which regulates the development and homeostasis of joints57. Activation of the Wnt signaling pathway has been noted in the cartilage, bone, and synovial membrane in OA patients58,59.
Canonical Wnt signaling starts with Wnt binding to Frizzled receptors, leading to the disruption of the β-catenin destruction complex. Stabilized β-catenin then translocates into the nucleus and interacts with the transcription factors T-cell factor (TCF) and lymphoid enhancer factor (LEF), activating the expression of Wnt target genes60,61. Interestingly, β-catenin levels are frequently upregulated in OA joint tissues, causing chondrocyte hypertrophy and synovial inflammation62,63,64,65,66. Canonical Wnt signaling plays an essential role in regulating bone remodeling and repair57, indicating that this signaling pathway needs to be carefully modulated in the joint when developing Wnt-targeting therapeutic strategies. Indeed, previous strategies targeting members of the Wnt pathway, such as β-catenin or upstream members, have not resulted in FDA-approved drugs67,68, suggesting that the selective regulation of Wnt target genes or approaches that spare β-catenin may be necessary.
Notably, lorecivivint (also known as SM04690) was identified through high-throughput screening for compounds targeting the Wnt signaling pathways and demonstrated efficacy in mitigating cartilage degeneration in a rat model of OA69. Later, the anti-inflammatory and chondroprotective effects of lorecivivint were found to be unrelated to β-catenin but were mediated by the inhibition of two intranuclear kinases, CLK2 and DYRK1A70. In a phase I trial (NCT02095548) involving 61 patients with moderate-to-severe knee OA, intra-articular administration of lorecivivint effectively restricted systemic exposure of lorecivivint and did not induce any severe adverse events, thus validating the safety of this compound71. In a phase IIa proof-of-concept study (NCT02536833) involving 455 patients, compared with placebo treatment, lorecivivint treatment did not meet the primary endpoint of improvement set by the WOMAC pain score by week 1372. However, at week 52, patients treated with the 0.07 mg dose showed significant improvements compared with those in the placebo group72. In a phase IIb study (NCT03122860), among the 695 patients treated with either of four different doses (0.03, 0.07, 0.15, and 0.23 mg), those treated with 0.07 and 0.23 mg showed statistically significant improvements in OA-associated pain according to the NRS and WOMAC pain score73. The phase II clinical trial (NCT03706521) was completed in December 2020, but the results had not yet been reported when this review was prepared. Other ongoing or scheduled clinical trials (Phase II: NCT03727022 and Phase III: NCT03928184, NCT04385303, and NCT04520607) are underway to test the efficacy of long-term administration of lorecivivint at the optimized dose of 0.07 mg.
Cartilage regeneration
DMOADs targeting catabolic factors are effective in delaying further cartilage degeneration but are insufficient in reconstructing degenerated tissue. Regenerative therapy aims to restore the normal architecture and function of a damaged joint. Cartilage regeneration is mediated by the chondrogenic differentiation of stem cells and the synthesis of cartilage matrix by chondrocytes3,74. However, the regenerative capacity of cartilage tissue in joints markedly declines with age and traumatic joint injuries.
Kartogenin is a small molecule that stimulates chondrogenic differentiation in mesenchymal stem cells (MSCs) and was developed by Johnson et al. in 2012 for the purpose of cartilage regeneration74. Kartogenin binds to filamin A and consequently interrupts the interaction of filamin A with the transcription factor core-binding factor β subunit, thereby upregulating type II collagen and aggrecan expression74. While kartogenin showed promise in stimulating cartilage regeneration, several challenges remain in its clinical applications. Recently, through extensive medicinal modifications, KA3475 was developed as an analog of kartogenin, and this variant significantly improved the potency and chemical stability of kartogenin. With an improved safety and efficacy profile, KA34 has recently finished a phase I clinical study (NCT03133676) with 60 OA patients, but the results have not yet been reported.
LNA043 is a novel angiopoietin-like protein 3 (ANGPTL3) agonist76. Human ANGPTL3 is a 460-amino-acid polypeptide that is mainly involved in regulating lipid metabolism and angiogenesis77. The current patent (US20160213748A1) claims a novel role of ANGPTL3 in facilitating the chondrogenic differentiation of MSCs. An ANGPTL3-variant polypeptide has been shown to enhance chondrogenesis, playing a chondroprotective role in a preclinical OA mouse model (US20160213748A1, WO2014138687A1). A phase I clinical trial (NCT03334812) of LNA043 in patients with knee cartilage defects was completed early based on favorable outcomes in terms of safety and tolerability. An additional phase I study (NCT02491281) of knee OA patients further confirmed the safety of LNA043 without eliciting any noticeable immune responses. The researchers also showed that the compound was effectively delivered by penetrating the cartilage layers, enhancing the anabolic activities of cartilage. Patients with cartilage lesions and knee OA are currently being recruited for the phase II trial (NCT03275064) of LNA043.
Tankyrase inhibition has been suggested as a potential strategy to simulate regenerative potentials in osteoarthritic cartilage3. Pharmacological inhibition of tankyrase induces chondrogenic differentiation in MSCs and stimulates the expression of cartilage-specific matrisome, collectively ameliorating osteoarthritic cartilage destruction in preclinical models of OA3. Recent accomplishments in fostering the regenerative capacity of adult cartilage suggest the clinical potential of regenerative therapy as an OA treatment.
OA-associated pain
Chronic pain is one of the prominent symptoms of OA, and the clinical management of OA largely aims pain relief. Molecular pathways eliciting chronic pain are regulated in a complex manner via the peripheral and central nervous systems. Although cartilage is an aneural tissue, nociceptors are abundant in other tissues of the joints, such as the joint capsule, synovium, subchondral bone, and ligaments78. Specific receptors on the peripheral terminal, such as heat receptors, chemoreceptors, and mechanoreceptors, detect diverse stimuli, including cytokines, chemokines, neuropeptides, and prostaglandins78,79. These factors form a biochemical milieu that elicit OA-associated pain in the joint. With the progression of peripheral sensitization, joint movement within the normal range becomes painful. Central sensitization also contributes to an abnormal state of responsiveness or increased gain in the nociceptive system80. Collectively, OA patients experience hypersensitivity to noxious stimuli, which is generally characterized by mechanical allodynia or hyperalgesia81,82. There have been recent advances in understanding the cellular and molecular basis of mechanical allodynia and hyperalgesia development in OA-affected joints. The critical role of nerve growth factor (NGF) in damaged joint environments has been linked to pain development in OA patients83,84.
NGF is a member of neurotrophins in the peripheral and central nervous system85. On peripheral nociceptors, the interaction of NGF and its receptor, tropomyosin-related kinase A (TrkA), activates transient receptor potential cation channel subfamily V member 1 (TRPV1) and contributes to pain hypersensitivity associated with tissue damage86,87. NGF expression is elevated in various cell types (e.g., synoviocytes, chondrocytes, osteoclasts, and some immune cells) in the synovium, cartilage, and subchondral bone in patients with knee OA85,88,89. Therefore, NGF has been suggested to be a rational target whose inhibition may effectively manage OA-associated pain in joints78.
Tanezumab is a humanized IgG2 monoclonal NGF antibody that effectively interferes with the binding of NGF to its corresponding receptors90. Phase II clinical trials (NCT00394563) showed that a single intravenous injection of tanezumab substantially reduced pain in patients with knee OA83. A randomized phase III clinical study (NCT02709486) with a 24-week follow-up period demonstrated the significant efficacy of subcutaneously injected tanezumab in controlling OA-associated pain in the hip or knee91. However, safety concerns have been raised recently, along with the report that tanezumab increases the onset of rapidly progressive OA and abnormal peripheral sensation92.
Fasinumab, a human monoclonal NGF antibody93, has been tested in multiple clinical phase trials involving patients with knee or hip OA. A phase IIb/III double‐blind clinical trial (NCT02447276) was conducted with 421 patients with moderate-to-severe knee or hip OA, and 346 patients completed the study94. Patients were randomized to receive 1, 3, 6, or 9 mg fasinumab or placebo which was administered subcutaneously every 4 weeks for 16 weeks with a 36-week follow-up. Fasinumab induced significant reductions in OA-associated pain and improvements in physical function for patients with OA. A phase III clinical trial (NCT02683239) has been conducted to test the long-term safety and efficacy of fasinumab in knee or hip OA patients, but the results have not yet been reported.
Fulranumab, another human monoclonal antibody against NGF95, underwent a phase II clinical trial (NCT01094262) involving 196 patients with moderate-to-severe chronic knee OA. Patients were subcutaneously injected with 3 or 9 mg fulranumab or placebo every 4 weeks for 12 weeks. Fulranumab administration improved the NRS knee pain score compared with that of the active comparator oxycodone96. In another phase II clinical trial (NCT00973141), patients with knee or hip OA were randomized to receive subcutaneous injections of placebo or various doses of fulranumab. Knee and hip pain, as assessed by the WOMAC score, were effectively alleviated by fulranumab administration (3 mg every 4 weeks or 10 mg every 8 weeks) as early as 4 weeks, and the effect was maintained for up to 53 weeks. However, rapidly progressive OA was observed as an adverse effect97. In phase III clinical trials (NCT02336685, NCT02336698, NCT02289716, and NCT02301234), patients with moderate-to-severe OA were randomized to receive subcutaneous injections of placebo or fulranumab (1 or 3 mg every 4 weeks) in the 16-week double-blind phase, followed by a 52-week posttreatment follow-up phase. Fulranumab improved pain management and physical function in patients with OA98.
Emerging approaches for DMOAD development
This section discusses new technologies and modalities emerging from the fundamental understanding of OA pathogenesis. Senescent chondrocytes accumulate in osteoarthritic cartilage and serve as a source of chronic inflammation in joints. Senolytic approaches aim to specifically remove these senescent cells. The versatility of noncoding RNAs (ncRNAs) in regulating a broad range of targets has stimulated the recent focus on RNA therapeutics, and there are now several FDA-approved RNA-based therapeutics in the pharmaceutical market. These therapeutics involve small interfering RNAs (siRNAs), microRNAs (miRNAs), or antisense oligonucleotides (ASOs) and will serve as new modalities of DMOADs, enabling the modulation of previously undruggable targets in joint tissues.
Targeting cellular senescence
Cellular senescence refers to a state in which the cell cycle is irreversibly arrested99,100. In cartilage, oxidative stress associated with aging and mechanical overload mainly cause the accumulation of senescent chondrocytes22. Senescent chondrocytes trigger the formation of an arthritic joint microenvironment through the secretion of pro-inflammatory cytokines and proteases, which are referred to as senescence-associated secretory phenotype (SASP) factors and collectively accelerate osteoarthritic cartilage degeneration and synovial inflammation2,22,100,101. Two possible strategies to modulate the detrimental effects of senescence involve the use of senolytics that selectively eliminate senescent cells100,102,103,104,105,106,107,108 and senomorphics (i.e., senostatics) that abrogate the inflammatory senescent secretome102. UBX0101, developed as the first in-class small molecule sensitizing the senolysis of senescent chondrocytes, has shown positive results in a posttraumatic OA mouse model100. This senolytic drug, which decouples p53 from the MDM2-mediated degradation pathway, was tested in a phase I, double-blind, randomized, placebo-controlled trial involving 48 OA patients (NCT03513016). The clinical outcome showed a reduction in OA-associated pain without notable adverse events when UBX0101 was administered at high doses of 1.0–4.0 mg109. Unfortunately, the recently completed phase II trial (NCT04129944) with 180 patients did not demonstrate sufficient clinical efficacy in terms of joint pain relief.
Navitoclax (ABT-263), the most well-established senolytic drug, inhibits Bcl-2 and Bcl-xL and has been shown to attenuate OA manifestations, including cartilage degeneration and subchondral bone sclerosis, in a posttraumatic OA rat model107. Interestingly, neither UBX0101 nor navitoclax injection exerted any protective effect against age-associated OA in mice, whereas the combination of these two drugs ameliorated OA progression in aged animals110. It appears that senolytic strategies should be refined before they can be used in the clinic. Senomorphics, which modulate the phenotypes of senescent cells without killing them, may serve as alternative options to eliminate SASP factor expression and thereby abrogate the detrimental effects of senescent chondrocytes on OA development.
RNA-based therapeutics
ncRNAs have emerged as regulators of inflammation111,112, chondrocyte apoptosis113, and ECM degradation114,115, which are related to OA-pathogenic mechanisms. To date, more than 50 ncRNAs, including circular RNAs, long noncoding RNAs (lncRNAs), and miRNAs, have been reported to be differentially regulated in OA, affecting the onset and progression of the disease111,116,117. RNA therapeutics have multiple benefits over traditional small-molecule- or antibody-based approaches, including versatility in their design to modulate target gene expression118. RNA therapeutics can be subcategorized into three major groups: siRNAs, miRNAs, and ASOs. These three groups use different mechanisms of action to silence their target genes but share common challenges in their clinical use: mainly in vivo delivery and stability issues119. Although RNAs are widely used to modulate target gene expression in vitro, their low stability and delivery efficiency in vivo limit their use as therapeutic agents120. The recent breakthrough in lipid nanoparticle (LNP) formulations has dramatically improved both the stability and delivery of RNA molecules, resulting in the first FDA-approved siRNA therapeutic in 2018121.
MMP-13 and ADAMTS-5, two critical catabolic enzymes responsible for the degradation of type II collagen and aggrecan, respectively, have been the prime targets of RNA-based therapies. Hoshi et al. examined the effect of chemically modified Mmp13 or Adamts5 siRNA, alone or in combination, in a posttraumatic OA mouse model122. Significant improvements in OA manifestations were observed in all three siRNA-treated groups (Mmp13 siRNA alone, Adamts5 siRNA alone, or combination) compared with the control-siRNA group. Furthermore, the combined treatment group displayed a better therapeutic outcome than the Adamts5–siRNA-only group.
The NF-κB pathway is the most well-known regulatory pathway governing inflammatory responses in OA14,123,124. Intra-articular delivery of a peptide nanoparticle containing an NF-κB siRNA alleviated cartilage degradation and synovitis in a surgically induced OA model125. Hypoxia-inducible factor-2α (HIF-2α) is another key transcription factor that controls the collective expression of matrix-degrading enzymes during OA development126,127. Intra-articular injection of an Epas1-targeting siRNA encapsulated in LNPs and the chondrocyte-affinity peptide DWRVIIPPRPSAC alleviated cartilage degeneration and synovial inflammation in a mouse model of OA128.
Compared with an siRNA, which is generally designed to exclusively knockdown a single target gene, a miRNA regulates the expression of hundreds of target genes simultaneously and has broader impacts on the transcriptome and chondrocyte physiology. There are currently no miRNAs in a clinical trials for OA treatment. Several miRNAs that can potentially delay osteoarthritic processes by modulating matrix degradation and synthesis or autophagy are listed in Table 1. In contrast, ASOs, which are short single-stranded oligodeoxynucleotides, can be used to degrade target RNAs that promote OA129. An ASO has been used to target miR-204, which suppresses the proteoglycan synthesis pathway and augments chronic inflammatory responses in senescent chondrocytes. This miR-204-targeting ASO effectively attenuated OA manifestations and pain development in a preclinical mouse model of OA2.
Challenges and future directions of newly developed drugs
An important aspect of OA treatment is the consideration of diverse clinical syndromes and pathological conditions associated with stages of disease progression130,131,132. There is emerging evidence of the heterogeneity and complexity of OA pathogenesis, which urges modifications to the current “one-fits-all” treatment guidelines. Distinct molecular-level mechanisms are being rapidly elucidated to account for the diversity of OA-associated symptoms and pathogenesis. Therefore, it is urgent to establish guidelines for personalized OA treatments.
There is another vital need for the development of biomarkers that enable the early diagnosis of OA. Many of the developed DMOADs aim to delay degenerative processes in articular cartilage. Evidently, these approaches can be particularly effective in treating patients in the early stage of OA when significant cartilage remains, rather than in the late stage of OA. Therefore, when coupled with early OA diagnosis, DMOADs can fully exert their designated effects, ensuring a superior prognosis in patients with OA.
Conclusions
With advances in the understanding of the basic molecular mechanisms underlying OA pathology, multiple DMOADs have been developed, resulting in several promising outcomes from clinical trials. In this review, we discussed multiple DMOAD options, such as those targeting inflammation, matrix-degrading enzymes and the Wnt pathway to ameliorate the degradation of cartilage matrix. Several regenerative DMOADs have shown promise in promoting the chondrogenic differentiation of stem cells and the reconstruction of cartilage matrix. Senolytic/senomorphic strategies and RNA therapeutics have been suggested to be new modalities of DMOADs, enabling the modulation of previously undruggable targets in joint tissues. DMOADs have certainly reached the point of clinical application. Their ultimate approval and availability on the pharmaceutical market are coming and will aid in the treatment of one of the most devastating joint diseases.
References
Kim, J. H. et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell 156, 730–743 (2014).
Kang, D. et al. Stress-activated miR-204 governs senescent phenotypes of chondrocytes to promote osteoarthritis development. Sci. Transl. Med. 11, eaar6659 (2019).
Kim, S. et al. Tankyrase inhibition preserves osteoarthritic cartilage by coordinating cartilage matrix anabolism via effects on SOX9 PARylation. Nat. Commun. 10, 4898 (2019).
Mobasheri, A. et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 13, 302–311 (2017).
Goldring, S. R. & Goldring, M. B. Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage-bone crosstalk. Nat. Rev. Rheumatol. 12, 632–644 (2016).
Crofford, L. J. Use of NSAIDs in treating patients with arthritis. Arthritis Res. Ther. 15, S2 (2013).
Frallonardo, P. et al. Basic calcium phosphate and pyrophosphate crystals in early and late osteoarthritis: relationship with clinical indices and inflammation. Clin. Rheumatol. 37, 2847–2853 (2018).
Kim, H., Kang, D., Cho, Y. & Kim, J. H. Epigenetic regulation of chondrocyte catabolism and anabolism in osteoarthritis. Mol. Cells 38, 677–684 (2015).
Chow, Y. Y. & Chin, K. Y. The role of inflammation in the pathogenesis of osteoarthritis. Mediators Inflamm. 2020, 8293921 (2020).
Martel-Pelletier, J. et al. Osteoarthritis. Nat. Rev. Dis. Prim. 2, 16072 (2016).
El Mansouri, F. E. et al. Contribution of H3K4 methylation by SET-1A to interleukin-1-induced cyclooxygenase 2 and inducible nitric oxide synthase expression in human osteoarthritis chondrocytes. Arthritis Rheumatol. 63, 168–179 (2011).
Hardy, M. M. et al. Cyclooxygenase 2-dependent prostaglandin E2 modulates cartilage proteoglycan degradation in human osteoarthritis explants. Arthritis Rheumatol. 46, 1789–1803 (2002).
Jones, S. W. et al. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) modulates key biological pathways associated with OA disease pathology. Osteoarthr. Cartil. 17, 124–131 (2009).
Goldring, M. B. & Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 23, 471–478 (2011).
Chevalier, X. et al. Intraarticular injection of anakinra in osteoarthritis of the knee: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheumatol. 61, 344–352 (2009).
Cohen, S. B. et al. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 13, R125 (2011).
Kloppenburg, M. et al. Phase IIa, placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1α and anti-interleukin-1β dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann. Rheum. Dis. 78, 413–420 (2019).
Fleischmann, R. M. et al. A phase II trial of lutikizumab, an anti-interleukin-1α/β dual variable domain immunoglobulin, in knee osteoarthritis patients with synovitis. Arthritis Rheumatol. 71, 1056–1069 (2019).
Aitken, D. et al. A randomised double-blind placebo-controlled crossover trial of HUMira (adalimumab) for erosive hand OsteoaRthritis—the HUMOR trial. Osteoarthr. Cartil. 26, 880–887 (2018).
Kloppenburg, M. et al. Etanercept in patients with inflammatory hand osteoarthritis (EHOA): a multicentre, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 77, 1757–1764 (2018).
Heinegard, D. & Saxne, T. The role of the cartilage matrix in osteoarthritis. Nat. Rev. Rheumatol. 7, 50–56 (2011).
Loeser, R. F., Collins, J. A. & Diekman, B. O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 12, 412–420 (2016).
Pap, T. & Korb-Pap, A. Cartilage damage in osteoarthritis and rheumatoid arthritis-two unequal siblings. Nat. Rev. Rheumatol. 11, 606–615 (2015).
Wang, M. et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 15, R5 (2013).
Kamekura, S. et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthr. Cartil. 13, 632–641 (2005).
Little, C. B. et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheumatol. 60, 3723–3733 (2009).
Sabatini, M. et al. Effect of inhibition of matrix metalloproteinases on cartilage loss in vitro and in a guinea pig model of osteoarthritis. Arthritis Rheumatol. 52, 171–180 (2005).
Johnson, A. R. et al. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J. Biol. Chem. 282, 27781–27791 (2007).
Krzeski, P. et al. Development of musculoskeletal toxicity without clear benefit after administration of PG-116800, a matrix metalloproteinase inhibitor, to patients with knee osteoarthritis: a randomized, 12-month, double-blind, placebo-controlled study. Arthritis Res. Ther. 9, R109 (2007).
Holmbeck, K. et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 99, 81–92 (1999).
Baragi, V. M. et al. A new class of potent matrix metalloproteinase 13 inhibitors for potential treatment of osteoarthritis: Evidence of histologic and clinical efficacy without musculoskeletal toxicity in rat models. Arthritis Rheumatol. 60, 2008–2018 (2009).
Glasson, S. S. et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434, 644–648 (2005).
Stanton, H. et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 434, 648–652 (2005).
Clement-Lacroix, P. et al. Glpg1972: a potent, selective, orally available adamts-5 inhibitor for the treatment of OA. Osteoarthr. Cartil. 25, S58–S59 (2017).
Deckx, H. M. et al. A safety, tolerability, pharmacokinetics (Pk) and pharmacodynamics (Pd) study with increasing oral doses of Glpg1972 administered daily for 29 days shows a strong biomarker effect in patients with knee and/or hip OA. Ann. Rheum. Dis. 77, 795–796 (2018).
van der Aar, E. M. et al. Adamts-5 inhibitor Glpg1972, a potential new treatment in osteoarthritis, shows favorable safety, pharmacokinetics and pharmacodynamics in healthy subjects. Osteoarthr. Cartil. 26, S310–S310 (2018).
Marques, S. IntradoGlobeNewswire https://www.globenewswire.com/news-release/2020/10/15/2109556/0/en/Galapagos-and-Servier-report-topline-results-for-ROCCELLA-Phase-2-clinical-trial-with-GLPG1972-S201086-in-knee-osteoarthritis-patients.html (2020).
Santamaria, S. ADAMTS-5: a difficult teenager turning 20. Int. J. Exp. Pathol. 101, 4–20 (2020).
Siebuhr, A. S. et al. The anti-ADAMTS-5 nanobody((R)) M6495 protects cartilage degradation ex vivo. Int. J. Mol. Sci. 21, 5992 (2020).
Guehring, H. et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of single ascending doses of the anti-ADAMTS-5 nanobody (R), M6495, in healthy male subjects: a phase I, placebo-controlled, first-in-human study. Arthritis Rheumatol. 71, 2175 (2019).
Joyce, J. A. et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 5, 443–453 (2004).
Turk, V., Turk, B., Guncar, G., Turk, D. & Kos, J. Lysosomal cathepsins: structure, role in antigen processing and presentation, and cancer. Adv. Enzym. Regul. 42, 285–303 (2002).
Bogyo, M., Verhelst, S., Bellingard-Dubouchaud, V., Toba, S. & Greenbaum, D. Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chem. Biol. 7, 27–38 (2000).
Patel, S., Homaei, A., El-Seedi, H. R. & Akhtar, N. Cathepsins: proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 105, 526–532 (2018).
Aguda, A. H. et al. Structural basis of collagen fiber degradation by cathepsin K. Proc. Natl Acad. Sci. USA 111, 17474–17479 (2014).
Kozawa, E. et al. Increased expression and activation of cathepsin K in human osteoarthritic cartilage and synovial tissues. J. Orthop. Res. 34, 127–134 (2016).
Ben-Aderet, L. et al. Detecting cathepsin activity in human osteoarthritis via activity-based probes. Arthritis Res. Ther. 17, 69 (2015).
Connor, J. R. et al. Protective effects of a cathepsin K inhibitor, SB-553484, in the canine partial medial meniscectomy model of osteoarthritis. Osteoarthr. Cartil. 17, 1236–1243 (2009).
Hayami, T., Zhuo, Y., Wesolowski, G. A., Pickarski, M. & Duong, L. T. Inhibition of cathepsin K reduces cartilage degeneration in the anterior cruciate ligament transection rabbit and murine models of osteoarthritis. Bone 50, 1250–1259 (2012).
Nwosu, L. N. et al. Analgesic effects of the cathepsin K inhibitor L-006235 in the monosodium iodoacetate model of osteoarthritis pain. Pain Rep. 3, e685 (2018).
McDougall, J. J., Schuelert, N. & Bowyer, J. Cathepsin K inhibition reduces CTXII levels and joint pain in the guinea pig model of spontaneous osteoarthritis. Osteoarthr. Cartil. 18, 1355–1357 (2010).
Lindstrom, E. et al. Nonclinical and clinical pharmacological characterization of the potent and selective cathepsin K inhibitor MIV-711. J. Transl. Med. 16, 125 (2018).
Conaghan, P. G. et al. Six months’ treatment with Miv-711, a novel cathepsin K inhibitor induces osteoarthritis structure modification: results from a randomized double-blind placebo-controlled phase IIA trial. Osteoarthr. Cartil. 26, S25–S26 (2018).
Conaghan, P. G. et al. Safety and efficacy of six months’ open label extension post-RCT using the novel cathepsin K inhibitor MIV-711 in patients with knee osteoarthritis. Osteoarthr. Cartil. 27, S501–S502 (2019).
Conaghan, P. G. et al. Disease-modifying effects of a novel cathepsin K inhibitor in osteoarthritis: a randomized controlled trial. Ann. Intern. Med. 172, 86–95 (2020).
Nusse, R. & Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Lories, R. J., Corr, M. & Lane, N. E. To Wnt or not to Wnt: the bone and joint health dilemma. Nat. Rev. Rheumatol. 9, 328–339 (2013).
Lambert, C. et al. Gene expression pattern of cells from inflamed and normal areas of osteoarthritis synovial membrane. Arthritis Rheumatol. 66, 960–968 (2014).
Nakamura, Y., Nawata, M. & Wakitani, S. Expression profiles and functional analyses of Wnt-related genes in human joint disorders. Am. J. Pathol. 167, 97–105 (2005).
Nguyen, D. X. et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 138, 51–62 (2009).
Anastas, J. N. & Moon, R. T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 13, 11–26 (2013).
Hartmann, C. & Tabin, C. J. Wnt-14 plays a pivotal role in inducing synovial joint formation in the developing appendicular skeleton. Cell 104, 341–351 (2001).
Tamamura, Y. et al. Developmental regulation of Wnt/β-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J. Biol. Chem. 280, 19185–19195 (2005).
Day, T. F., Guo, X., Garrett-Beal, L. & Yang, Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750 (2005).
Kim, S. J. et al. β-Catenin regulates expression of cyclooxygenase-2 in articular chondrocytes. Biochem. Biophys. Res. Commun. 296, 221–226 (2002).
Corr, M. Wnt-β-catenin signaling in the pathogenesis of osteoarthritis. Nat. Clin. Pract. Rheumatol. 4, 550–556 (2008).
Lu, B., Green, B. A., Farr, J. M., Lopes, F. C. & Van Raay, T. J. Wnt drug discovery: weaving through the screens, patents and clinical trials. Cancers (Basel) 8, 82 (2016).
Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 (2014).
Deshmukh, V. et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 26, 18–27 (2018).
Deshmukh, V. et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr. Cartil. 27, 1347–1360 (2019).
Yazici, Y. et al. A novel Wnt pathway inhibitor, SM04690, for the treatment of moderate to severe osteoarthritis of the knee: results of a 24-week, randomized, controlled, phase 1 study. Osteoarthr. Cartil. 25, 1598–1606 (2017).
Yazici, Y. et al. Lorecivivint, a novel intraarticular CDC-like kinase 2 and dual-specificity tyrosine phosphorylation-regulated kinase 1A inhibitor and Wnt pathway modulator for the treatment of knee osteoarthritis: a phase II randomized trial. Arthritis Rheumatol. 72, 1694–1706 (2020).
Yazici, Y. et al. Efficacy and safety from a phase 2b trial of Sm04690, a novel, intra-articular, Wnt pathway inhibitor for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 27, S503–S503 (2019).
Johnson, K. et al. A stem cell-based approach to cartilage repair. Science 336, 717–721 (2012).
Johnson, K. A. et al. Development of Ka34 as a cartilage regenerative therapy for osteoarthritis. Osteoarthr. Cartil. 28, S518–S518 (2020).
Scotti, C. et al. LNA043, a novel cartilage regenerative treatment for osteoarthritis: results from a first-in-human trial in patients with knee osteoarthritis. Arthritis Rheumatol. 72, 1485 (2020).
Jiang, S. et al. ANGPTL3: a novel biomarker and promising therapeutic target. J. Drug Target 27, 876–884 (2019).
Malfait, A. M. & Schnitzer, T. J. Towards a mechanism-based approach to pain management in osteoarthritis. Nat. Rev. Rheumatol. 9, 654–664 (2013).
Miller, R. J., Jung, H., Bhangoo, S. K. & White, F. A. Cytokine and chemokine regulation of sensory neuron function. Handb. Exp. Pharm. 194, 417–449 (2009).
Latremoliere, A. & Woolf, C. J. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain. 10, 895–926 (2009).
Fu, K., Robbins, S. R. & McDougall, J. J. Osteoarthritis: the genesis of pain. Rheumatology (Oxf.) 57, iv43–iv50 (2018).
Arendt-Nielsen, L. et al. Sensitization in patients with painful knee osteoarthritis. Pain 149, 573–581 (2010).
Lane, N. E. et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N. Engl. J. Med. 363, 1521–1531 (2010).
Lolignier, S., Eijkelkamp, N. & Wood, J. N. Mechanical allodynia. Pflug. Arch. 467, 133–139 (2015).
Wise, B. L., Seidel, M. F. & Lane, N. E. The evolution of nerve growth factor inhibition in clinical medicine. Nat. Rev. Rheumatol. 17, 34–46 (2021).
Caterina, M. J., Rosen, T. A., Tominaga, M., Brake, A. J. & Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398, 436–441 (1999).
Huang, J., Zhang, X. & McNaughton, P. A. Inflammatory pain: the cellular basis of heat hyperalgesia. Curr. Neuropharmacol. 4, 197–206 (2006).
Stoppiello, L. A. et al. Structural associations of symptomatic knee osteoarthritis. Arthritis Rheumatol. 66, 3018–3027 (2014).
Walsh, D. A. et al. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology (Oxf.) 49, 1852–1861 (2010).
Abdiche, Y. N., Malashock, D. S. & Pons, J. Probing the binding mechanism and affinity of tanezumab, a recombinant humanized anti-NGF monoclonal antibody, using a repertoire of biosensors. Protein Sci. 17, 1326–1335 (2008).
Berenbaum, F. et al. Subcutaneous tanezumab for osteoarthritis of the hip or knee: efficacy and safety results from a 24-week randomised phase III study with a 24-week follow-up period. Ann. Rheum. Dis. 79, 800–810 (2020).
Yu, Y., Lu, S. T., Sun, J. P. & Zhou, W. Safety of low-dose tanezumab in the treatment of hip or knee osteoarthritis: a systemic review and meta-analysis of randomized phase III clinical trials. Pain. Med. 22, 585–595 (2021).
Tiseo, P. J., Kivitz, A. J., Ervin, J. E., Ren, H. & Mellis, S. J. Fasinumab (REGN475), an antibody against nerve growth factor for the treatment of pain: results from a double-blind, placebo-controlled exploratory study in osteoarthritis of the knee. Pain 155, 1245–1252 (2014).
Dakin, P. et al. The efficacy, tolerability, and joint safety of fasinumab in osteoarthritis pain: a phase IIb/III double-blind, placebo-controlled, randomized clinical trial. Arthritis Rheumatol. 71, 1824–1834 (2019).
Sanga, P. et al. Efficacy, safety, and tolerability of fulranumab, an anti-nerve growth factor antibody, in the treatment of patients with moderate to severe osteoarthritis pain. Pain 154, 1910–1919 (2013).
Mayorga, A. J., Wang, S., Kelly, K. M. & Thipphawong, J. Efficacy and safety of fulranumab as monotherapy in patients with moderate to severe, chronic knee pain of primary osteoarthritis: a randomised, placebo- and active-controlled trial. Int. J. Clin. Pract. 70, 493–505 (2016).
Sanga, P. et al. Long-term safety and efficacy of fulranumab in patients with moderate-to-severe osteoarthritis pain: a phase II randomized, double-blind, placebo-controlled extension study. Arthritis Rheumatol. 69, 763–773 (2017).
Kelly, K. M. et al. Safety and efficacy of fulranumab in osteoarthritis of the hip and knee: results from four early terminated phase III randomized studies. Curr. Med. Res. Opin. 35, 2117–2127 (2019).
Kang, C. et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612 (2015).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Zhang, M. et al. Induced superficial chondrocyte death reduces catabolic cartilage damage in murine posttraumatic osteoarthritis. J. Clin. Invest 126, 2893–2902 (2016).
Kang, C. Senolytics and senostatics: a two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol. Cells 42, 821–827 (2019).
Cao, X. et al. Intraarticular senescent chondrocytes impair the cartilage regeneration capacity of mesenchymal stem cells. Stem Cell Res. Ther. 10, 86 (2019).
Fuhrmann-Stroissnigg, H. et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 8, 422 (2017).
Dai, H. et al. Eliminating senescent chondrogenic progenitor cells enhances chondrogenesis under intermittent hydrostatic pressure for the treatment of OA. Stem Cell Res. Ther. 11, 199 (2020).
Siebelt, M. et al. Hsp90 inhibition protects against biomechanically induced osteoarthritis in rats. Arthritis Rheumatol. 65, 2102–2112 (2013).
Yang, H. et al. Navitoclax (ABT263) reduces inflammation and promotes chondrogenic phenotype by clearing senescent osteoarthritic chondrocytes in osteoarthritis. Aging (Albany NY) 12, 12750–12770 (2020).
Wu, D. & Prives, C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 25, 169–179 (2018).
Hsu, B. et al. Safety, tolerability, pharmacokinetics, and clinical outcomes following single-dose IA administration of UBX0101, a senolytic MDM2/p53 interaction inhibitor, in patients with knee OA. Arthritis Rheumatol. 71, L05 (2019).
Faust, H. J. et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J. Clin. Invest 130, 5493–5507 (2020).
Miyaki, S. & Asahara, H. Macro view of microRNA function in osteoarthritis. Nat. Rev. Rheumatol. 8, 543–552 (2012).
Santini, P., Politi, L., Vedova, P. D., Scandurra, R. & Scotto d’Abusco, A. The inflammatory circuitry of miR-149 as a pathological mechanism in osteoarthritis. Rheumatol. Int. 34, 711–716 (2014).
Yan, S. et al. MicroRNA-34a affects chondrocyte apoptosis and proliferation by targeting the SIRT1/p53 signaling pathway during the pathogenesis of osteoarthritis. Int. J. Mol. Med. 38, 201–209 (2016).
Akhtar, N. et al. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheumatol. 62, 1361–1371 (2010).
Miyaki, S. et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 24, 1173–1185 (2010).
Jiang, S., Liu, Y., Xu, B., Zhang, Y. & Yang, M. Noncoding RNAs: new regulatory code in chondrocyte apoptosis and autophagy. Wiley Interdiscip. Rev. RNA 11, e1584 (2020).
Wu, Y., Lu, X., Shen, B. & Zeng, Y. The therapeutic potential and role of miRNA, lncRNA, and circRNA in osteoarthritis. Curr. Gene Ther. 19, 255–263 (2019).
Lieberman, J. Tapping the RNA world for therapeutics. Nat. Struct. Mol. Biol. 25, 357–364 (2018).
Zhou, L. B., Rubin, L. E., Liu, C. J. & Chen, Y. P. Short interfering RNA (siRNA)-based therapeutics for cartilage diseases. Regen. Eng. Transl. Med 7, 283–290 (2020).
Kole, R., Krainer, A. R. & Altman, S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 11, 125–140 (2012).
Setten, R. L., Rossi, J. J. & Han, S. P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 18, 421–446 (2019).
Hoshi, H. et al. Effect of inhibiting MMP13 and ADAMTS5 by intra-articular injection of small interfering RNA in a surgically induced osteoarthritis model of mice. Cell Tissue Res. 368, 379–387 (2017).
Marcu, K. B., Otero, M., Olivotto, E., Borzi, R. M. & Goldring, M. B. NF-κB signaling: multiple angles to target OA. Curr. Drug Targets 11, 599–613 (2010).
Rigoglou, S. & Papavassiliou, A. G. The NF-κB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 45, 2580–2584 (2013).
Yan, H. et al. Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc. Natl Acad. Sci. USA 113, E6199–E6208 (2016).
Yang, S. et al. Hypoxia-inducible factor-2 α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 16, 687–693 (2010).
Saito, T. et al. Transcriptional regulation of endochondral ossification by HIF-2α during skeletal growth and osteoarthritis development. Nat. Med. 16, 678–686 (2010).
Pi, Y. et al. Intra-articular delivery of anti-Hif-2α siRNA by chondrocyte-homing nanoparticles to prevent cartilage degeneration in arthritic mice. Gene Ther. 22, 439–448 (2015).
Rinaldi, C. & Wood, M. J. A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21 (2018).
Bierma-Zeinstra, S. M. & Verhagen, A. P. Osteoarthritis subpopulations and implications for clinical trial design. Arthritis Res. Ther. 13, 213 (2011).
Felson, D. T. Identifying different osteoarthritis phenotypes through epidemiology. Osteoarthr. Cartil. 18, 601–604 (2010).
Deveza, L. A., Nelson, A. E. & Loeser, R. F. Phenotypes of osteoarthritis: current state and future implications. Clin. Exp. Rheumatol. 37, 64–72 (2019).
Zhou, X. et al. MiR-132-3p regulates ADAMTS-5 expression and promotes chondrogenic differentiation of rat mesenchymal stem cells. J. Cell Biochem. 119, 2579–2587 (2018).
Qian, J. et al. miR-107 affects cartilage matrix degradation in the pathogenesis of knee osteoarthritis by regulating caspase-1. J. Orthop. Surg. Res. 16, 40 (2021).
Ren, T. et al. MiR-140-3p ameliorates the progression of osteoarthritis via targeting CXCR4. Biol. Pharm. Bull. 43, 810–816 (2020).
Wang, Z. et al. miR-140-5p/miR-149 affects chondrocyte proliferation, apoptosis, and autophagy by targeting FUT1 in osteoarthritis. Inflammation 41, 959–971 (2018).
Xue, H. et al. miR-93-5p attenuates IL-1β-induced chondrocyte apoptosis and cartilage degradation in osteoarthritis partially by targeting TCF4. Bone 123, 129–136 (2019).
Zhong, G. et al. miRNA-335-5p relieves chondrocyte inflammation by activating autophagy in osteoarthritis. Life Sci. 226, 164–172 (2019).
Ji, Q. et al. Cryptotanshinone protects cartilage against developing osteoarthritis through the miR-106a-5p/GLIS3 axis. Mol. Ther. Nucleic Acids 11, 170–179 (2018).
Gu, R. et al. MicroRNA-9 regulates the development of knee osteoarthritis through the NF-κB1 pathway in chondrocytes. Medicine (Baltim.) 95, e4315 (2016).
Zhang, G. et al. MiR-502-5p inhibits IL-1β-induced chondrocyte injury by targeting TRAF2. Cell Immunol. 302, 50–57 (2016).
Hu, G. et al. MicroRNA-145 attenuates TNF-α-driven cartilage matrix degradation in osteoarthritis via direct suppression of MKK4. Cell Death Dis. 8, e3140 (2017).
Hu, J. et al. MiR-26a and miR-26b mediate osteoarthritis progression by targeting FUT4 via NF-κB signaling pathway. Int. J. Biochem. Cell Biol. 94, 79–88 (2018).
Wang, G. et al. MicroRNA-411 inhibited matrix metalloproteinase 13 expression in human chondrocytes. Am. J. Transl. Res. 7, 2000–2006 (2015).
Yang, F., Huang, R., Ma, H., Zhao, X. & Wang, G. miRNA-411 regulates chondrocyte autophagy in osteoarthritis by targeting hypoxia-inducible factor 1 α (HIF-1α). Med. Sci. Monit. 26, e921155 (2020).
Liu, W., Zha, Z. & Wang, H. Upregulation of microRNA-27a inhibits synovial angiogenesis and chondrocyte apoptosis in knee osteoarthritis rats through the inhibition of PLK2. J. Cell Physiol. 234, 22972–22984 (2019).
Cai, C. et al. MiR-27a promotes the autophagy and apoptosis of IL-1β treated-articular chondrocytes in osteoarthritis through PI3K/AKT/mTOR signaling. Aging (Albany NY) 11, 6371–6384 (2019).
Funding
This work was supported by grants from the National Research Foundation of Korea (NRF-2015M3A9E6028674, NRF-2020R1A2C2012300, NRF-2016R1A5A1010764, NRF-2017M3A9D8064193, NRF-2021R1I1A1A01059252), the Institute for Basic Science from the Ministry of Science, ICT and Future Planning of Korea (IBS-R008-D1), and Suh Kyungbae foundation.
Author information
Authors and Affiliations
Contributions
Y.C. and S.J. researched data for the article. Y.C., S.J., S.-B.K., and J.-H.K. wrote the article. All authors provided substantial contribution to discussion of content and reviewed the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cho, Y., Jeong, S., Kim, H. et al. Disease-modifying therapeutic strategies in osteoarthritis: current status and future directions. Exp Mol Med 53, 1689–1696 (2021). https://doi.org/10.1038/s12276-021-00710-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00710-y
This article is cited by
-
Gene therapy for polygenic or complex diseases
Biomarker Research (2024)
-
Mechanisms of treatment effects using allogeneic, umbilical cord-derived mesenchymal stromal stem cells (MSCs) in knee osteoarthritis: a pharmacological clinical study protocol
Trials (2024)
-
Inhibition of the MALT1-LPCAT3 axis protects cartilage degeneration and osteoarthritis
Cell Communication and Signaling (2024)
-
Trachelogenin alleviates osteoarthritis by inhibiting osteoclastogenesis and enhancing chondrocyte survival
Chinese Medicine (2024)
-
Chondroitin sulfate-modified tragacanth gum–gelatin composite nanocapsules loaded with curcumin nanocrystals for the treatment of arthritis
Journal of Nanobiotechnology (2024)
