
Abstract
The microbiota of human breast milk (HBM) contribute to infant gut colonization; however, whether bacterial extracellular vesicles (EVs) are present in HBM or might contribute to this process remains unknown. In this study, we characterized the HBM microbiota of healthy Korean mothers and measured the key bacteria likely affecting infant gut colonization by analyzing both the microbiota and bacterial EVs. A total of 22 HBM samples were collected from lactating mothers. The DNA of bacteria and bacteria-derived EVs was extracted from each sample. In alpha-diversity analyses, bacterial samples showed higher richness and evenness than bacterial EV samples, and beta-diversity analyses showed significant differences between bacteria and bacterial EVs within identical individual samples. Firmicutes accounted for the largest proportion among the phyla, followed by Proteobacteria, Bacteroidetes, and Actinobacteria, in both bacteria and bacterial EV samples. At the genus level, Streptococcus (25.1%) and Staphylococcus (10.7%) were predominant in bacterial samples, whereas Bacteroides (9.1%), Acinetobacter (6.9%), and Lactobacillaceae(f) (5.5%) were prevalent in bacterial EV samples. Several genera, including Bifidobacterium, were significantly positively correlated between the two samples. This study revealed the diverse bacterial communities in the HBM of healthy lactating mothers, and found that gut-associated genera accounted for a high proportion in bacterial EV samples. Our findings suggest the existence of key bacteria with metabolic activity that are independent of the major bacterial populations that inhabit HBM, and the possibility that EVs derived from these bacteria are involved in the vertical transfer of gut microbiota.
Similar content being viewed by others

Introduction
More than 100 trillion microbes dwell within the human gut and interact with the host in a symbiotic relationship to aid the development of the host immune system. The early acquisition of the gut microbiome during infancy affects health throughout human life. Several factors, such as the type of delivery, antibiotic therapy, environment of care, and nutritional exposure, are assumed to influence the early colonization of the infant gut1. Diet plays a key role in the development of the newborn’s immune system. Human breast milk (HBM) is not only the best source of nutrition for infants, but is also known to contain immune components, such as secretory antibodies, immune cells, antimicrobial proteins (such as lactoferrin and lysozyme), cytokines, and human milk oligosaccharide2. Before the 2000s, the presence of bacteria in HBM was thought to indicate contamination or infection, but several researchers have demonstrated that HBM contains commensal bacteria by using culture-dependent techniques3,4. The development of the non-culture-based sequencing technique has led to more thorough investigation of the diversity of the HBM microbiota5. Currently, it is recognized that HBM contains abundant commensal bacteria that can affect the colonization of the infant gut6.
Extracellular vesicles (EVs) are nanometer-sized membrane vesicles that contain various bioactive molecules, such as transmembrane proteins, cytosolic proteins, nucleic acids, and lipids. It is widely accepted that all kinds of bacteria release EVs. Bacteria are classified into Gram-positive (G + ) and Gram-negative (G–) bacteria based on membrane characteristics, and the G + bacteria-derived EVs are usually called bacterial membrane vesicles, while the G-bacteria-derived EVs are called outer membrane vesicles7. Bacteria-derived EVs can be detected in body fluids, such as blood, urine, or stool, indicating that they can affect host cells by directly activating host receptors, delivering various bioactive molecules, or integrating EVs into host cells8,9,10. However, there has been no study on the analysis of bacteria-derived EVs in HBM until now.
In this context, the objective of this study was to characterize the HBM microbiota of healthy lactating mothers in Korea, and to measure the abundance of key bacteria that can affect infant gut colonization by analyzing both the microbiota and bacterial EVs using a culture-independent technique.
Materials and methods
Subjects and sample collection
This study was conducted according to the guidelines proposed in the Declaration of Helsinki and approved by the Institutional Review Board (IRB) of Chung-Ang University Hospital, Seoul, South Korea (IRB No.: 1810-004-309). Healthy lactating mothers who had delivered a full-term baby were recruited within 1 week–4 months of childbirth for this study (July 2017–June 2018).
Samples of breast milk were collected from 22 mothers in sterile bags by manual or pump expression. Before milk expression, hands were washed, and nipples were cleaned with sterile water. The first ten drops were discarded. The fresh samples were delivered to the laboratory within 24 h and then stored at −80 °C until further processing. If the samples did not reach the laboratory within 24 h, they were excluded. Changes in bacterial diversity based on the lactation stage, especially the difference between the microbiomes of the colostrum and mature HBM, are well known1. As we tried to analyze the bacteria that infants actually consume while feeding on stabilized mature milk, we excluded the colostrum samples obtained within 1 week of childbirth. Samples from mothers who developed disease or were administered antibiotics during lactation were also excluded.
EV isolation and DNA extraction from human milk samples
To separate the EVs from HBM, EVs in milk samples were isolated using differential centrifugation at 10,000×g for 10 min at 4 °C. After centrifugation, the pellet contained bacteria, and the supernatant contained EVs. Bacteria and foreign particles were thoroughly eliminated by sterilizing the supernatant through a 0.22-µm filter. To extract the DNA from bacteria and bacterial EVs, bacteria and EVs were boiled for 40 min at 100 °C. To eliminate the remaining floating particles and waste, the sample was centrifuged at 13,000 rpm for 30 min at 4 °C, and the supernatant was collected. DNA was extracted using a DNA isolation kit (DNeasy PowerSoil Kit, QIAGEN, Germany) in accordance with the standard protocol provided along with the kit. The DNA from bacteria and EVs in each sample was quantified using the QIAxpert system (QIAGEN, Germany).
Bacterial metagenomic analysis using DNA from human milk samples
Bacterial genomic DNA was amplified with the primers 16S_V3_F (5′-TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG-3′) and 16S_V4_R (5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C-3′), which are specific for the V3–V4 hypervariable regions of the 16S rDNA gene. The libraries were prepared using PCR products according to the MiSeq System guide (Illumina, USA) and quantified using QIAxpert (QIAGEN, Germany). Each amplicon was then quantified, set to an equimolar ratio, pooled, and sequenced on a MiSeq instrument (Illumina, USA) according to the manufacturer’s recommendations.
Analysis of bacterial composition in the microbiota
Paired-end reads that matched the adapter sequences were trimmed by cutadapt version 1.1.611. The resulting FASTQ files containing paired-end reads were merged with CASPER and then quality- filtered using the Phred (Q) score-based criteria described by Bokulich12,13. Any reads shorter than 350 bp and longer than 550 bp after merging were also discarded. To identify the chimeric sequences, a reference-based chimera-detection step was performed using VSEARCH against the SILVA gold database14,15. Next, the sequence reads were clustered into operational taxonomic units (OTUs) using VSEARCH with a de novo clustering algorithm with a sequence similarity threshold of 97%. The representative sequences of the OTUs were finally classified using the GREENGENES database with UCLUST (parallel_assign_taxonomy_uclust.py script on QIIME version 1.9.1) with default parameters16.
Statistical analysis
The richness (rarefaction curves) was analyzed using Chao 1, and alpha-diversity indices (Chao 1, ACE, Shannon index, Simpson index, and Fisher’s index) were calculated. Significance between subgroups was tested using a t test for continuous variables. Principal component analysis (PCA) based on Euclidean distance was performed to quantify the relationships between samples. In addition, networks were constructed using the Pearson correlation coefficient. All statistical analyses were performed using R version 3.3.2.
Results
Characteristics of the lactating mothers
A total of 22 lactating mothers were enrolled in this study; the mean age was 33.1 ± 3.4 years. Of the 22 lactating mothers, 16 (72.7%) were primiparous, and 6 (27.3%) were multiparous. Eight (36.4%) mothers had vaginal deliveries and 14 (63.6%) mothers had delivered by cesarian section (CS). Samples were collected 11.1 ± 2.4 days after delivery on average.
Diversity
When Chao 1 and ACE were determined to analyze the alpha diversity and richness, bacteria showed significantly higher alpha diversity than bacterial EVs (p < 0.001 and p = 0.005, respectively). Bacteria showed higher Fisher’s index values than the EVs, indicating higher evenness in the bacterial samples (p < 0.001). However, EVs and bacteria did not show any significant difference in the Shannon diversity index (p = 0.51), which signifies richness, and the Simpson index (p = 0.05), which signifies evenness (Fig. 1). The number of reads for bacteria, 14966.6 (SD 10189.1), was higher than that for EVs, 9267.8 (SD 6507.0). Bacteria also showed significantly higher levels of operational taxonomic units (OTUs), with 1202.3 (SD 789.1) reads compared with 501.0 (SD 446.8) reads in EVs.

Alpha diversity analysis showed that microbial richness and evenness were significantly higher in bacterial samples than in bacterial EV samples when analyzed using Chao 1 (p = 0.002), ACE (p = 0.005), and Fisher’s exact test (p < 0.001). Bacteria and bacterial EV samples did not show any significant difference in Shannon diversity index (p = 0.51) or Simpson’s index (p = 0.05).
The beta diversity of all samples was analyzed using PCA based on the Euclidean distance (Fig. 2). The Euclidean distance value and cluster in the breast milk samples did not differ significantly between different individual samples at the EV level. However, the Euclidean distance values showed a significant difference in different individual samples at the bacterial level. Furthermore, the Euclidean distance of bacteria and EVs from an identical sample showed a significant difference.

(a) at the phylum level and (b) at the genus level.
Bacterial composition
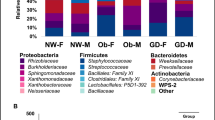
We detected 23 phyla by analyzing the microbiota originating from breast milk. Among the microbes, Firmicutes accounted for the largest proportion, with 56.4% (SD 19.4) among the phyla, followed by Proteobacteria (19.6% (SD 19.1)), Bacteroidetes (9.8% (SD 10.5)), and Actinobacteria (9.0% (SD 7.9)). Verrucomicrobia showed an average proportion below 5%, while other phyla accounted for <1% of the bacterial composition of breast milk (Fig. 3a). Three-hundred and ninety-two genera were detected in total at the genus level, with Streptococcus accounting for the highest proportion at 25.1% (SD 20.9), followed by Staphylococcus at 10.7% (SD 12.3). Bacteroides, Acinetobacter, Enterobacteriaceae(f), Ruminococcaceae(f), Bifidobacterium, Prevotella, Clostridiales(o), Corynebacterium, Akkermansia, Lactobacillus, Pseudomonas, Dialister, Stenotrophomonas, Blautia, Sphingomonas, Haemophilus, Neisseria, Lachnospiraceae(f), Rothia, and Faecalibacterium showed average proportions below 5%, while other genera accounted for <1% of the bacterial composition on average (Fig. 3b).

(a) Bacteria at the phylum level, (b) bacteria at the genus level, (c) bacterial extracellular vesicles (EVs) at the phylum level, and (d) bacterial EVs at the genus level.
Meanwhile, bacterial EV analysis revealed a total of 22 phyla. Firmicutes accounted for the largest proportion (35.8% (SD 12.9)), while Proteobacteria (24.5% (SD 13.3)), Bacteroidetes (15.5% (SD 8.8)), and Actinobacteria (13.1% (SD 8.3)) accounted for the remaining majority of the bacterial EV composition. Verrucomicrobia and Cyanobacteria accounted for less than 5% of the bacterial EV microbiome on average, and other phyla accounted for less than 1% on average (Fig. 3c). A total of 291 genera were detected at the genus level in bacterial EV samples. Bacteroides, Acinetobacter, and Lactobacillaceae(f) accounted for high proportions of EV samples (9.1% (SD 5.4), 6.9% (SD 8.2), and 5.5% (SD 9.1), respectively). Meanwhile, Streptococcus, Staphylococcus, Enterobacteriaceae(f), Ruminococcaceae(f), Bifidobacterium, Prevotella, Clostridiales(o), Corynebacterium, Akkermansia, Lactobacillus, Dialister, Stenotrophomonas, Blautia, Sphingomonas, Streptophyta(o), Comamonadaceae(f), Collinsella, Actinomyces, Clostridiaceae(f), and Burkholderia accounted for less than 5% of the bacterial EV composition on average. The rest of the detected genera accounted for <1% of the bacterial EV composition on average (Fig. 3d).
Correlation between bacteria and bacterial EVs
We analyzed the correlation between bacteria isolated from HBM (bacteria) and bacterial EVs at the phylum level, and determined that there were no taxa with a significant correlation between the bacteria and bacterial EV samples. However, at the genus level, we did observe significant differences in the proportions of several genera. We analyzed these correlations to determine any trends, and found that Acinetobacter, Ruminococcaceae(f), Bifidobacterium, Comamonadaceae(f), Rothia, and Clostridiaceae(f) showed a significant positive correlation between bacteria derived from HBM and bacterial EV samples (p < 0.05) (Fig. 4; Supplementary Table S1).

Correlation analysis between bacteria and bacterial EV samples showed a significant positive correlation for Acinetobacter, Ruminococcaceae (f), Bifidobacterium, Comamonadaceae (f), Rothia, and Clostridiaceae (f) (p < 0.05) (see also Supplementary Table S1).
Comamonadaceae(f) showed a particularly high correlation between the two sample groups (r = 0.95). Although they account for less than 1% of the total proportion, Comamonas and Diaphorobacter also showed a significantly high correlation between bacteria and bacterial EV samples (r = 0.94). Among the bacteria accounting for <1% of the total proportion, 24 genera showed a significant positive correlation. Meanwhile, Bacteroides, Prevotella, Lachnospiraceae(f), Collinsella, and Burkholderia showed a negative correlation trend, although this trend was not significant. Although some genera accounted for less than 1% of the average proportion with a negative correlation, this result was not significant.
Differences between subgroups
We also analyzed the bacterial composition of HBM at the genus level based on parity. We determined 0.31- and 0.18-fold changes between bacteria and bacterial EV samples with respect to Staphylococcus and Collinsella, respectively, which are significantly lower than the values obtained when the breast milk was obtained from multiparous mothers. Meanwhile, the abundance of Haemophilus was shown to be 2.86-fold higher in the HBM of multiparous mothers than in that of primiparous mothers (p < 0.05) (Fig. 5a). No significant difference in the microbiome based on parity was observed in bacterial EV samples. When we compared the microbial composition of HBM of mothers who delivered by CS with that of mothers who had vaginal delivery, the abundance of Streptophyta(o) was significantly higher (6.24-fold) in bacterial samples from mothers who delivered by CS (Fig. 5b). The abundance of Actinomycetales was significantly lower (0.19-fold) in bacterial EV samples from mothers who delivered by CS than in those from mothers who had vaginal delivery (Fig. 5b). A comparative analysis was conducted comparing calories; however, none of the microbiomes showed any significant difference.

Analysis of bacterial composition based on (a) parity and (b) mode of delivery.
Discussion
To the best of our knowledge, this is the first study to analyze both the microbiota and bacteria-derived EVs in HBM using next-generation sequencing (NGS) worldwide. In the present study, we found that HBM of healthy lactating mothers contained diverse and complex bacterial communities, with Firmicutes as the predominant phylum. An analysis at the genus level showed significant differences in proportions between the microbiota and bacteria-derived EV samples, and several genera showed a significant positive correlation between the microbiota and bacterial EVs.
An initial study that analyzed the HBM using NGS suggested that there was a common core microbiome consisting of nine bacterial genera17. Several subsequent studies also identified the bacteria that make up the core HBM microbiome. However, the results differed from study to study6,18,19. The core HBM microbiome has been the subject of controversy, as it is thought to play a major role in the colonization of the infant gut. A systematic review analyzed 242 studies and found that most of the bacterial species isolated from breast tissue and milk belonged to four bacterial phyla, namely, Firmicutes, Proteobacteria, Actinobacteria, and Bacteroidetes20. A recent study, which reviewed 44 studies that investigated 3105 breast milk samples from 2655 women, showed that the most frequently found genera were Staphylococcus, Streptococcus, Lactobacillus, Pseudomonas, Bifidobacterium, Corynebacterium, Enterococcus, Acinetobacter, Rothia, Cutibacterium, Veillonella, and Bacteroides21. Another systematic review of 12 studies that used only culture-independent methods for healthy mothers reported that Staphylococcus and Streptococcus were the predominant genera, and that these genera may be universally present in HBM5. In our study, Firmicutes accounted for the largest proportion among the phyla, followed by Proteobacteria, Bacteroidetes, and Actinobacteria, in both bacteria and bacterial EV samples. At the genus level, Streptococcus (25.1%) and Staphylococcus (10.7%) were predominant in bacterial samples. The results of the present study are in accordance with those of previous studies. However, analysis at the genus level showed significant differences in proportions between bacteria and bacterial EVs.
As no research has been done to analyze bacteria-derived EVs in HBM, EVs released from the HBM microbiota are bound to be a part of the findings. A study has reported that intraperitoneally injected bacterial EVs spread throughout the mouse body, accumulating in the liver, lung, spleen, and kidney within 3 h of administration22. Several studies have also reported that bacteria-derived EVs can be detected in human blood serum, urine, or stool, and have claimed that bacteria-derived EVs are related to the immune response or development of disease23,24,25,26,27. For example, studies have shown that EVs derived from Staphylococcus aureus contribute to atopic dermatitis-like skin inflammation28,29. In the present study, we were able to detect bacterial EVs in HBM, and unlike the dominant genera in the bacterial sample, Bacteroides (9.1%), Acinetobacter (6.9%), and Lactobacillaceae(f) (5.5%) accounted for a high proportion. When we analyzed the alpha diversity, the present study showed that the number of reads and OTUs in bacterial samples were higher than those in bacterial EVs. Bacterial samples also showed higher richness and evenness than bacterial EV samples. Beta-diversity analyses showed significant differences between bacteria and bacterial EVs within identical individual samples. The results of the present study indicate that while abundant bacterial communities are distributed evenly in HBM, the bacteria that release EVs do not necessarily match the bacteria present in HBM in high proportion and are not distributed evenly.
The origins of bacteria in HBM are still unclear. It has been suggested that commensal bacteria from maternal skin (such as Staphylococcus, Corynebacterium, and Cutibacterium) or the infant’s mouth (such as Streptococcus) can enter the mammary duct during breastfeeding30. Another hypothesis is that the maternal gut bacteria enter the mammary glands through the entero-mammary pathway, which would require the cells to penetrate the intestinal epithelium and reach the mammary glands through the bloodstream31,32,33. The observation that anaerobic bacteria such as Bacteroides, Bifidobacterium, Parabacteroides, and Clostridium that are not found on the skin are detected and shared between HBM and infant feces supports this hypothesis3,6. Other observations showing that there are bacteria in the maternal gut, infant’s gut, and HMB that share the same strains also support the vertical transmission of maternal commensal bacteria to the infant’s gut through HBM31,34,35, but no exact mechanism has been revealed. It is thought that not only pathogens but also commensal bacteria can secrete EVs. One study revealed that commensal bacteria can produce EVs containing immunomodulatory molecules and affect host immunity and health36, while others suggested that EVs derived from commensal bacteria can be involved in gut colonization37. In our study, gut-associated genera such as Bacteroides and Lactobacillaceae(f) accounted for a high proportion in the bacterial EV samples, unlike the predominant bacteria in the bacterial samples. Our findings suggest that EVs derived from gut bacteria may contribute to the vertical transfer of the commensal microbiota from mothers to infants via HBM.
Evaluation of the correlation between HBM-derived bacteria and bacterial EV samples revealed that several genera were significantly positively correlated. As a strong correlation can be interpreted as being indicative of high metabolic activity of the bacteria, the effect of these bacteria on gut colonization is worth considering. In particular, we highlighted Bifidobacterium, as the gut microbiota of infant feces usually exhibits a high abundance of this bacterium, with exclusively breastfed infants in particular exhibiting a relatively high abundance of Bifidobacterium in feces38,39. Several lines of evidence suggest that the relative level of Bifidobacterium abundance is associated with the development of disease or immunity. For example, a systematic review compared 101 studies on healthy controls with 147 studies on infected patients, all of which included molecular analysis of HBM. Some species were found only in infected patients, whereas others were never detected in infected patients and were present only in healthy controls; specifically, Bifidobacterium and Lactobacillus were associated with the absence of infection20. In this study, Lactobacillus was among the bacteria present at high abundance in bacterial EV samples, and Bifidobacterium was identified as being highly correlated between bacteria and bacterial EVs. Our findings suggest the presence of key bacteria with significant metabolic activity that are independent of the major bacterial populations that inhabit HBM, and the possibility of measuring the association of these bacteria with EVs. Together, these findings warrant additional research to explore the effects and functions of bacterial EVs on infant gut colonization and immune development.
Various factors, such as mode of delivery, maternal weight, antibiotics, lactation stage, gestational age, geographical location, and maternal health, have been found to contribute to the diversity of the HBM microbiota1. In the present study, subgroup analysis revealed that the abundance of Staphylococcus in the bacterial samples and Collinsella in the bacterial EV samples was significantly lower, and that of Haemophilus in the bacterial samples was significantly higher, when HBM was obtained from multiparous mothers compared with primiparous mothers. In contrast, a large study did not identify an effect of parity on the composition of the HBM microbiota40. Our study also showed that the abundance of Streptophyta(o) was significantly higher in the bacterial samples, and that of Actinomycetales was significantly lower in the bacterial EV samples in the CS than in the vaginal delivery group. It has been suggested that labor is associated with the composition of the HBM microbiota via increased intestinal permeability, and by facilitating a greater degree of entero-mammary transfer of maternal gut bacteria41. Women who delivered by CS were reported to have a higher relative abundance of Proteobacteria and Carnobacteriaceae, but a lower relative abundance of Bifidobacterium, Leuconostocaceae, and Lactobacillus1. Alternatively, some studies have found no association between the composition of the HBM microbiota and delivery mode40,42. We therefore postulate that the diversity of bacterial communities is affected by a number of variables, such as host immunity, environmental factors, and microbial factors, resulting in quantitative and qualitative differences.
Our study has some limitations. First, the sample size was relatively small. Second, we did not analyze communities from maternal and infant mouths, skin, and stool simultaneously, so the origins of the HBM bacteria are unclear. Third, we also did not demonstrate the process by which the HBM microbiota and EVs are involved in the colonization of the infant gut. Considering that bacteria reaching the infant’s gut can affect immunity, it is more important to know which bacteria have active biological functions than to simply describe the composition of the HBM microbiota. It is necessary to determine whether EV analysis can predict the key bacteria that play a major role in the colonization of the infant gut, and to identify the factors that influence the release of bacterial EVs in HBM. Studies are also needed to explain how bacterial EVs in HBM affect the composition of the infant gut microbiota and change infant health outcomes. This will be the next task in our future study. Despite these limitations, the present study was the first to analyze bacterial EVs in HBM, suggesting the possibility that bacterial EVs in HBM contribute to the colonization of the infant gut, and will be the basis for further study.
In conclusion, the present study revealed the diverse bacterial communities in HBM from healthy lactating mothers along with those of bacterial EVs and the relationship between these factors. Our findings suggest the existence of key bacteria with metabolic activity in HBM, and the possibility that EVs derived from these bacteria are involved in the vertical transfer of the gut microbiota from mothers to their progeny. Future studies will be needed to confirm the role of bacterial EVs in HBM colonization of the infant gut, and to ultimately promote colonization with beneficial bacteria in HBM.
References
Ojo-Okunola, A., Nicol, M. & du Toit, E. Human breast milk bacteriome in health and disease. Nutrients 10, 1643 (2018).
Kim, S. Y. & Yi, D. Y. Components of human breast milk: from macronutrient to microbiome and microRNA. Clin. Exp. Pediatr. (2020). Online ahead of print.
Martin, R. et al. Human milk is a source of lactic acid bacteria for the infant gut. J. Pediatr. 143, 754–758 (2003).
Heikkila, M. P. & Saris, P. E. Inhibition of Staphylococcus aureus by the commensal bacteria of human milk. J. Appl. Microbiol. 95, 471–478 (2003).
Fitzstevens, J. L. et al. Systematic review of the human milk microbiota. Nutr. Clin. Pr. 32, 354–364 (2017).
Murphy, K. et al. The composition of human milk and infant faecal microbiota over the first three months of life: a pilot study. Sci. Rep. 7, 40597 (2017).
Brown, L., Wolf, J. M., Prados-Rosales, R. & Casadevall, A. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 13, 620–630 (2015).
O’Donoghue, E. J. & Krachler, A. M. Mechanisms of outer membrane vesicle entry into host cells. Cell. Microbiol. 18, 1508–1517 (2016).
Perez-Cruz, C., Delgado, L., Lopez-Iglesias, C. & Mercade, E. Outer-inner membrane vesicles naturally secreted by gram-negative pathogenic bacteria. PLoS ONE 10, e0116896 (2015).
Kang, C. S. et al. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PLoS ONE 8, e76520 (2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Kwon, S., Lee, B. & Yoon, S. CASPER: context-aware scheme for paired-end reads from high-throughput amplicon sequencing. BMC Bioinforma. 15(Suppl 9), S10 (2014).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59 (2013).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahe, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Hunt, K. M. et al. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS ONE 6, e21313 (2011).
Jimenez, E. et al. Metagenomic analysis of milk of healthy and mastitis-suffering women. J. Hum. Lact. 31, 406–415 (2015).
Urbaniak, C., Angelini, M., Gloor, G. B. & Reid, G. Human milk microbiota profiles in relation to birthing method, gestation and infant gender. Microbiome 4, 1 (2016).
Togo, A. et al. Repertoire of human breast and milk microbiota: a systematic review. Future Microbiol. 14, 623–641 (2019).
Zimmermann, P. & Curtis, N. Breast milk microbiota: A review of the factors that influence composition. J. Infect 8, 17–47 (2020).
Jang, S. C. et al. In vivo kinetic biodistribution of nano-sized outer membrane vesicles derived from bacteria. Small 11, 456–461 (2015).
Yoo, J. Y. et al. 16S rRNA gene-based metagenomic analysis reveals differences in bacteria-derived extracellular vesicles in the urine of pregnant and non-pregnant women. Exp. Mol. Med. 48, e208 (2016).
Choi, Y., Park, H., Park, H.-S. & Kim, Y.-K. Extracellular vesicles, a key mediator to link environmental microbiota to airway immunity. Allergy Asthma Immunol. Res. 9, 101–106 (2017).
Lee, Y. et al. Rapid assessment of microbiota changes in individuals with autism spectrum disorder using bacteria-derived membrane vesicles in urine. Exp. Neurobiol. 26, 307–317 (2017).
Choi, Y. et al. Gut microbe-derived extracellular vesicles induce insulin resistance, thereby impairing glucose metabolism in skeletal muscle. Sci. Rep. 5, 15878 (2015).
Kim, M. H. et al. A metagenomic analysis provides a culture-independent pathogen detection for atopic dermatitis. Allergy Asthma Immunol. Res. 9, 453–461 (2017).
Hong, S. W. et al. Extracellular vesicles derived from Staphylococcus aureus induce atopic dermatitis-like skin inflammation. Allergy 66, 351–359 (2011).
Hong, S. W. et al. An important role of alpha-hemolysin in extracellular vesicles on the development of atopic dermatitis induced by Staphylococcus aureus. PLoS ONE 9, e100499 (2014).
Ramsay, D. T., Kent, J. C., Owens, R. A. & Hartmann, P. E. Ultrasound imaging of milk ejection in the breast of lactating women. Pediatrics 113, 361–367 (2004).
Jost, T., Lacroix, C., Braegger, C. P., Rochat, F. & Chassard, C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ. Microbiol. 16, 2891–2904 (2014).
Perez, P. F. et al. Bacterial imprinting of the neonatal immune system: lessons from maternal cells? Pediatrics 119, e724–e732 (2007).
Kordy, K. et al. Contributions to human breast milk microbiome and enteromammary transfer of Bifidobacterium breve. PLoS ONE 15, e0219633 (2020).
Milani, C. et al. Exploring vertical transmission of bifidobacteria from mother to child. Appl. Environ. Microbiol. 81, 7078–7087 (2015).
Asnicar, F. et al. Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems 2, e00164-16 (2017).
Shen, Y. et al. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 12, 509–520 (2012).
Bryant, W. A. et al. In silico analysis of the small molecule content of outer membrane vesicles produced by Bacteroides thetaiotaomicron indicates an extensive metabolic link between microbe and host. Front. Microbiol. 8, 2440 (2017).
Turroni, F. et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS ONE 7, e36957 (2012).
Zimmermann, P. & Curtis, N. Factors influencing the intestinal microbiome during the first year of life. Pediatr. Infect. Dis. J. 37, e315–e335 (2018).
Moossavi, S. et al. Composition and variation of the human milk microbiota are influenced by maternal and early-life factors. Cell Host Microbe 25, 324–335.e324 (2019).
Cabrera-Rubio, R. et al. The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am. J. Clin. Nutr. 96, 544–551 (2012).
Williams, J. E. et al. Human milk microbial community structure is relatively stable and related to variations in macronutrient and micronutrient intakes in healthy lactating women. J. Nutr. 147, 1739–1748 (2017).
Acknowledgements
The authors are grateful to the volunteers who participated in this study. This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of Science and ICT, No. NRF-2019R1F1A1059569).
Author information
Authors and Affiliations
Contributions
Conceptualization, S.Y.K. and D.Y.Y.; methodology, D.Y.Y.; validation, D.Y.Y.; formal analysis, S.Y.K. and D.Y.Y.; investigation, S.Y.K. and D.Y.Y; resources, S.Y.K. and D.Y.Y.; data curation, S.Y.K. and D.Y.Y.; writing—original draft preparation, S.Y.K. and D.Y.Y.; writing—review and editing, S.Y.K. and D.Y.Y.; supervision, D.Y.Y.; project administration, D.Y.Y. All authors have read and approved the published version of the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, S.Y., Yi, D.Y. Analysis of the human breast milk microbiome and bacterial extracellular vesicles in healthy mothers. Exp Mol Med 52, 1288–1297 (2020). https://doi.org/10.1038/s12276-020-0470-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0470-5
