
Abstract
As an essential nutrient and trace element, selenium is required for living organisms and its beneficial roles in human health have been well recognized. The role of selenium is mainly played through selenoproteins synthesized by the selenium metabolic system. Selenoproteins have a wide range of cellular functions including regulation of selenium transport, thyroid hormones, immunity, and redox homeostasis. Selenium deficiency contributes to various diseases, such as cardiovascular disease, cancer, liver disease, and arthropathy—Kashin–Beck disease (KBD) and osteoarthritis (OA). A skeletal developmental disorder, KBD has been reported in low-selenium areas of China, North Korea, and the Siberian region of Russia, and can be alleviated by selenium supplementation. OA, the most common form of arthritis, is a degenerative disease caused by an imbalance in matrix metabolism and is characterized by cartilage destruction. Oxidative stress serves as a major cause of the initiation of OA pathogenesis. Selenium deficiency and dysregulation of selenoproteins are associated with impairments to redox homeostasis in cartilage. We review the recently explored roles of selenium metabolism and selenoproteins in cartilage with an emphasis on two arthropathies, KBD and OA. Moreover, we discuss the potential of therapeutic strategies targeting the biological functions of selenium and selenoproteins for OA treatment.
Similar content being viewed by others


Introduction
Selenium (Se) is an essential trace element in humans1,2. Selenium is generally taken up from the diet through food or other forms of external supplementation. Dietary selenium is obtained in the form of selenomethionine (SeMet), selenocysteine (Sec), selenite, and selenate. Significant health benefits have been attributed to selenium metabolic systems that play major physiological roles in thyroid hormone metabolism, immunity, and antioxidant defense2,3. Selenium is required for the production of thyroid hormone-metabolizing enzymes and selenium supplementation is thought to improve the function of thyrocytes and immune cells4. Selenium supplementation demonstrated immunostimulant effects, such as enhanced proliferation of activated T cells, activation of natural killer cells, and tumor cytotoxicity mediated by cytotoxic lymphocytes5,6. In contrast, selenium deficiency is associated with the occurrence, virulence, and disease progression of viral infections7.
Selenium inadequacy can lead to various types of diseases, most notably cardiovascular disease8,9,10,11,12, cancer13,14,15, hepatopathy16,17, and arthropathy. Cardiovascular diseases are associated with systemic selenium level, with a higher risk at <55 or >145 μg/L selenium concentration in the blood10. A type of endemic cardiomyopathy, Keshan disease is linked to selenium deficiency8,11. Keshan disease occurs in low-selenium areas in China and is prevented by sodium selenite supplementation12. Low-selenium status is correlated with a significantly increased risk of cancer incidence and mortality13,14,15. Epidemiological studies have provided evidence on the cancer-preventing effects of selenium18,19,20. Selenium deficiency is also characterized by elevated levels of oxidative stress markers in the liver21, which significantly contribute to liver injury17. The oxidative stress caused by selenium deficiency further plays a detrimental role in joint development. Selenium deficiency is the main cause of endemic Kashin–Beck disease (KBD), which is mainly reported in low-selenium areas of China, North Korea, and the Siberian region of Russia. Moreover, there is a growing body of evidence suggesting that the pathogenesis of osteoarthritis (OA), the most common form of arthritis, may be associated with selenium deficiency by resulting in oxidative stress22,23,24,25,26,27,28. However, it is noteworthy that excessive selenium intake can also cause selenosis29,30, which accompanies adverse symptoms including fatigue, diarrhea, nausea, increased heart rate, necrosis in liver and kidney, and neurological damage. Chronic selenosis eventually compromises immune and reproductive systems in humans.
OA is characterized by progressive loss of cartilage extracellular matrix (ECM) and pathological changes in other joint tissues such as subchondral bone sclerosis, osteophyte formation, and synovial inflammation31. Cartilage destruction is considered a hallmark of OA and is a result of increased production of catabolic effectors32,33,34,35 and reduced matrix biosynthesis by chondrocytes36. OA is associated with multiple etiologies involving systemic factors such as age37 as well as local factors such as mechanical stress38 driven by weight-bearing and joint instability. Both OA-causing factors have been found to cause oxidative stress in chondrocytes. Oxidative stress results from the abnormal production of reactive oxygen species (ROS) and the loss of cellular antioxidant capacity. Many preclinical and clinical studies have indicated the accumulation of oxidative burden in chondrocytes undergoing osteoarthritic changes39,40. Emerging evidence suggests that oxidative stress is mechanistically linked to the initiation of osteoarthritic changes in chondrocytes through the acquisition of senescent phenotypes36. Therefore, restoring redox homeostasis can serve as a rational therapeutic strategy to alleviate OA progression. Here, we review the role of selenium metabolism in cartilage and bone and the significance of maintaining its homeostasis in the context of joint diseases such as KBD and OA.
Overview of the selenium metabolic system
The selenium metabolic system and the biosynthesis of selenoproteins
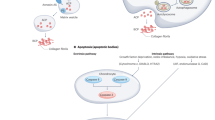
Selenium metabolism is a systemic process that includes the absorption, transportation, transformation, and excretion of selenium (Fig. 1). Selenium is obtained in organic forms—SeMet and Sec—and inorganic forms—selenite and selenate—from diet. Selenium is taken up by the liver that synthesizes and exports SELENOP which eventually circulates through the bloodstream. SELENOP, with multiple Sec residues41, transports selenium to other tissues and organs42 and the transported selenium is converted to selenophosphate by intracellular selenium metabolic pathways. Selenium is excreted through exhalation and urine in the form of small-molecule metabolites formed by sequential methylation43,44.

Selenium is absorbed from the diet, undergoes several conversion steps, and is incorporated into polypeptide chains, completing selenoprotein synthesis. Dietary sources of selenium uptake exist in inorganic form such as selenate and selenite and organic form such as Sec and SeMet. Inorganic forms are reduced by TXNRD/TRX or GRX/GSH systems and organic forms are cleaved by SCLY, forming selenide. Selenophosphate is synthesized from selenide by SEPHS2, and the subsequent reaction with PSer-tRNA[Ser]Sec mediated by SEPSECS yields Sec-tRNA[Ser]Sec. Sec-tRNA[Ser]Sec is transferred to the A-site of ribosome mediated by SBP2, which binds to SECIS located in the 3′UTR of a selenoprotein mRNA. Finally, the UGA codon is recognized as the Sec integration codon. Abbreviations: SeMet, selenomethionine; Sec, selenocysteine; GRX, glutathione reductase; TRX, thioredoxin; TXNRD, thioredoxin reductase; GSH, glutathione; MGL, methionine gamma-lyase; SCLY, selenocysteine lyase; SEPHS2, selenophosphate synthetase 2; SARS, seryl-tRNA synthetase; PSTK, phosphoseryl(Sep)-tRNA kinase; SEPSECS, Sep-tRNA:Sec-tRNA synthase; EEFSEC, Sec-specific eukaryotic elongation factor; SBP2, SECIS binding protein 2.
Selenium plays biological roles predominantly in the form of selenoproteins synthesized by the selenium metabolic system. Ingested inorganic selenium is first reduced to hydrogen selenide (H2Se) via glutathione (GSH) and thioredoxin (TXN) systems. Selenide is further converted to Sec amino acids for incorporation into specific sites of selenoproteins such as the catalytic sites of a selenoenzyme. Mechanistically, selenophosphate synthetase 2 (SEPHS2) catalyzes the production of selenophosphate through the reduction of hydrogen selenide. The subsequent reaction with phosphoseryl-tRNA (PSer-tRNA[Ser]Sec) yields Sec-tRNA[Ser]Sec. Sec amino acids are incorporated into polypeptide chains through the machinery utilizing the UGA codon. Selenocysteine insertion sequence binding protein 2 (SBP2) binds to selenocysteine insertion sequence (SECIS) element which is located in the 3′-untranslated region (3′UTR) of selenoprotein mRNA, and mediates the transfer of Sec-tRNA[Ser]Sec to the A-site of ribosome which recognizes the UGA codon as the Sec integration codon. Collectively, the selenoprotein translation machinery consists of SECIS element, SBP2, Sec-specific eukaryotic elongation factor (EEFSEC), and aminoacylated Sec-tRNA[Ser]Sec, thereby enabling UGA to be recognized as a Sec codon and utilized for translation into the growing polypeptide.
Selenoproteins
Selenoprotein is defined as a protein containing Sec amino acid residue. The biological functions of selenium are mostly exerted through selenoprotein domains that contain Sec residues. Twenty-five selenoprotein genes have been identified in the human genome45. In mice, a total of 24 selenoproteins have been characterized46 and targeted deletion of some of these selenoproteins demonstrated their essential roles in developmental processes and in disease pathogenesis. Selenoproteins can be classified into subfamilies based on their cellular functions such as those implicated in antioxidation (GPX1, GPX2, GPX3, GPX4), redox regulation (TXNRD1, TXNRD2, TXNRD3, MSRB1, SELENOH, SELENOM, SELENOW), thyroid hormone metabolism (DIO1, DIO2, DIO3), selenium transport and storage (SELENOP), selenophosphate synthesis (SEPHS2), calcium metabolism (SELENOK, SELENOT), myogenesis (SELENON), protein folding (SELENOF, SELENOI, SELENOS), and protein AMPylation (SELENOO)47,48. The functions of other selenoproteins such as GPX6 and SELENOV still remain unclear. Glutathione peroxidases (GPXs) such as GPX1 (cytosolic GPX), GPX2 (gastrointestinal GPX), and GPX4 (phospholipid hydroperoxide GPX) catalyze the decomposition of a great variety of peroxides, thus protecting cells against oxidative damage49,50. Thioredoxin reductases (TXNRDs) employ NADPH as an electron donor to revert oxidized TXN to a reduced dithiol, the oxidation status of which is critically implicated in regulating various cell behaviors including proliferation and apoptosis51. The physiological significance of TXNRDs is further supported by the embryonic lethality of Txnrd1 or Txnrd2 knockout mice52,53. Deiodinases (DIOs) regulate thyroid hormone metabolism by catalyzing the conversion of thyroid hormones from precursor thyroxine (T4) to biologically active triiodothyronine (T3) or inactive reverse T3 (rT3)54. The expression levels of several selenoproteins are influenced by the extent of selenium uptake. For example, selenium-deficient animals and human cell lines exhibit reduced transcription of selenoproteins such as GPX1, DIOs, SELENOI, and SELENOW55,56,57. A subset of selenoproteins such as GPX1 and SELENOW is more sensitive to selenium supplementation or deficiency. The hierarchy of selenoprotein expression is more apparent when the intracellular level of selenium is limited1.
Selenium-responsive genes
Selenium-responsive genes are the genes whose expression patterns are influenced by supplementation with selenium or selenium-containing compounds. Treatment of a cancer cell line with methylseleninic acid induced expression changes in 951 genes58. These responsive genes were closely associated with annotations related to cell cycle regulation, androgen-responsive genes, and phase II detoxification pathway. Selenium supplementation of macrophages diminished the expression of lipopolysaccharide (LPS)-induced pro-inflammatory genes such as cyclooxygenase-2 (COX-2) and tumor necrosis factor-α (TNF-α)59, suggesting that selenium has anti-inflammatory effects on the immune system. The CTD database (http://ctdbase.org/) reports the effect of environmental chemicals including selenium on gene expression profiles in various human tissues.
The role of selenium and selenoproteins in cartilage development and KBD
Selenium levels and its role in joint tissues
Joints are composed of various types of connective tissues including cartilage, bone, synovium, meniscus, and ligament. Among these tissues, cartilage is the main component that absorbs mechanical stress, cushioning bones from impacting each other during various weight-bearing activities. In the human knee joint, the selenium concentration in cartilage is approximately 80 μg/kg dry weight, whereas the selenium concentrations in ligament and meniscus are 270 and 307 μg/kg dry weight, respectively60,61. The requirement of adequate physiological selenium levels for maintaining cartilage homeostasis has been recognized. Selenium deficiency retards the growth and development of cartilage and bone62,63,64,65,66. Growth retardation was observed in rats after two generations of selenium deficiency62. Mice fed a diet deficient in selenium resulted in fibrocartilage formation at the articular surface, ultimately showing degeneration of articular cartilage63. Selenium deficiency induced the expression of the chondrocyte hypertrophy marker gene type X collagen (COLX) in articular cartilage64. The expression of parathyroid hormone-related protein (PTHrP), which controls chondrocyte maturation during endochondral ossification, was enhanced in both articular cartilage and hypertrophic growth plate following selenium deficiency. These changes were in line with the phenotypic changes observed in the cartilage of KBD patients64. However, it should be noted that growth retardation caused by selenium deficiency may also be associated with the deregulation of bone metabolism65. In a study by Cao et al., selenium deficiency severely compromised bone microarchitecture as a result of increased bone resorption66.
Abnormalities in selenium metabolism and skeletal development diseases
Selenium deficiency is regarded as one of the initiating factors of KBD, which is an endemic osteoarthropathy caused by the premature closure of epiphyseal plate and the impaired skeletal development. Skeletal deformities in hands, fingers, knees, and elbows, and in severe cases, dwarfism and movement disorders are the symptoms of KBD22. The KBD area roughly coincides with low-selenium areas including a geological belt extending from northeast to southwest China, North Korea, and eastern Siberia22. A meta-analysis showed that selenium levels in the water, soil, cereal, and corn in KBD endemic regions were lower than they were in non-endemic regions, supporting the fact that the level of selenium in tissue is predominantly affected by dietary intake23. In line with this finding, selenium levels in the whole blood, serum, hair, and urine of KBD patients were markedly lower than those of healthy controls24.
Selenoprotein gene polymorphisms are associated with increased susceptibility to KBD. There were significant differences in the allelic frequency of GPX1 Pro198Leu (rs1050450) between the KBD and control group67. In addition, the mRNA level of GPX1 and enzyme activity of total GPX in blood were lower in the KBD group than they were in the control group67. Haplotypes of TCC, TTC, and TTT of rs1050450, rs3811699, and rs1800668 in GPX1 gene also had a significant link to KBD68. A single-nucleotide polymorphism (SNP) in the promoter region of SELENOS rs28665122 (−105G > A) was related to the increased risk of KBD and upregulation of PI3K/Akt signaling in patients with KBD69. In this study, tert-butyl hydroperoxide (tBHP) treatment-induced chondrocyte apoptosis was mitigated by selenium supplementation via sodium selenite treatment, which suppressed the PI3K/Akt pathway. The minor A-allele of SELENOF rs5859 was associated with a significantly higher incidence of KBD70.
The animals fed a selenium-deficient diet recapitulated some of the pathological manifestations of KBD, strongly supporting the notion that selenium deficiency is critically associated with the development of this endemic arthropathy. Selenium deficiency impaired bone and cartilage growth with the exhibition of premature chondrocyte hypertrophy as evidenced by an increased expression of COLX, compatible with the phenotypes in KBD cartilage64. The low-selenium condition in combination with three mycotoxins, deoxynivalenol (DON), nivalenol (NIV), and T-2, yielded pro-catabolic changes and hypertrophic phenotype of chondrocytes, as evidenced by the loss of aggrecan and type II collagen (COLII) and the increase in COLX and matrix metalloproteinases (MMPs) expression, respectively71. In contrast, selenium supplementation partially alleviated these mycotoxin-induced damages in chondrocytes71. In rats, dietary selenium deficiency over two generations caused the onset of physiological selenium insufficiency72. In this condition, pathological changes in the epiphyseal plate were observed with the decreased expression of COLII and GPX1 in the chondrocytes, suggesting a possible association of reduced chondrocyte anabolism and antioxidant capacity with the epiphyseal plate lesions observed in KBD72. The relevance of impaired selenium metabolism to the onset of KBD was further validated using a mouse genetic deletion model. Targeted deletion of Sec-tRNA[Ser]Sec (Trsp) gene in osteochondroprogenitor cells from embryonic stage caused the depletion of selenoproteins in skeletal systems, causing growth retardation, abnormalities in the epiphyseal growth plate, delayed endochondral ossification, and chondronecrosis, which recapitulated the major pathological features of KBD73.
As a prophylactic treatment, selenium supplementations were given to children living in a KBD area. The supplemented group showed elevated physiological selenium levels in their hair samples and exhibited a substantially lower prevalence of KBD74. A meta-analysis including five randomized controlled trials (RCTs) and ten prospective non-RCTs statistically demonstrated the benefits of selenium supplementation in preventing KBD in children75.
Selenium metabolism and OA
Physiological significance of oxidative stress in chondrocytes
OA is the most common form of arthritis and is primarily characterized by the loss of cartilage-specific ECM and other pathological changes in joints including subchondral bone sclerosis, osteophyte formation, and synovial inflammation31. Articular cartilage is composed of abundant proteoglycans in which sulfated glycosaminoglycan chains such as chondroitin sulfates are bound to a core protein such as aggrecan. Loss of cartilage matrix during OA progression is a combined result of increased catabolic process in cartilage and reduced anabolic activity of chondrocytes. The molecular-level understanding of OA pathogenesis has led to the identification of major catabolic enzymes, ADAMTS576, MMP377, and MMP1378, which mediate the degradation of cartilage matrix. Pro-inflammatory cytokines drive the expression of these catabolic factors in chondrocytes through the activation of transcription factors such as HIF-2α32 and NF-κB79. Abnormalities in various metabolic pathways such as glucose80 or amino acid metabolic system81 in chondrocytes have been implicated in activating catabolic cascades in osteoarthritic cartilage82. Moreover, increased cellular uptake of Zn2+ through the upregulation of zinc transporter ZIP8 activates metal-regulatory transcription factor-1 (MTF1), which in turn induces the expression of matrix-degrading enzymes in chondrocytes33,83. Regulation of catabolism by the cholesterol–CH25H–CYP7B1–RORα axis further showed the association of metabolic abnormalities with the catabolic process of OA34.
Meanwhile, the upstream regulatory mechanism eliciting an imbalance in OA matrix homeostasis needs further investigation. OA-causing factors such as age and mechanical stress lead to excessive oxidative stress in chondrocytes37,38. Consistently, clinical and preclinical OA studies indicated a cumulative oxidative burden in osteoarthritic chondrocytes39,40. Emerging evidence suggests that oxidative stress plays a significant role in OA development and the disease progression can be mitigated by counteracting oxidative stress36,84,85,86. In general, oxidative stress results from the abnormal production of ROS and the loss of cellular antioxidant capacity. Synovial fluid from patients with late-stage OA who were undergoing knee joint replacement had a lower level of oxidoreductases than that from healthy controls87. In part, the increase in oxidative stress is attributable to mitochondrial dysfunction in OA chondrocytes88,89. OA chondrocytes displayed reduced mitochondrial DNA content, mitochondrial dysfunction, and diminished expression of NRF2 which regulates the transcription of oxidoreductase genes89. Similarly, chondrocytes from aged individuals exhibited increased ROS burden and mitochondrial and genomic DNA damage90,91,92. Therefore, the proper maintenance of redox homeostasis can potentially serve as a rational therapeutic strategy to protect against OA progression.
Potential roles of selenium metabolism in OA
The protective effect of selenium in OA has been explored in a large number of epidemiological and genetic studies (Table 1). The concentration of selenium in serum was significantly lower in OA patients than that of normal controls25. Similarly, the results from a population-based cohort study demonstrated the linkage between low-selenium levels in toenails with OA-associated pain and disease severity26,27. Several studies have indicated that cartilage matrix homeostasis is impaired in selenium deficiency. Low-selenium status diminished COLII expression level regulated by SOX9, which is known as a master regulator required for maintaining cartilage matrix homeostasis. In fact, SOX9 was destabilized by the downregulation of selenium-responsive PRMT5 that sustains SOX9 stability via methylation93. In another study, rats fed a selenium-deficient diet exhibited low sulfotransferase activity, which resulted in diminished contents of sulfated glycosaminoglycan essential for mechanical stress-absorbing property of cartilage matrix28. In contrast, selenium supplementation ameliorated the spontaneous degeneration of articular cartilage in STR/1 N mice by increasing the expression of GPXs94. In cultured chondrocytes, pretreatment with SeMet markedly inhibited nitric oxide (NO) and prostaglandin E2 (PGE2) production in response to pro-inflammatory cytokine IL-1β95. Expression of SBP2, a factor recognizing SECIS element, had a positive correlation with GPX1 and GPX4 expression and antioxidant capacity in chondrocytes96. Oxidation resistance mediated by SBP2 was diminished in response to IL-1β treatment in vitro and in damaged regions of cartilage in OA patients96. Downregulation of selenoprotein mRNAs including GPX397, GPX1, and GPX496,98, and Selenop99 was observed in human and mouse OA chondrocytes.
Genetic factors such as SNPs in selenoproteins were identified to be risk factors for OA development. A GAG haplotype in SELENOS gene was significantly associated with increased levels of inflammatory factors in OA patient plasma100. SNPs in DIO2, which converts precursor thyroid hormone T4 to its active form T3, were also related to genetic susceptibility to OA development. Levels of DIO2 mRNA and protein were markedly upregulated in OA cartilage101. A common DIO2 haplotype composed of the minor C-allele of SNP rs225014 and the common C-allele of SNP rs12885300 was significantly associated with advanced hip OA, as indicated by a higher odds ratio101,102,103. Locus rs225014, which confers risk to OA, was associated with the differential methylation of CpG located in the upstream region of DIO2 gene and was correlated with upregulated DIO2 expression in OA104. Meanwhile, DIO3 depletes the resources that can be utilized for the production of active thyroid molecules by catalyzing the conversion of T4 and T3 into inactive metabolites. The minor G-allele of the DIO3 variant rs945006 was associated with a protective effect against OA development105.
However, a few aspects regarding the relationship between selenium and OA remain controversial. First, several studies indicate that there are no differences in selenium levels between OA and normal tissues. The selenium concentrations in synovial fluid and plasma of 25 OA patients were not significantly different from those of 25 healthy controls106. Similarly, no significant difference in selenium concentration was noted between six dogs with post-traumatic OA and six control dogs107. Second, the beneficial effect of selenium supplementation in alleviating OA symptoms has been debated. The results from a controlled double-blind trial of 30 patients revealed that the supplementation of a formulation containing selenium with vitamins A, C, and E (Se-ACE) did not have any remarkable curative effect compared to a placebo108. In a study with an independent cohort, the prevalence of radiographic knee OA was not significantly associated with dietary selenium intake109.
Nonetheless, it is apparent that selenium deficiency, dysregulation of selenoproteins, and genetic variations in selenoprotein genes serve as potential risk factors for OA. The vital role of selenium metabolism in maintaining cartilage homeostasis is expected, considering its critical involvement in regulating cellular processes such as chondrogenic differentiation of progenitor cells, maintenance of redox homeostasis and DNA damage repair in chondrocytes, which are covered in the next section.
Intracellular roles of selenium metabolism and selenoproteins in cartilage
Chondrogenic differentiation programs of progenitor cells
Selenium exerts various beneficial effects to promote proliferation and differentiation of chondrogenic progenitor cells110,111. Selenium supplementation stimulated the proliferation of ATDC5 chondrogenic cells even under serum deprivation by inducing cyclin D1 expression110. Deficiency of SELENOO interfered with the chondrogenic differentiation of ATDC5 cells by suppressing the expression of chondrogenic genes SOX9, COLII, and aggrecan and decreasing the activity of alkaline phosphatase112. Knockdown of Gpx1 reduced the chondrogenic differentiation of ATDC5 cells by modulating intracellular GSH/oxidized GSH (GSSG) ratio113. Selenop was differentially upregulated during the chondrogenic differentiation of micromass culture of mesenchymal cells isolated from mouse limb buds114. In line with the effects of selenium metabolism and selenoproteins in chondrogenic progenitor cells observed in vitro, deficient uptake of selenium severely affected chondrogenic differentiation of mesenchymal lineage cells and thus endochondral ossification in mice64. Osteochondroprogenitor-specific deletion of Trsp gene significantly impaired chondrogenic programs, causing abnormalities in bone and cartilage development in mice73.
Antioxidant defense and redox homeostasis
The protective effects of selenium on cartilage are primarily attributed to the function of antioxidant defense115,116,117. The metabolism and survival of chondrogenic progenitors and chondrocytes are greatly compromised by ROS including free radicals, peroxides, and superoxide anions118,119,120. Recent studies strongly support the notion that mitochondrial dysfunction and oxidative stress are the main drivers of OA pathogenesis37. Although ROS play essential roles in the maintenance of basal cellular activities such as chondrocyte proliferation and matrix remodeling in cartilage, excessive oxidative stress causes detrimental events such as cellular senescence36,121, dedifferentiation122, and apoptosis123. ROS cause oxidative damage to various cellular components and disrupt the balance between ECM catabolism and anabolism119. ROS suppress mitochondrial oxidative phosphorylation and ATP production, which are essentially required to sustain cartilage matrix synthesis124. In addition, ROS induce matrix degeneration through the upregulation of matrix-degrading enzyme expression while this effect is abolished by antioxidant treatment123,125. The detrimental effects of ROS on cartilage homeostasis can be effectively alleviated by augmenting cellular antioxidant activity under stress conditions, and several attempts have been made to treat OA by targeting the regulators involved in oxidative stress production in cartilage84,85,86.
The protective role of selenium metabolism is thought to be exerted through the neutralization of ROS via antioxidant activities of selenoproteins including GPXs and TXNRDs. Bone marrow stromal cells cultured in medium supplemented with low selenite concentration exhibited ROS accumulation along with the reduced expression of GPXs, TXNRDs, and other selenium-independent oxidoreductase enzymes, resulting in micronuclei generation which is an indication of chromosome damage126. Both GPX1 expression and activity were substantially lower in mice fed a selenium-deficient diet than those in mice fed a normal diet, leading to decreased trabecular number, reduced femoral trabecular volume/total bone volume ratio, and trabecular separation66. The rats exposed to a selenium-deficient diet with T-2 toxin showed increased lipid peroxidation level and decreased antioxidant GPX activity in their serum and cartilage127. A selenium-deficient diet induced the expression of miR-138-5p, which in turn suppressed the expression of SELENOM that has antioxidant function, and caused mitochondrial dysfunction and apoptosis of chondrocytes128. Lead (Pb)-induced oxidative stress and toxicity reduced the expression of selenoprotein mRNAs, and the effect was mitigated by selenium supplementation129. In summary, the antioxidant properties of selenoproteins showed therapeutic potential by counteracting the accumulation of damage induced by oxidative stress in cartilage.
DNA damage repair
It is well known that DNA damage pathways play substantial roles in the progression of arthropathies119. The expression of genes related to DNA damage was changed in the cartilage of KBD patients130,131. Chronic DNA damage induces the initiation of apoptosis or cellular senescence in chondrocytes36,132,133. Selenium has a potential to reduce DNA damage and increase DNA repair capacity134. In part, the beneficial effect of selenium on genomic stability is associated with the antioxidation effect of selenoproteins such as GPXs and TXNRDs, which remove ROS before they cause DNA damage134. Cancer cells supplemented with selenium (30 nM sodium selenite or 10 μM SeMet) showed elevated levels of GPX1 and TXNRD1 enzyme activity, effectively protecting against DNA strand breaks induced by ultraviolet A- or H2O2-induced oxidative stress135. SeMet reduced the extent of DNA damage and enhanced DNA repair capacity by inducing repair complex formation in DNA-damaged cells through UV radiation exposure136. SeMet treatment elevated the levels of p53 and REF1 proteins and induced their interaction with BRCA1137, resulting in the activation of DNA repair pathways. Cells treated with SeMet showed significantly enhanced DNA repair capacity under exposure to various DNA damaging agents such as UV radiation or cisplatin treatment138.
Conclusion and perspectives: selenium metabolism-based therapeutic strategies for treating arthropathies
This review discussed the roles of selenium metabolism in cartilage development and arthropathies such as KBD and OA, and highlighted its crucial functions in maintaining cartilage homeostasis. Considering the physiological role of selenoproteins in antioxidation and the detrimental effects of oxidative stress in chondrocytes, aberrant selenium metabolism is likely to disrupt cartilage homeostasis and cause arthropathic diseases via dysregulation of redox homeostasis (Fig. 2). Besides the protective role against oxidative stress, selenium appears to exert pro-anabolic effects to augment the regeneration capacity of cartilage. Selenium is essentially required to induce the proliferation and chondrogenic differentiation of mesenchymal stem cells. However, the mechanisms by which selenium metabolism regulates chondrogenic programs remain still unclear and require further investigation. Despite the overall beneficial effects of selenium in maintaining cartilage homeostasis, strategies to supplement selenium or selenoproteins should be considered with care to avoid adverse health effects such as selenosis. Strategies aimed at optimizing the benefits of selenium and selenoproteins should be considered for the therapeutic treatment and prevention of arthropathies.

Oxidative stress induced by systemic and local factors causes arthropathies, and selenoproteins play protective roles in the maintenance of redox homeostasis. OA, one of the most common forms of arthropathies, is related to multiple etiologies involving systemic factors and local factors such as aging, metabolic abnormality, and mechanical stress associated with overuse, injury, and misalignment. These OA-causing factors disrupt the balance between ROS production and antioxidation, thereby resulting in accumulation of oxidative stress. The dysregulation of redox homeostasis causes the disruption of cartilage homeostasis and leads to the development of arthropathies such as KBD and OA. Restoring redox homeostasis through the activation of selenium metabolism and supplementation with selenoproteins can be a rational therapeutic strategy to treat arthropathies.
References
Hatfield, D. L. & Gladyshev, V. N. How selenium has altered our understanding of the genetic code. Mol. Cell Biol. 22, 3565–3576 (2002).
Rayman, M. P. Selenium and human health. Lancet 379, 1256–1268 (2012).
Arthur, J. R., McKenzie, R. C. & Beckett, G. J. Selenium in the immune system. J. Nutr. 133, 1457S–1459S (2003).
Schomburg, L. Selenium, selenoproteins and the thyroid gland: interactions in health and disease. Nat. Rev. Endocrinol. 8, 160–171 (2011).
Hawkes, W. C., Kelley, D. S. & Taylor, P. C. The effects of dietary selenium on the immune system in healthy men. Biol. Trace Elem. Res. 81, 189–213 (2001).
Wood, S. M., Beckham, C., Yosioka, A., Darban, H. & Watson, R. R. beta-Carotene and selenium supplementation enhances immune response in aged humans. Integr. Med. 2, 85–92 (2000).
Broome, C. S. et al. An increase in selenium intake improves immune function and poliovirus handling in adults with marginal selenium status. Am. J. Clin. Nutr. 80, 154–162 (2004).
Nève, J. Selenium as a risk factor for cardiovascular diseases. J. Cardiovascular Risk 3, 42–47 (1996).
Alissa, E. M., Bahijri, S. M. & Ferns, G. A. The controversy surrounding selenium and cardiovascular disease: a review of the evidence. Med. Sci. Monitor 9, RA9–RA18 (2003).
Zhang, X., Liu, C., Guo, J. & Song, Y. Selenium status and cardiovascular diseases: meta-analysis of prospective observational studies and randomized controlled trials. Eur. J. Clin. Nutr. 70, 162–169 (2016).
Chen, X. et al. Studies on the relations of selenium and Keshan disease. Biol. Trace Elem. Res. 2, 91–107 (1980).
Loscalzo, J. Keshan disease, selenium deficiency, and the selenoproteome. N. Engl. J. Med. 370, 1756–1760 (2014).
Hatfield, D. L., Tsuji, P. A., Carlson, B. A. & Gladyshev, V. N. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem. Sci. 39, 112–120 (2014).
Patrick, L. Selenium biochemistry and cancer: a review of the literature. Alternative Med. Rev. 9, 239–258 (2004).
Willett, W. et al. Prediagnostic serum selenium and risk of cancer. Lancet 322, 130–134 (1983).
Navarro-Alarcon, M., de la Serrana, H. L.-G., Perez-Valero, V. & López-Martı́nez, M. Selenium concentrations in serum of individuals with liver diseases (cirrhosis or hepatitis): relationship with some nutritional and biochemical markers. Sci. Total Environ. 291, 135–141 (2002).
Guo, C. H., Chen, P. C. & Ko, W. S. Status of essential trace minerals and oxidative stress in viral hepatitis C patients with nonalcoholic fatty liver disease. Int. J. Med. Sci. 10, 730–737 (2013).
Davis, C. D., Tsuji, P. A. & Milner, J. A. Selenoproteins and cancer prevention. Annu. Rev. Nutr. 32, 73–95 (2012).
Clark, L. C. et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin: a randomized controlled trial. JAMA 276, 1957–1963 (1996).
Zhuo, H., Smith, A. H. & Steinmaus, C. Selenium and lung cancer: a quantitative analysis of heterogeneity in the current epidemiological literature. Cancer Epidemiol. Biomarkers Prev. 13, 771–778 (2004).
Lawrence, R. A. & Burk, R. F. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem. Biophys. Res. Commun. 71, 952–958 (1976).
Li, S., Cao, J., Caterson, B. & Hughes, C. E. Proteoglycan metabolism, cell death and Kashin-Beck disease. Glycoconj. J. 29, 241–248 (2012).
Wang, Q. et al. Correlation between selenium and Kaschin-Beck disease: a meta-analysis. Clin. J. Evid. Based Med. 13, 1421–1430 (2013).
Yang, L., Zhao, G.-h, Yu, F.-f, Zhang, R.-q & Guo, X. Selenium and iodine levels in subjects with Kashin-Beck disease: a meta-analysis. Biol. Trace Elem. Res. 170, 43–54 (2016).
Wang, L. et al. Serious selenium deficiency in the serum of patients with Kashin-Beck disease and the effect of nano-selenium on their chondrocytes. Biological Trace Elem. Res. https://doi.org/10.1007/s12011-019-01759-7 (2019).
Jordan, J. M. An ongoing assessment of osteoarthritis in African Americans and Caucasians in North Carolina: The Johnston County Osteoarthritis Project. Trans. Am. Clin. Climatol. Assoc. 126, 77–86 (2015).
Jordan, J. et al. 34 Low selenium levels are associated with increased odds of radiographic hip osteoarthritis in African American and white women. Osteoarthr. Cartil. 15, C33 (2007).
Sasaki, S., Iwata, H., Ishiguro, N., Habuchi, O. & Miura, T. Low-selenium diet, bone, and articular cartilage in rats. Nutrition 10, 538–543 (1994).
Raisbeck, M. F. Selenosis. Vet. Clin. North Am.—Food Anim. Pract. 16, 465–480 (2000).
Sutter, M. E., Thomas, J. D., Brown, J. & Morgan, B. Selenium toxicity: a case of selenosis caused by a nutritional supplement. Ann. Intern. Med. 148, 970–971 (2008).
Loeser, R. F., Goldring, S. R., Scanzello, C. R. & Goldring, M. B. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheumatism 64, 1697–1707 (2012).
Yang, S. et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 16, 687–693 (2010).
Kim, J. H. et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell 156, 730–743 (2014).
Choi, W. S. et al. The CH25H-CYP7B1-ROR alpha axis of cholesterol metabolism regulates osteoarthritis. Nature 566, 254 (2019).
Kim, S. et al. Tankyrase inhibition preserves osteoarthritic cartilage by coordinating cartilage matrix anabolism via effects on SOX9 PARylation. Nat. Commun. 10, 4898 (2019).
Kang, D. et al. Stress-activated miR-204 governs senescent phenotypes of chondrocytes to promote osteoarthritis development. Sci. Transl. Med. 11, https://doi.org/10.1126/scitranslmed.aar6659 (2019).
Loeser, R. F., Collins, J. A. & Diekman, B. O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 12, 412 (2016).
Felson, D. T. Osteoarthritis as a disease of mechanics. Osteoarthr. Cartil. 21, 10–15 (2013).
Hui, W. et al. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 75, 449–458 (2016).
Yudoh, K. et al. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res. Ther. 7, R380–R391 (2005).
Hill, K. E., Lloyd, R. S., Yang, J. G., Read, R. & Burk, R. F. The cDNA for rat selenoprotein P contains 10 TGA codons in the open reading frame. J. Biol. Chem. 266, 10050–10053 (1991).
Hill, K. E. et al. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 278, 13640–13646 (2003).
Mc, C. K. & Portman, O. W. Excretion of dimethyl selenide by the rat. J. Biol. Chem. 195, 277–282 (1952).
Byard, J. L. Trimethyl selenide. A urinary metabolite of selenite. Arch. Biochem. Biophys. 130, 556–560 (1969).
Lu, J. & Holmgren, A. Selenoproteins. J. Biol. Chem. 284, 723–727 (2009).
Kryukov, G. V. et al. Characterization of mammalian selenoproteomes. Science 300, 1439–1443 (2003).
Pitts, M. W. & Hoffmann, P. R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 70, 76–86 (2018).
Sreelatha, A. et al. Protein AMPylation by an evolutionarily conserved pseudokinase. Cell 175, 809–821 e819 (2018).
Brigelius-Flohe, R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 387, 1329–1335 (2006).
Ingold, I. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422. e421 (2018).
Arnér, E. S. & Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–6109 (2000).
Conrad, M. et al. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell Biol. 24, 9414–9423 (2004).
Jakupoglu, C. et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol. Cell Biol. 25, 1980–1988 (2005).
Gereben, B. et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. 29, 898–938 (2008).
Liang, Y. et al. Effect of selenium on selenoprotein expression in the adipose tissue of chickens. Biol. Trace Elem. Res. 160, 41–48 (2014).
Christensen, M. J. & Burgener, K. W. Dietary selenium stabilizes glutathione peroxidase mRNA in rat liver. J. Nutr. 122, 1620–1626 (1992).
Avissar, N., Kerl, E. A., Baker, S. S. & Cohen, H. Extracellular glutathione peroxidase mRNA and protein in human cell lines. Arch. Biochem. Biophys. 309, 239–246 (1994).
Zhao, H., Whitfield, M. L., Xu, T., Botstein, D. & Brooks, J. D. Diverse effects of methylseleninic acid on the transcriptional program of human prostate cancer cells. Mol. Biol. Cell 15, 506–519 (2004).
Vunta, H. et al. Selenium attenuates pro‐inflammatory gene expression in macrophages. Mol. Nutr. Food Res. 52, 1316–1323 (2008).
Kosik-Bogacka, D. I. et al. Effects of biological factors and health condition on mercury and selenium concentrations in the cartilage, meniscus and anterior cruciate ligament. J. Trace Elem. Med. Biol. 44, 201–208 (2017).
Bissardon, C. et al. Sub-ppm level high energy resolution fluorescence detected X-ray absorption spectroscopy of selenium in articular cartilage. Analyst 144, 3488–3493 (2019).
Thompson, K. M., Haibach, H. & Sunde, R. A. Growth and plasma triiodothyronine concentrations are modified by selenium deficiency and repletion in second-generation selenium-deficient rats. J. Nutr. 125, 864–873 (1995).
Yang, C., Wolf, E., Roser, K., Delling, G. & Muller, P. K. Selenium deficiency and fulvic acid supplementation induces fibrosis of cartilage and disturbs subchondral ossification in knee joints of mice: an animal model study of Kashin-Beck disease. Virchows Arch. A Pathol. Anat. Histopathol. 423, 483–491 (1993).
Ren, F. L. et al. Effects of selenium and iodine deficiency on bone, cartilage growth plate and chondrocyte differentiation in two generations of rats. Osteoarthr. Cartil. 15, 1171–1177 (2007).
Moreno-Reyes, R., Egrise, D., Neve, J., Pasteels, J. L. & Schoutens, A. Selenium deficiency-induced growth retardation is associated with an impaired bone metabolism and osteopenia. J. Bone Miner. Res. 16, 1556–1563 (2001).
Cao, J. J., Gregoire, B. R. & Zeng, H. Selenium deficiency decreases antioxidative capacity and is detrimental to bone microarchitecture in mice. J. Nutr. 142, 1526–1531 (2012).
Xiong, Y. M. et al. Association study between polymorphisms in selenoprotein genes and susceptibility to Kashin-Beck disease. Osteoarthr. Cartil. 18, 817–824 (2010).
Huang, L. et al. Association study of polymorphisms in selenoprotein genes and kashin–beck disease and serum selenium/iodine concentration in a tibetan population. PLoS ONE 8, e71411 (2013).
Du, X. et al. Role of selenoprotein S (SEPS1)-105G> A polymorphisms and PI3K/Akt signaling pathway in Kashin-Beck disease. Osteoarthr. Cartil. 23, 210–216 (2015).
Wu, R. et al. The study on polymorphisms of sep15 and TrxR2 and the expression of AP-1 signaling pathway in Kashin-Beck disease. Bone 120, 239–245 (2019).
Lu, M. L. et al. The effects of mycotoxins and selenium deficiency on tissue-engineered cartilage. Cells Tissues Organs 196, 241–250 (2012).
Min, Z. et al. Abnormality of epiphyseal plate induced by selenium deficiency diet in two generation DA rats. Apmis 123, 697–705 (2015).
Downey, C. M. et al. Osteo-chondroprogenitor-specific deletion of the selenocysteine tRNA gene, Trsp, leads to chondronecrosis and abnormal skeletal development: a putative model for Kashin-Beck disease. PLoS Genet. 5, e1000616 (2009).
Guo, X. et al. Recent advances in the research of an endemic osteochondropathy in China: Kashin-Beck disease. Osteoarthr. Cartil. 22, 1774–1783 (2014).
Zou, K., Liu, G., Wu, T. & Du, L. Selenium for preventing Kashin-Beck osteoarthropathy in children: a meta-analysis. Osteoarthr. Cartil. 17, 144–151 (2009).
Glasson, S. S. et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434, 644–648 (2005).
Blom, A. B. et al. Crucial role of macrophages in matrix metalloproteinase-mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis Rheumatism 56, 147–157 (2007).
Little, C. B. et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheumatism 60, 3723–3733 (2009).
Marcu, K. B. et al. NF-kappa B signaling: multiple angles to target OA. Curr. Drug Targets 11, 599–613 (2010).
Berenbaum, F. Diabetes-induced osteoarthritis: from a new paradigm to a new phenotype. Ann. Rheum. Dis. 70, 1354–1356 (2011).
Choi, W. S. et al. Critical role for arginase II in osteoarthritis pathogenesis. Ann. Rheum. Dis. 78, 421–428 (2019).
Mobasheri, A. et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 13, 302–311 (2017).
Won, Y. et al. Pleiotropic roles of metallothioneins as regulators of chondrocyte apoptosis and catabolic and anabolic pathways during osteoarthritis pathogenesis. Ann. Rheum. Dis. 75, 2045–2052 (2016).
Matsuzaki, T. et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci. Transl. Med. 10, https://doi.org/10.1126/scitranslmed.aan0746 (2018).
Cornelis, F. M. F. et al. ANP32A regulates ATM expression and prevents oxidative stress in cartilage, brain, and bone. Sci. Transl. Med. 10, https://doi.org/10.1126/scitranslmed.aar8426 (2018).
Coleman, M. C. et al. Targeting mitochondrial responses to intra-articular fracture to prevent posttraumatic osteoarthritis. Sci. Transl. Med. 10, https://doi.org/10.1126/scitranslmed.aan5372 (2018).
Regan, E. A., Bowler, R. P. & Crapo, J. D. Joint fluid antioxidants are decreased in osteoarthritic joints compared to joints with macroscopically intact cartilage and subacute injury. Osteoarthr. Cartil. 16, 515–521 (2008).
Blanco, F. J., Lopez-Armada, M. J. & Maneiro, E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion 4, 715–728 (2004).
Wang, Y., Zhao, X., Lotz, M., Terkeltaub, R. & Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol. 67, 2141–2153 (2015).
McCulloch, K., Litherland, G. J. & Rai, T. S. Cellular senescence in osteoarthritis pathology. Aging Cell 16, 210–218 (2017).
Akhmedov, A. T. & Marin-Garcia, J. Mitochondrial DNA maintenance: an appraisal. Mol. Cell Biochem 409, 283–305 (2015).
Goyns, M. H. Genes, telomeres and mammalian ageing. Mech. Ageing Dev. 123, 791–799 (2002).
Sun, M. et al. Maintenance of SOX9 stability and ECM homeostasis by selenium-sensitive PRMT5 in cartilage. Osteoarthr. Cartil. 27, 932–944 (2019).
Kurz, B., Jost, B. & Schünke, M. Dietary vitamins and selenium diminish the development of mechanically induced osteoarthritis and increase the expression of antioxidative enzymes in the knee joint of STR/1N mice. Osteoarthr. Cartil. 10, 119–126 (2002).
Cheng, A. W., Stabler, T. V., Bolognesi, M. & Kraus, V. B. Selenomethionine inhibits IL-1beta inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX2) expression in primary human chondrocytes. Osteoarthr. Cartil. 19, 118–125 (2011).
Xue, J. et al. The hsa-miR-181a-5p reduces oxidation resistance by controlling SECISBP2 in osteoarthritis. BMC Musculoskelet. Disord. 19, 355 (2018).
Aigner, T. et al. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheumatism 54, 3533–3544 (2006).
Hosseinzadeh, A., Jafari, D., Kamarul, T., Bagheri, A. & Sharifi, A. M. Evaluating the protective effects and mechanisms of diallyl disulfide on interlukin-1beta-induced oxidative stress and mitochondrial apoptotic signaling pathways in cultured chondrocytes. J. Cell Biochem. 118, 1879–1888 (2017).
Bateman, J. F. et al. Transcriptomics of wild-type mice and mice lacking ADAMTS-5 activity identifies genes involved in osteoarthritis initiation and cartilage destruction. Arthritis Rheumatism 65, 1547–1560 (2013).
Bos, S. et al. The role of plasma cytokine levels, CRP and Selenoprotein S gene variation in OA. Osteoarthr. Cartil. 17, 621–626 (2009).
Bos, S. D. et al. Increased type II deiodinase protein in OA-affected cartilage and allelic imbalance of OA risk polymorphism rs225014 at DIO2 in human OA joint tissues. Ann. Rheumatic Dis. 71, 1254–1258 (2012).
Waarsing, J. H. et al. Osteoarthritis susceptibility genes influence the association between hip morphology and osteoarthritis. Arthritis Rheumatism 63, 1349–1354 (2011).
Meulenbelt, I. et al. Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum. Mol. Genet. 17, 1867–1875 (2008).
Bomer, N. et al. Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann. Rheum. Dis. 74, 1571–1579 (2015).
Meulenbelt, I. et al. Meta-analyses of genes modulating intracellular T3 bio-availability reveal a possible role for the DIO3 gene in osteoarthritis susceptibility. Ann. Rheum. Dis. 70, 164–167 (2011).
Yazar, M., Sarban, S., Kocyigit, A. & Isikan, U. Synovial fluid and plasma selenium, copper, zinc, and iron concentrations in patients with rheumatoid arthritis and osteoarthritis. Biol. Trace Elem. Res. 106, 123–132 (2005).
de Oliveira El-Warrak, A., Rouma, M., Amoroso, A., Boysen, S. R. & Chorfi, Y. Measurement of vitamin A, vitamin E, selenium, and L-lactate in dogs with and without osteoarthritis secondary to ruptured cranial cruciate ligament. Can. Vet. J. 53, 1285 (2012).
Hill, J. & Bird, H. Failure of selenium-ace to improve osteoarthritis. Rheumatology 29, 211–213 (1990).
Li, H. et al. Associations between dietary antioxidants intake and radiographic knee osteoarthritis. Clin. Rheumatol. 35, 1585–1592 (2016).
Yan, J. D., Tian, J., Zheng, Y. W., Han, Y. & Lu, S. M. Selenium promotes proliferation of chondrogenic cell ATDC5 by increment of intracellular ATP content under serum deprivation. Cell Biochem. Funct. 30, 657–663 (2012).
Ahmed, H. H., Aglan, H. A., Mabrouk, M., Abd-Rabou, A. A. & Beherei, H. H. Enhanced mesenchymal stem cell proliferation through complexation of selenium/titanium nanocomposites. J. Mater. Sci. Mater. Med. 30, 24 (2019).
Yan, J. D., Fei, Y., Han, Y. & Lu, S. M. Selenoprotein O deficiencies suppress chondrogenic differentiation of ATDC5 cells. Cell Biol. Int. 40, 1033–1040 (2016).
Yan, J. et al. GPx1 knockdown suppresses chondrogenic differentiation of ATDC5 cells through induction of reductive stress. Acta Biochim. Biophys. Sin. (Shanghai) 49, 110–118 (2017).
James, C. G., Appleton, C. T. G., Ulici, V., Underhill, T. M. & Beier, F. Microarray analyses of gene expression during chondrocyte differentiation identifies novel regulators of hypertrophy. Mol. Biol. Cell 16, 5316–5333 (2005).
Hawkes, W. C. & Alkan, Z. Regulation of redox signaling by selenoproteins. Biol. Trace Elem. Res. 134, 235–251 (2010).
Ramakrishnan, P. et al. Oxidant conditioning protects cartilage from mechanically induced damage. J. Orthop. Res. 28, 914–920 (2010).
Henrotin, Y., Kurz, B. & Aigner, T. Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarthr. Cartil. 13, 643–654 (2005).
Bigarella, C. L., Liang, R. & Ghaffari, S. Stem cells and the impact of ROS signaling. Development 141, 4206–4218 (2014).
Loeser, R. F. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 17, 971–979 (2009).
Vaillancourt, F. et al. 4-Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: the protective role of glutathione-S-transferase. Arthritis Res Ther. 10, R107 (2008).
Brandl, A. et al. Oxidative stress induces senescence in chondrocytes. J. Orthop. Res 29, 1114–1120 (2011).
Cha, B. H., Lee, J. S., Kim, S. W., Cha, H. J. & Lee, S. H. The modulation of the oxidative stress response in chondrocytes by Wip1 and its effect on senescence and dedifferentiation during in vitro expansion. Biomaterials 34, 2380–2388 (2013).
Henrotin, Y. E., Bruckner, P. & Pujol, J. P. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthr. Cartil. 11, 747–755 (2003).
Johnson, K. et al. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheumatism 43, 1560–1570 (2000).
Tiku, M. L., Gupta, S. & Deshmukh, D. R. Aggrecan degradation in chondrocytes is mediated by reactive oxygen species and protected by antioxidants. Free Radic. Res. 30, 395–405 (1999).
Ebert, R. et al. Selenium supplementation restores the antioxidative capacity and prevents cell damage in bone marrow stromal cells in vitro. Stem Cells 24, 1226–1235 (2006).
Chen, J. H. et al. Oxidant damage in Kashin‐Beck disease and a rat Kashin‐Beck disease model by employing T‐2 toxin treatment under selenium deficient conditions. J. Orthop. Res. 30, 1229–1237 (2012).
Chi, Q., Luan, Y., Zhang, Y., Hu, X. & Li, S. The regulatory effects of miR-138-5p on selenium deficiency-induced chondrocyte apoptosis are mediated by targeting SelM. Metallomics 11, 845–857 (2019).
Gao, H., Liu, C., Song, S. & Fu, J. Effects of dietary selenium against lead toxicity on mRNA levels of 25 selenoprotein genes in the cartilage tissue of broiler chicken. Biol. Trace Elem. Res. 172, 234–241 (2016).
Yu, F.-F. et al. Identified molecular mechanism of interaction between environmental risk factors and differential expression genes in cartilage of Kashin-Beck disease. Medicine 95, e5669 (2016).
Wang, W.-Z. et al. Comparative analysis of gene expression profiles between the normal human cartilage and the one with endemic osteoarthritis. Osteoarthr. Cartil. 17, 83–90 (2009).
Rose, J. et al. DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthr. Cartil. 20, 1020–1028 (2012).
Grishko, V. I., Ho, R., Wilson, G. L. & Pearsall, A. W. Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthr. Cartil. 17, 107–113 (2009).
Baliga, M. S., Wang, H., Zhuo, P., Schwartz, J. L. & Diamond, A. M. Selenium and GPx-1 overexpression protect mammalian cells against UV-induced DNA damage. Biol. Trace Elem. Res. 115, 227–241 (2007).
de Rosa, V. et al. Low doses of selenium specifically stimulate the repair of oxidative DNA damage in LNCaP prostate cancer cells. Free Radic. Res. 46, 105–116 (2012).
Seo, Y. R., Sweeney, C. & Smith, M. L. Selenomethionine induction of DNA repair response in human fibroblasts. Oncogene 21, 3663–3669 (2002).
Fischer, J. L., Lancia, J. K., Mathur, A. & Smith, M. L. Selenium protection from DNA damage involves a Ref1/p53/Brca1 protein complex. Anticancer Res. 26, 899–904 (2006).
Fischer, J. L., Mihelc, E. M., Pollok, K. E. & Smith, M. L. Chemotherapeutic selectivity conferred by selenium: a role for p53-dependent DNA repair. Mol. Cancer Ther. 6, 355–361 (2007).
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (NRF-2015M3A9E6028674, NRF-2016R1A5A1010764, and NRF-2017M3A9D8064193), the Institute for Basic Science from the Ministry of Science, ICT, and Future Planning of Korea (IBS-R008-D1), and Suh Kyungbae Foundation. C.W. was supported by Chey Institute for Advanced Studies—International Scholar Exchange Fellowship 2018–19.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, D., Lee, J., Wu, C. et al. The role of selenium metabolism and selenoproteins in cartilage homeostasis and arthropathies. Exp Mol Med 52, 1198–1208 (2020). https://doi.org/10.1038/s12276-020-0408-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0408-y
This article is cited by
-
PRDX6 augments selenium utilization to limit iron toxicity and ferroptosis
Nature Structural & Molecular Biology (2024)
-
Harmful Free Radicals in Aging: A Narrative Review of Their Detrimental Effects on Health
Indian Journal of Clinical Biochemistry (2024)
-
Comparative Analysis of Differentially Expressed Genes in Chondrocytes from Rats Exposed to Low Selenium and T-2 Toxin
Biological Trace Element Research (2024)
-
Selenium-SelK-GPX4 axis protects nucleus pulposus cells against mechanical overloading-induced ferroptosis and attenuates senescence of intervertebral disc
Cellular and Molecular Life Sciences (2024)
-
Products of Selenite/Thiols Interaction Have Reducing Properties, Cleave Plasmid DNA and Decrease Rat Blood Pressure and Tension of Rat Mesenteric Artery
Biological Trace Element Research (2024)
