
Abstract
N6-methyladenosine (m6A) is the most prevalent modification of mammalian cellular RNAs. m6A methylation is linked to epigenetic regulation of several aspects of gene expression, including RNA stability, splicing, nuclear export, RNA folding, and translational activity. m6A modification is reversibly catalyzed by methyltransferases (m6A writers) and demethylases (m6A erasers), and the dynamics of m6A-modified RNA are regulated by m6A-binding proteins (m6A readers). Recently, several studies have shown that m6A methylation sites have been identified in hepatitis B virus (HBV) transcripts and the hepatitis C virus (HCV) RNA genome. Here, we review the role of m6A modification in HBV/HCV replication and its contribution to liver disease pathogenesis. A better understanding of the functions of m6A methylation in the life cycles of HBV and HCV is required to establish the role of these modifications in liver diseases associated with these viral infections.
Similar content being viewed by others


Introduction
Eukaryotic cellular RNAs contain diverse chemical modifications, including N6-methyladenosine (m6A), 5-methylcytidine (m5C), uridine to pseudouridine (U to Ψ), adenosine to inosine (A to I), and addition to N7-methylguanosine (m7G)1. Among the diverse RNA modifications, m6A methylation, methylation of the adenosine base at the nitrogen 6 position, is the most well-characterized and the most common modification of cellular RNAs2,3. This modification has been linked to various biological processes, including innate immune responses, sex determination, stem cell differentiation, circadian clock regulation, meiosis, stress responses, and cancer development3. m6A methylation was first identified in the 1970s but the technology to map individual-specific m6A sites in a given RNA became available only recently4. The development of highly sensitive detection methods with high-throughput sequencing revealed the topology of m6A in the cellular transcriptome2,5. Over 25% of mammalian transcripts contain m6A modifications and m6A modification occurs within the consensus DRACH/RRACH motif (D = A, G, or U; R = G or A; H = A, C, or U)2. Furthermore, this modification is typically enriched near the stop codon and the 3′-untranslated region (UTR)2. Similar to DNA methylation, m6A methylation is reversibly catalyzed by various methyltransferases and demethylases (Fig. 1). The cellular m6A methyltransferase machinery is composed of methyltransferase-like 3 (METTL3), METTL14, and WT1-associated protein (WTAP)6,7. Other additional subunits, such as Vir like m6A methyltransferase associated (VIRMA), zinc finger CCCH-type containing 13 (ZC3H13), and RNA-binding motif protein 15/15B (RBM15/15B), are also components of the m6A machinery8,9,10,11. WTAP regulates the recruitment of the optimal substrate and nuclear localization of METTL3/148. RBM15/15B interacts with the U-rich RNA regions, ZC3H13 is required for nuclear import of the METTL3/14 complex, and VIRMA is necessary for writing m6A in the 3′-UTR9,10,11. Fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5) are m6A demethylases that remove m6A from cellular RNA (Fig. 1)12,13. The dynamics of m6A modified RNAs are regulated by m6A readers, YT521-B homology (YTH) domain family proteins (YTHDF1/2/3 and YTHDC1/2)14. YTHDF3 first recognizes m6A-modified RNA and recruits the YTHDF1 or 2 protein15. The YTHDF1-YTHDF3 complex induces the translation of m6A-methylated mRNA, while the YTHDF2-YTHDF3 complex causes the degradation of its target mRNA degradation16,17. Thus, YTHDF3 regulates mRNA degradation and translation by cooperating with YTHDF1 or 2. Because YTHDF2 has no RNase activity, it interacts with the CCR4-NOT (C-C motif chemokine receptor 4 negative on TATA-less) deadenylase complex to promote the degradation of its target RNA17. YTHDC1 regulates mRNA nuclear export in cooperation with the major export receptor NXF1, as well as RNA splicing18,19. YTHDC2 is the only m6A reader protein containing an RNA helicase domain and induces the translation of m6A-modified mRNA by interacting with a small ribosomal subunit20,21. The helicase activity of YTHDC2 is essential for YTHDC2-mediated mRNA translation, implying that YTHDC2 helps to resolve mRNA secondary structure22. Thus, m6A-methylated RNAs are epigenetically regulated by diverse m6A-binding proteins. However, the mechanism by which the m6A site recruits specific m6A binding proteins remains to be elucidated.

m6A modification occurs in consensus DRACH motifs of cellular and viral RNAs. This modification is reversibly catalyzed by an m6A ‘writer’ or ‘eraser’. The m6A ‘writer’ (methyltransferase) complex is composed of METTL3, METTL14, and WTAP, and FTO or ALKBH5 is m6A ‘eraser’ (demethylase). The dynamics of m6A-modified RNAs are regulated by the m6A ‘reader’ proteins, including YTHDF1/2/3, YTHDC1/2, and IGF2BP1/2/3.
Several recent reports highlighted the role of m6A in the genomes of RNA viruses as well as in the transcripts of DNA viruses23,24,25,26,27,28,29,30,31,32. m6A modification can affect viral life cycles in a complex way. Viral RNAs can be m6A methylated; therefore, m6A can play an antiviral or proviral role in the viral life cycle through the recruitment of different m6A-binding proteins. In addition, m6A can indirectly affect viral replication by regulating the expression of specific genes involved in the viral life cycle. A better understanding of the biological functions of m6A modification in viruses is important to establish their role in viral pathogenesis and to design innovative prevention measures to affect viral infection. In this review, we will summarize the emerging roles of m6A modifications in HBV and HCV infections and discuss their functions and associated mechanisms related to the biological processes of viral infection.
The role of m6A during hepatitis B and C virus infections
The role of m6A in the HBV life cycle
HBV infection leads to chronic hepatitis and carries a risk for the development of hepatocellular carcinoma (HCC)33,34. HBV belongs to the Hepadnaviridae family and contains a partially double-stranded DNA genome. Although HBV is a DNA virus, it replicates by reverse transcription of an RNA intermediate termed pregenome RNA (pgRNA) to ultimately produce viral genomic DNA in a covalently closed circular conformation termed cccDNA35. Initially, pgRNA is reverse transcribed to relaxed circular DNA (rcDNA) in the cytoplasmic core particles, and rcDNA is subsequently converted to cccDNA in the nucleus, where it functions as a template for transcription34. Transcription from cccDNA is achieved through the cellular polymerase II machinery to synthesize viral RNAs. Synthesis of HBV transcripts is initiated from different transcription start sites in the HBV genome, but it terminates at a common transcription termination signal34. Hence, HBV transcripts have different 5′ termini but share a common 3′ terminal sequence. These HBV transcripts encode the following proteins: surface (HBs), precore or ‘e’ (HBe), and core (HBc) antigen, polymerase, and X (HBx) proteins.
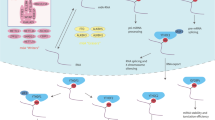
We first reported that HBV transcripts were m6A methylated at an m6A consensus motif (A1907) located within the epsilon stem-loop region present in all HBV RNAs23. pgRNA of HBV acquires this m6A motif at two locations-at the 5′ and 3′ termini due to terminal redundancy, but this motif is presented only once in the 3′ termini of the other subgenomic viral transcripts34. Importantly, m6A modification of HBV RNAs differentially regulates the viral life cycle depending on its position in the viral RNAs (Fig. 2)23. m6A modification at the 3′ epsilon stem-loop of HBV RNA transcripts reduces their RNA stability, leading to decreased viral protein expression23. The reduction in viral RNA stability resulting from m6A is mediated by the recognition of the m6A site at the 3′ epsilon stem-loop by YTHDF2 and 3 (m6A binding proteins). On the other hand, the m6A site located in the 5′ epsilon stem-loop of pgRNA positively regulates reverse transcriptase activity, but the exact mechanism remains to be characterized. Therefore, these results reveal that m6A modification in the epsilon stem-loop structure of HBV regulates effects on HBV RNA stability and reverse transcription.

HBV transcripts are cotranscriptionally m6A-methylated at a consensus DRACH motif in the epsilon stem-loop region. HBV pgRNA contains two such motifs at the 5′ and 3′ termini owing to terminal redundancy, but other viral transcripts contain only one such motif, in the 3′ terminal sequence. m6A methylation of the 5′ terminus occurs in the area surrounding the priming site for reverse transcription initiation and induces reverse transcription of HBV DNA from pgRNA, whereas m6A at the 3′ terminus in all viral transcripts reduces RNA stability by interacting with YTHDF2.
We have recently discovered that HBV utilizes a specific mechanism to guide m6A modification on viral RNAs36. During HBV infection, HBx interacts with m6A methyltransferases, which in turn stimulates their nuclear import and thereby delivers the m6A methyltransferases to HBV cccDNA to achieve cotranscriptional m6A modification of HBV RNAs. On the other hand, infection with HBx-defective HBV fails to produce m6A-modified viral transcripts36. In this role, HBx regulates the HBV life cycle by modulating m6A modification of viral RNAs. These findings highlight the unique role of HBx in the cotranscriptional RNA modification at the sites of transcription initiation, in addition to its transactivating function affecting the Smc5/6 complex and HBx-DDB-mediated degradation activity37,38,39.
In addition, m6A modification plays an important role in interferon (IFN)-mediated inhibition of HBV replication40. IFN treatment of HBV-infected cells promotes the reduction of viral replication through the degradation of viral RNAs by the exonuclease activity of the IFN-stimulated gene 20 (ISG20)41. ISG20 induced by IFN treatment is recognized by YTHDF2, and YTHDF2 then deliveries ISG20 to the m6A-methylated HBV RNAs for their degradation40. Mutation of the m6A site of HBV RNA abrogates ISG20-mediated viral RNA degradation. This study shows a new function of m6A reader proteins in IFN-mediated HBV RNA degradation.
The role of m6A in the HCV life cycle
Hepatitis C virus (HCV) belongs to the Flaviviridae family42. HCV is a positive-sense single-stranded RNA virus and encodes a polyprotein of ~3010 amino acids that is cleaved by cellular and viral proteases into structural and nonstructural proteins. The viral polymerase has RNA-dependent RNA polymerase activity to replicate viral RNA from a template RNA.
Horner and colleagues reported that the HCV RNA genome is m6A methylated at approximately 19 regions and that all YTHDF proteins broadly interact with the HCV genome24. Interestingly, m6A modification in the HCV genome decreased extracellular viral RNA levels and viral particle production without affecting viral replication or protein translation. YTHDF1-3 proteins recognized the m6A-methylated HCV genome and relocalized HCV RNAs to the lipid droplet fraction to inhibit HCV RNA packaging into virions (Fig. 3)24. To elucidate the functional relevance of a specific m6A site in the HCV genome, Gokhale et al. mutated m6A sites within the HCV E1 coding region. Mutations of these m6A sites in the HCV E1 gene increased HCV virion production by abolishing HCV E1 recognition by YTHDF1-3 proteins. These results suggest that m6A modifications of the HCV E1 gene regulate viral RNA packaging into virions via interactions with YTHDF1-3 proteins24.

The HCV genome is m6A-methylated in several regions (~19 regions), including the HCV E1 region. m6A methylations in the HCV E1 region decrease extracellular viral RNA and virion production via recognition by YTHDF proteins. YTHDF proteins sequester the m6A-methylated HCV genome to inhibit interaction with the HCV core protein in the lipid droplets.
Gokhale et al. further analyzed m6A motifs in the RNA genomes of other members of the Flaviviridae family, including dengue, yellow fever, West Nile, and Zika viruses24. Among these viruses, some m6A sites were enriched within the NS3 and NS5 regions. Furthermore, HCV, Zika virus, and dengue virus contained similar m6A sites in the E1 region. Therefore, these data suggest that potentially conserved m6A sites in flaviviruses could regulate the virion maturation process.
The role of m6A in the modulation of host response by HBV and HCV infections
Activation of host pattern recognition receptors (PRRs) by viral infection allows the detection of pathogen-associated molecular patterns and initiates innate immune responses to ultimately eliminate viral infection43. PRRs, which detect foreign RNAs, rely on specific molecular signatures and structures to distinguish these RNAs from cellular RNAs and this recognition of foreign RNA is an important cellular surveillance strategy44. Interestingly, the ability to use m6A to distinguish self from non-self RNAs has been recently highlighted during HBV and HCV infection based on the finding that m6A suppresses recognition by retinoic acid-inducible gene I (RIG-I)-like PRRs45. The 5′ epsilon structure of HBV pgRNA and the 3′-end poly(U/UC) region of HCV are high-affinity RIG-I ligands46,47. m6A modifications at the 5′ epsilon stem-loop of HBV pgRNA and the adenosine nucleotide at position 8766 of HCV reduced the sensing activity of RIG-I, while abrogation of these m6A sites in HBV and HCV enhanced RIG-I signaling45. YTHDF2 interacted with m6A sites within RIG-I ligand regions of the HBV and HCV RNAs and inhibited RIG-I signaling. Thus, YTHDF2 may inhibit the sensing of m6A-modified viral RNAs by RIG-I by sequestering these RNAs from RIG-I. A similar role of m6A in preventing the sensing of viral RNAs by PRRs was also studied in human metapneumovirus (HMPV)48. The genome and antigenome of HMPV were m6A-methylated and m6A modification of the HMPV genomes suppressed RIG-I sensing and subsequent IFN production. In contrast, deficient m6A modification in the HMPV genomes increased the recognition by RIG-I, leading to enhanced IFN synthesis. Together, these studies indicate that m6A modification of viral RNAs contributes to inhibiting RIG-I sensing through its sequestration by m6A binding proteins.
In addition to regulating the host immune response, viral infection can regulate host gene expression and cellular processes to optimize long-term survival49,50,51,52. As m6A methylation can regulate many cellular pathways, including stress responses and cancer development, its role in viral infection-related cellular gene expression is an important aspect of virus–host interactions3. Indeed, several studies have shown that diverse viral infections modulate the m6A profile within the host transcriptome53,54,55,56. We recently analyzed changes in the m6A profile of cellular RNAs during HBV infection53. Among the host genes whose m6A status was dramatically altered by HBV infection was the phosphatase and tensin homolog (PTEN) transcript, which exhibited enhanced levels of m6A methylation during HBV infection. Importantly, increased m6A modification of PTEN mRNA by HBV decreased its stability, affecting its protein expression. PTEN is a phosphatase of both proteins and lipids that functions as a metabolic regulator as well as a tumor suppressor57,58. Chronic HBV infection causes HCC via diverse pathways, including inflammation and oxidative stress pathways59. Thus, the decreased PTEN expression by HBV partially explains its role in HBV-associated hepatocarcinogenesis. In addition to its role as a tumor suppressor, PTEN plays an important role in the innate immune response during viral infections60. PTEN promotes IRF-3 nuclear translocation to activate the IFN signaling pathway by inducing dephosphorylation at the Ser96 residue of IRF-3. Based on these findings, HBV inhibits the host immune response through upregulation of m6A modification of PTEN53. Interestingly, the HBx protein cotranscriptionally regulates m6A modification of cellular RNA, including that of PTEN36. HBx promoted the recruitment of m6A methyltransferases (METTL3/14) to the PTEN chromosomal locus to add m6A to PTEN transcripts. In addition to its role as a viral protein, the HBx protein is widely acknowledged to be indirectly involved in the development of HCC and viral immune evasion61,62. These studies highlight the unique role of the HBx protein in regulating virus/host gene expression, immune responses, and HBV-associated hepatocarcinogenesis by modulating m6A modification of cellular RNAs.
HCV infection also regulates host gene expression by modulating m6A modification of cellular mRNAs54. HCV infection increased the m6A level of cellular RIOK3 mRNA, promoting its translation54. RIOK3 is a serine/threonine kinase that may be involved in antiviral signaling63. Importantly, viral activation of the innate immune response contributed to the increased m6A levels of RIOK3, and the increase in RIOK3 expression by m6A promoted the production of IFN, leading to inhibition of HCV replication. In addition, the m6A level of CIRBP, a stress-induced RNA-binding protein, was changed during HCV infection, although this transcript lost m6A modification54,64. In the case of CIRBP, m6A deficiency promoted alternative splicing to its shorter isoform. Interestingly, endoplasmic stress induced by viral infection promoted the loss of m6A in CIRBP, and the expression of the short isoform of CIRBP positively regulated HCV replication54,65. The precise mechanisms by which HCV infection changes the m6A status of individual transcripts are not clear, but these data suggest that activation of host cell pathways during infection may affect the m6A status of individual cellular RNAs.
Conclusion and future perspectives
New roles of m6A in epigenetically regulating cellular RNAs and viral RNAs are constantly emerging. Recently, reports have demonstrated that the genomes of several RNA viruses, as well as the RNA transcripts of DNA viruses, are modified by m6A methylation and that this modification of viral transcripts regulates various aspects of the viral life cycle and the development of pathogenesis23,24,25,26,27,28,29,30,31. In this review, we discussed the recently identified functions of m6A modification during HBV and HCV infections. m6A modification regulates the HBV and HCV life cycles in a complex way because it can differentially affect both viral and host RNAs depending on their location in the genome. Eventually, the regulation of HBV and HCV infections by m6A affects the development of liver disease, suggesting that m6A modification plays previously undefined roles in regulating the hepatitis B and C virus life cycles.
Generally, histone H3 trimethylation at lysine 36 (H3K36me3) is bound directly by the cellular m6A machinery, which in turn promotes the binding of the m6A machinery to adjacent RNA polymerase II molecules, thereby transporting the m6A machinery to the transcribed nascent RNA to add m6A cotranscriptionally66. Importantly, m6A methyltransferases are present in the cytoplasm as well as the nucleus67. Because HCV replication occurs in the cytoplasmic fraction42, it is conceivable that m6A methylation of the HCV genome may be accomplished by the cytoplasmic methyltransferases. However, the functional roles of cytoplasmic methyltransferases in mammalian cells are not yet clear. To gain a broad understanding of the mechanism by which the m6A machinery and its bound cellular proteins regulate viral infection, future research must address the roles of both the cytoplasmic and nuclear m6A machinery in the regulation of viral infection and cellular pathways. Furthermore, an understanding of how and whether viral infections regulate the function of the cellular m6A machinery and the m6A profiles of host RNAs are needed to enhance our understanding of the role of m6A in virus–host interactions. This understanding may offer novel avenues for possible m6A-based therapeutic interventions to promote viral genome clearance from infected cells. In addition, m6A reader proteins are known to interact with many RNA-binding proteins, suggesting that these interactions can affect viral replication and translation17,21. Hence, the interactome of the m6A binding proteins during viral infection needs to be identified, which may reveal the unique roles of the RNA-binding protein network that affects the viral life cycle.
Recent studies have highlighted the distinct role of m6A methylation in differentiating self RNAs from non-self RNAs based on the findings that m6A modification reduces recognition by Toll-like receptor 3 (TLR3), TLR7, and RIG-I68,69,70. In this respect, m6A methylation may allow self RNAs to be distinguished from non-self RNAs to evade recognition by cellular RNA sensor proteins, which trigger the immune response. In addition to m6A modification, several other chemical modifications, including 5-methylcytosine (m5C), uridine to pseudouridine (U to Ψ) editing, and adenosine to inosine (A to I) editing, occur in viral transcripts, and the functions of these modifications are being characterized70,71,72,73. These modifications can also be used by viruses to mimic self RNA and disrupt the host immune response. This interesting issue is currently under further investigation.
References
Roundtree, I. A., Evans, M. E., Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Yue, Y., Liu, J. & He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355 (2015).
Desrosiers, R., Friderici, K. & Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl Acad. Sci. USA 71, 3971–3975 (1974).
Linder, B. et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 (2015).
Wang, P., Doxtader, K. A. & Nam, Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell 63, 306–317 (2016).
Wang, X. et al. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex (vol 534, pg 575, 2016). Nature 542, 260–260 (2017).
Ping, X. L. et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189 (2014).
Yue, Y. A. et al. VIRMA mediates preferential m(6)A mRNA methylation in 3 ‘ UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4, Article number 10. https://doi.org/10.1038/s41421-018-0019-0 (2018).
Wen, J. et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol. Cell 69, 1028–1038.e6 (2018).
Patil, D. P. et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 (2016).
Fu, Y. et al. FTO-mediated formation of N-6-hydroxymethyladenosine and N-6-formyladenosine in mammalian RNA. Nat Commun. 4, Article number 1798. https://doi.org/10.1038/ncomms2822 (2013).
Zheng, G. Q. et al. ALKBH5 Is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Patil, D. P., Pickering, B. F. & Jaffrey, S. R. Reading m(6)A in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. 28, 113–127 (2018).
Shi, H. L. et al. YTHDF3 facilitates translation and decay of N-6-methyladenosine-modified RNA. Cell Res. 27, 315–328 (2017).
Wang, X. et al. N-6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015).
Du, H. et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun 7, Article no. 12626. https://doi.org/10.1038/ncomms12626 (2016).
Xiao, W. et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519 (2016).
Roundtree, I. A. et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 6. https://doi.org/10.7554/eLife.31311 (2017).
Hsu, P. J. et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 27, 1115–1127 (2017).
Kretschmer, J. et al. The m(6)A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5’-3’ exoribonuclease XRN1. RNA 24, 1339–1350 (2018).
Mao, Y. H. et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. 10, Article number 5332. https://doi.org/10.1038/s41467-019-13317-9 (2019).
Imam, H. et al. N6-methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc. Natl Acad. Sci. USA 115, 8829–8834 (2018).
Gokhale, N. S. et al. N6-methyladenosine in flaviviridae viral RNA genomes regulates infection. Cell Host Microbe 20, 654–665 (2016).
Kennedy, E. M. et al. Posttranscriptional m(6)A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe 19, 675–685 (2016).
Tirumuru, N. et al. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife 5. https://doi.org/10.7554/eLife.15528 (2016).
Hesser, C. R., Karijolich, J., Dominissini, D., He, C. & Glaunsinger, B. A. N6-methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog. 14, e1006995 (2018).
Tan, B. & Gao, S. J. The RNA epitranscriptome of DNA viruses. J. Virol. 92. https://doi.org/10.1128/JVI.00696-18 (2018).
Imam, H., Kim, G. W. & Siddiqui, A. Epitranscriptomic(N6-methyladenosine) modification of viral RNA and virus-host interactions. Front. Cell. Infect. Microbiol. 10, 584283 (2020).
Kane, S. E. & Beemon, K. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol. Cell Biol. 5, 2298–2306 (1985).
Gonzales-van Horn, S. R. & Sarnow, P. Making the mark: the role of adenosine modifications in the life cycle of RNA viruses. Cell Host Microbe 21, 661–669 (2017).
Kim, G. W., & Siddiqui, A. N6-methyladenosine modification of HCV RNA genome regulates cap-independent IRES-mediated translation via YTHDC2 recognition. Proc. Natl Acad. Sci. USA 118. https://doi.org/10.1073/pnas.2022024118 (2021).
Seeger, C. & Mason, W. S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64, 51–68 (2000).
Seeger, C. & Mason, W. S. Molecular biology of hepatitis B virus infection. Virology 479-480, 672–686 (2015).
Hu, J., Protzer, U. & Siddiqui, A. Revisiting hepatitis B virus: challenges of curative therapies. J. Virol. 93, e01032–19 (2019).
Kim, G. W. & Siddiqui, A. Hepatitis B virus X protein recruits methyltransferases to affect cotranscriptional N6-methyladenosine modification of viral/host RNAs. Proc. Natl Acad. Sci. USA. https://doi.org/10.1073/pnas.2019455118 (2021).
Maguire, H. F., Hoeffler, J. P. & Siddiqui, A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science 252, 842–844 (1991).
Decorsiere, A. et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531, 386–389 (2016).
Minor, M. M. et al. Hepatitis B virus HBx protein mediates the degradation of host restriction factors through the cullin 4 DDB1 E3 ubiquitin ligase complex. Cells 9. https://doi.org/10.3390/cells9040834 (2020).
Imam, H., Kim, G. W., Mir, S. A., Khan, M. & Siddiqui, A. Interferon-stimulated gene 20 (ISG20) selectively degrades N6-methyladenosine modified Hepatitis B virus transcripts. PLoS Pathog. 16, e1008338 (2020).
Liu, Y. et al. Interferon-inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem-loop structure of viral RNA. PLoS Pathog. 13, e1006296 (2017).
Paul, D., Madan, V. & Bartenschlager, R. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe 16, 569–579 (2014).
Takeuchi, O. & Akira, S. Innate immunity to virus infection. Immunol. Rev. 227, 75–86 (2009).
Schlee, M. & Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 16, 566–580 (2016).
Kim, G. W., Imam, H., Khan, M. & Siddiqui, A. N (6)-Methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J. Biol. Chem. 295, 13123–13133 (2020).
Sato, S. et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 (2015).
Saito, T., Owen, D. M., Jiang, F., Marcotrigiano, J. & Gale, M. Jr Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527 (2008).
Lu, M. et al. N(6)-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 5, 584–598 (2020).
Lyles, D. S. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol Mol. Biol. Rev. 64, 709–724 (2000).
Kim, G. W. et al. Hepatitis C Virus Core Protein Promotes miR-122 Destabilization by Inhibiting GLD-2. PLoS Pathog. 12, e1005714 (2016).
Blackham, S. et al. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 84, 5404–5414 (2010).
Lamontagne, J., Mell, J. C. & Bouchard, M. J. Transcriptome-wide analysis of hepatitis B virus-mediated changes to normal hepatocyte gene expression. PLoS Pathog. 12, e1005438 (2016).
Kim, G. W. et al. HBV-induced increased N6 methyladenosine modification of PTEN RNA affects innate immunity and contributes to HCC. Hepatology. https://doi.org/10.1002/hep.31313 (2020).
Gokhale, N. S. et al. Altered m(6)A modification of specific cellular transcripts affects flaviviridae infection. Mol. Cell 77, 542–555.e548 (2020).
Lichinchi, G. et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 1, 16011 (2016).
Lichinchi, G. et al. Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe 20, 666–673 (2016).
Chen, C. Y., Chen, J., He, L. & Stiles, B. L. PTEN: tumor suppressor and metabolic regulator. Front. Endocrinol. 9, 338 (2018).
Song, M. S., Salmena, L. & Pandolfi, P. P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 (2012).
Ringelhan, M., McKeating, J. A. & Protzer, U. Viral hepatitis and liver cancer. Philos Trans R Soc Lond B Biol Sci 372. https://doi.org/10.1098/rstb.2016.0274 (2017).
Li, S. et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 17, 241–249 (2016).
Wang, C. et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology 55, 108–120 (2012).
Khan, M., Syed, G. H., Kim, S. J., Siddiqui, A. & Hepatitis, B. Virus-induced Parkin-dependent recruitment of linear ubiquitin assembly complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 12, e1005693 (2016).
Feng, J. et al. RIOK3 Is an adaptor protein required for IRF3-mediated antiviral type i interferon production. J. Virol. 88, 7987–7997 (2014).
Liao, Y., Tong, L., Tang, L. & Wu, S. The role of cold-inducible RNA binding protein in cell stress response. Int. J. Cancer 141, 2164–2173 (2017).
Tardif, K. D., Waris, G. & Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 13, 159–163 (2005).
Huang, H. et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 567, 414–419 (2019).
Scholler, E. et al. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. RNA 24, 499–512 (2018).
Kariko, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Sioud, M., Furset, G. & Cekaite, L. Suppression of immunostimulatory siRNA-driven innate immune activation by 2’-modified RNAs. Biochem Biophys. Res Commun. 361, 122–126 (2007).
Durbin, A. F., Wang, C., Marcotrigiano, J. & Gehrke, L. RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. mBio 7. https://doi.org/10.1128/mBio.00833-16 (2016).
Courtney, D. G. et al. Epitranscriptomic addition of m(5)C to HIV-1 transcripts regulates viral gene expression. Cell Host Microbe 26, 217–227 e216 (2019).
Uzri, D. & Gehrke, L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J. Virol. 83, 4174–4184 (2009).
Samuel, C. E. ADARs: viruses and innate immunity. Curr. Top. Microbiol. Immunol. 353, 163–195 (2012).
Acknowledgements
This work is supported by NIH grants AI125350, AI139234, and AI085087 to A.S.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, GW., Siddiqui, A. The role of N6-methyladenosine modification in the life cycle and disease pathogenesis of hepatitis B and C viruses. Exp Mol Med 53, 339–345 (2021). https://doi.org/10.1038/s12276-021-00581-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00581-3
This article is cited by
-
Novel methylation mark and essential hypertension
Journal of Genetic Engineering and Biotechnology (2022)
