
Abstract
Damage-associated molecular patterns (DAMPs) are danger signals (or alarmins) alerting immune cells through pattern recognition receptors (PRRs) to begin defense activity. Moreover, DAMPs are host biomolecules that can initiate a noninflammatory response to infection, and pathogen-associated molecular pattern (PAMPs) perpetuate the inflammatory response to infection. Many DAMPs are proteins that have defined intracellular functions and are released from dying cells after tissue injury or chemo-/radiotherapy. In the tumor microenvironment, DAMPs can be ligands for Toll-like receptors (TLRs) expressed on immune cells and induce cytokine production and T-cell activation. Moreover, DAMPs released from tumor cells can directly activate tumor-expressed TLRs that induce chemoresistance, migration, invasion, and metastasis. Furthermore, DAMP-induced chronic inflammation in the tumor microenvironment causes an increase in immunosuppressive populations, such as M2 macrophages, myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs). Therefore, regulation of DAMP proteins can reduce excessive inflammation to create an immunogenic tumor microenvironment. Here, we review tumor-derived DAMP proteins as ligands of TLRs and discuss their association with immune cells, tumors, and the composition of the tumor microenvironment.
Similar content being viewed by others
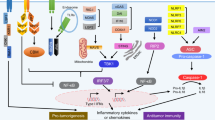
Introduction
Damage-associated molecular patterns (DAMPs), also known as danger-associated molecular patterns, are endogenous molecules released from dying cells that activate the immune system by interacting with pattern recognition receptors (PRRs)1. DAMPs contribute to host defense through inflammation induced by immune cells, though they are also related to autoimmune disease when chronically induced2. After chemotherapy or radiotherapy, tumor-derived DAMPs interact with Toll-like receptors (TLRs) that activate immune cells and produce inflammatory cytokines3. These pro-inflammatory cytokines mediate innate and adaptive immunity through autocrine secretion by immune cells. Moreover, these inflammatory cytokines directly induce tumor-derived secretory proteins that also stimulate paracrine secretion of inflammatory cytokines by immune cells. However, chronic inflammation ultimately increases the population of immunosuppressive cells in the tumor microenvironment and elevates the expression of immune checkpoint molecules that allow tumors to evade immune responses. Recently, it has been reported that tumor-derived DAMP proteins directly increase endothelial-to-mesenchymal transition (EMT), which is responsible for tumor metastasis1,4. Furthermore, TLR-overexpressing tumors have been shown to result in a poor prognosis for cancer patients5. Indeed, tumor-derived DAMP proteins bind to TLRs expressed on tumors and activate downstream signaling that is associated with tumor migration, invasion, and metastasis6,7. Here, we suggest that DAMPs are known to be associated with immune cells, but they could be important mediators between immune cells and tumors because tumor-derived secretory proteins can induce the production of pro-inflammatory cytokines by immune cells and directly increase the aggressive characteristics of tumors through TLRs (Fig. 1).

Anticancer drugs and irradiation induce apoptosis or necrosis in tumor cells. Several DAMP proteins (HMGB1, S100, HSP, API5, PAUF, RPS3, etc.) released from dying tumor cells stimulate immune cells or tumor cells after binding Toll-like receptors expressed on both of these cell types. Activated mature immune cells such as dendritic cells initiate adaptive immunity by educating tumor-specific T cells and secreting pro-inflammatory cytokines (TNF-α, IL-6, IL-12, IL-1β, IFN-β, etc.) that induce the secretion of DAMP proteins from tumor cells. TLR axis signaling in tumor cells induces the transcription of genes related to cell proliferation, chemotherapeutic resistance, invasion, and metastasis in tissues (e.g., lung tissue). In addition, tumors usually escape the immune response by altering the tumor microenvironment with immunosuppressive populations (Treg cells, MDSCs, M2 macrophages, etc.) and secretion of anti-inflammatory cytokines (IL-10, TGF-β, etc.).
DAMP proteins released from dying cells
DAMPs are danger signals that are released from dying or necrotic cells and are also actively secreted from cells by external stimulation (e.g., high-mobility group box 1 (HMGB1) protein)8,9,10,11. Chemotherapy or radiotherapy usually induces the release of DAMP proteins following tumor apoptosis or necrosis12,13,14,15. Tumor-derived DAMP proteins are expressed in various tumor types (Table 1). As a well-known DAMP molecule, HMGB1 can be passively released from damaged cells or actively secreted from activated immune cells through a leaderless secretory pathway16,17,18,19. Tang et al.19 reported that HMGB1, present in almost all metazoans and plants and first identified as a chromatin-associated protein, is a highly conserved nuclear protein that acts as a chromatin-binding factor that binds DNA and facilitates the assembly of transcriptional proteins on specific DNA targets. Moreover, HMGB1 has been implicated in disease states, including sepsis, ischemia-reperfusion, arthritis, meningitis, neurodegeneration, aging, and cancer20,21,22,23,24,25,26. In addition, it is associated with all of the central hallmarks of cancer and is released from tumor cells when induced by chemotherapy or radiotherapy27,28,29. Napolitano et al.30 assessed the serum levels of HMGB1 to reliably distinguish malignant mesothelioma patients, asbestos-exposed individuals, and unexposed controls. Total HMGB1 was significantly increased in malignant mesothelioma patients and asbestos-exposed individuals compared with healthy controls. Naumnik et al.31 investigated the prognostic role and effects of chemotherapy on HMGB1 serum levels in patients with advanced-stage non-small cell lung cancer (NSCLC). After four cycles of chemotherapy, the mean serum HMGB1 levels in peripheral blood samples were significantly higher in patients with advanced NSCLC than in controls. Moreover, it has been shown that a DAMP protein, HMGB1, functions in a manner depending on the oxidative status. Tang et al.32 noted that HMGB1 is integral to oxidative stress and downstream apoptosis or survival. When HMGB1 accumulates at sites of oxidative DNA damage, it can lead to DNA repair. Both reduced and oxidized HMGB1 have been shown to perform different functions in extracellular signaling and regulation of the immune response through Toll-like receptors. HMGB1 released from injured or necrotic cells or secreted from inflammatory cells triggers immune responses after binding receptor for advanced glycation end products (RAGE) or Toll-like receptor (TLR)-2 or TLR4, triggering receptor expressed on myeloid cells-1 (TREM-1) and CD24, which stimulates cell migration, cell proliferation, and cell differentiation.
Bresnick et al.33 reported that S100 protein family members, 21 proteins with a high degree of structural similarity, act as Ca2+ sensors that can translate fluctuations in intracellular Ca2+ levels into a cellular response and participate in a wide range of biological processes, such as proliferation, migration and/or invasion, inflammation, and differentiation. The function of S100 protein family members as DAMPs has been detected in the extracellular space, as well as in serum, urine, sputum, cerebrospinal fluid, and feces, and is generally associated with specific diseases, such as cancer33,34,35,36. The exact secretion mechanism of S100 proteins is still unclear, as they lack a sequence responsible for secretion through the classical endoplasmic reticulum (ER)-Golgi pathway33. Harpio et al.34 noted that circulating S100B plays a role in the decision to switch treatment regimens. The concentration of circulating S100B predicts the duration of survival in melanoma patients and are very sensitive for the detection of metastatic growth of malignant melanoma, particularly at stage IV of the disease. Pedrocchi et al.37 reported that the S100 protein level is elevated in the human breast cancer cell lines MDA-MD-231 and HS-578T. Cross et al.38 described how S100 protein family members, including S100A6, S100A8, S100A9, and S100A11, are expressed in breast cancer as well as other common cancers. In addition, S100A11 may have an effect on the proliferation of cancer cells through nucleocytoplasmic translocation.
Heat shock proteins (HSPs), a large family of molecular chaperones that are involved in protein folding, are overexpressed in various types of tumors and indicate a poor prognosis and increased resistance to therapies39,40,41,42. Released from dying tumor cells, they can also function as DAMP proteins to activate the immune system. In fact, the serum concentration of HSPs is elevated in cancer patients43,44,45. Fanelli et al.39 reported that the serum levels of the HSP Mr 27,000 (Hsp27), in breast cancer patients were higher than those in control patients. Chen et al.46 reported that serum concentrations of HSP90α in colorectal cancer patients were significantly higher than those in normal volunteers.
Histones are intranuclear cationic proteins that are present in all eukaryotic cells47. They act as DAMP proteins by activating the immune system through TLRs after being released into the extracellular space in response to injury or sepsis14,48,49,50,51. Chen et al.48 reported that histones are important in chromatin remodeling and gene transcription via posttranslational modifications. In addition, histones function as DAMPs when they translocate from the nucleus to the extracellular space, binding to receptors and activating various signaling pathways in a monomeric or multimeric manner. Moreover, ischemia/reperfusion (I/R) or drug-induced tissue injury results in sterile inflammation, and serum histone levels are significantly elevated in animal models of liver, kidney, lung, and brain injury. In addition, serum histone levels were found to be elevated in sepsis, and the H3 and H4 elements were the major components responsible for toxicity. In fact, the concentration of intracellular calcium is increased by the transient presence of exogenous histones in endothelial cells; histone administration in vivo causes neutrophil migration, endothelial injury and dysfunction, hemorrhage, and thrombosis, which finally results in animal death. Histones such as DAMPs can be released from dying or necrotic tumor cells52. Patwa et al.49 identified histone H4 in serum from pancreatic cancer patients.
Moreover, pancreatic adenocarcinoma upregulated factor (PAUF) is a ligand of TLRs that is involved in pancreatic tumor development5,7,53. PAUF is overexpressed in pancreatic cancer and acts as a DAMP protein, with resulting active secretion54. Kim et al.54 determined that PAUF is a novel upregulated secretory protein in pancreatic ductal adenocarcinoma—enhanced expression of PAUF mRNA and protein was observed in pancreatic cancer cell lines compared to normal cells. Furthermore, this group confirmed PAUF expression and secretion in lysates and culture medium by immunoreaction with anti-rhPAUF antibodies. Expression of PAUF was also confirmed in human pancreatic cancer tissues by immunohistochemical staining. As a ligand of TLR4, apoptosis inhibitor 5 (API5) is an apoptosis inhibitory protein that prevents apoptosis induced by the transcription factor E2F1 and interacts with a nuclear factor involved in apoptotic DNA fragmentation55,56,57. For instance, API5 is associated with diseases, including adenocarcinoma and cervical cancer58,59. It can be released from tumor cells and functions as a DAMP protein activating immune cells57. Cho et al.58 established that API5 is overexpressed in cervical cancer, with prognostic significance. By generating Kaplan–Meier plots, this group indicated that API5 expression is an important prognostic factor in human cervical cancer; high API5 expression reflected significantly shorter disease-free survival and overall survival times.
40S ribosomal protein S3 (RPS3) is a ribosomal protein component of the 40S subunit, which contains the domain responsible for translation initiation60,61,62. The RPS3 protein is involved in the repair of UV-induced DNA damage63,64,65. The expression levels of RPS3 in adenocarcinomas and adenomatous polyps were found to be higher than those in adjacent normal colonic mucosa66. Kim et al.66 reported that RPS3 is overexpressed in colorectal cancer cells, suggesting the relationship between this protein and tumorigenesis. This group showed that the secretion of dimeric RPS3 into the extracellular space and the secretion levels of RPS3 were obviously higher in malignant cancer cells than in normal cells. They demonstrated that RPS3 is secreted into cell culture medium via the ER-Golgi-dependent pathway, as detected by enzyme-linked immunosorbent assay (ELISA). Park et al.67 reported that RPS3 is a housekeeping protein that is expressed in all eukaryotic cells and is released from tumor cells by exposure to anticancer drugs. To this end, they specifically confirmed the release of RPS3 from B16F1 and B16F10 melanoma cancer cell lines into culture medium through western blotting.
In summary, various DAMP proteins are released from dying cells by tissue injury or chemo-/radiotherapy-induced apoptosis, and these DAMP proteins are associated with various diseases and functions through multiple signaling pathways.
Interaction of tumor-derived DAMP proteins with TLRs
DAMPs have been described as ligands for TLRs, known as PRRs, which initiate an inflammatory response68,69,70,71,72. The biological structure of TLRs comprises an extracellular transmembrane domain composed of multiple leucine-rich repeats (LRRs) and an intracellular signaling domain featuring a Toll-IL-1 receptor (TIR) domain73,74,75. Regarding intracellular signaling, myeloid differentiation factor 88 (MyD88), MyD88 adapter-like (Mal) protein, TIR-related adapter protein inducing IFN-β (TRIF), and TRIF-related adapter molecule (TRAM) are involved in activating immune cell responses76. TLRs are expressed in immune cells, such as macrophages and DCs, which are involved in innate immunity77,78,79 (Table 2). Innate immune cells produce inflammatory cytokines, and costimulatory molecules are increasingly expressed after a ligand, such as a DAMP protein, activates TLR signaling80,81. In particular, TLR2 and TLR4 are receptors triggered by a variety of endogenous DAMPs to induce an inflammatory response81. As mentioned earlier, HMGB1 is actively secreted from inflammatory immune cells or passively released by necrotic cells to signal tissue damage and initiate immune responses by binding to receptor for advanced glycation end products (RAGE), TLR2, or TLR4. Gary et al.18 reported that HMGB1 is a DNA-binding nuclear protein that is released actively by cytokine stimulation and passively during cell death and has been implicated in several inflammatory diseases, as it is a prototypical DAMP protein. Furthermore, this group identified the relationship between HMGB1 and TLR4, in which HMGB1 plays an important role in the activation of DCs through TLR4 for efficient presentation of tumor antigens from dying cells. Moreover, stimulation of RAGE-transfected HT1080 cells with HMGB1 induces the activation of ERK1/2 signaling downstream of RAGE. Wang et al.82 described how HMGB1 is associated with TLR4 in drug-induced damage-associated lethal hepatitis. In addition, the serum HMGB1 concentration was found to rise after treatment with acetaminophen. The interaction between HMGB1 and TLR4 was confirmed by stimulating macrophages from TLR4+/+ or TLR4−/− mice with soluble HMGB1; the production of IL-23 increased only with TLR4+/+ macrophages. Curtin et al.83 reported that HMGB1 mediates endogenous TLR2 activation in brain tumors. The supernatants from dying Ad-TK + GCV-treated GL26 tumor cells indicated that TLR2-dependent NFκB activation was inhibited when tumor cells were treated with glycyrrhizin, an antagonist of HMGB1.
Alarmin eosinophil-derived neurotoxin (EDN) is known to use TLR2 as its receptor for activation of immune cells84. Yang et al.84 reported that EDN acts as an alarmin and activates TLR2-MyD88 signaling in dendritic cells. Furthermore, EDN was observed to induce an increase in luciferase activity in HEK293 cells expressing TLR2. In addition, the production of IL-6 in response to EDN was decreased in supernatants from TLR2−/− DCs.
HSPs, as described previously, are released by necrotic cells or secreted via nonclassical pathways to convey a danger signal to surrounding cells, which is followed by binding to TLR2 or TLR4 to induce inflammation. Zhao et al.85 determined that heat-shock protein 60 (hsp60) induces IL-8 via the TLR2 and MAPK pathways in human monocytes. The concentration of NK-κB in NOMO1 cells was significantly reduced by pretreatment with an anti-TLR2 antibody before stimulation with recombinant hsp60 protein. Roelof et al.86 identified heat shock protein B8 (HSP22) as a novel TLR4 ligand and its involvement in the pathogenesis of rheumatoid arthritis. DC maturation by HSPB8 stimulation was inhibited in the presence of a TLR4 antagonist, and the expression levels of CD80, CD83, CD86, and MHC-II were decreased.
PAUF has been identified as an endogenous ligand of TLR2 and TLR4. Park et al.7 confirmed the specificity of the PAUF-TLR2 interaction, where PAUF induces ERK phosphorylation and activates the IKK-β-mediated TPL2/MEK/ERK signaling pathway through TLR2. Furthermore, TLR2 coimmunoprecipitated with PAUF formed by glycosylation, and preincubation of THP-1 cells with an anti-TLR2 antibody resulted in a significant decrease in the level of PAUF protein binding to TLR2. Kang et al.87 reported that PAUF functions as an adjuvant by activating DCs through TLR4. This group demonstrated PAUF-induced activation and maturation of DCs with activation of NF-κB through the TLR signaling pathway. To confirm the dependency of TLR4 and PAUF, the binding affinity of the PAUF protein for TLR4 was evaluated by a Blitz assay, and the levels of pro-inflammatory cytokines secreted from DCs lacking TLR4 were found to be reduced after treatment with PAUF.
Apoptosis inhibitor 5 (API5), which is overexpressed in many types of tumors, interacts with TLR4 and induces an inflammatory response. Kim et al.57 established that API5 activates antigen-presenting cells in a TLR4-dependent manner. Via western blotting, this group confirmed the release of API5 from chemically stressed cells of various murine and human cancer cell lines. Immunoprecipitation analysis revealed that API5 binds to TLR4 but not TLR2, and luciferase activity was increased in HEK293 cells after PAUF treatment. Moreover, activation and maturation of mouse and human DCs were observed after treatment with the PAUF protein, but the results were reversed in DCs lacking TLR4.
Furthermore, ribosomal family proteins have been shown to activate immune cells after binding to TLR4 and to induce inflammation within the tumor microenvironment. Park et al.67 reported that RPS3, a DAMP protein, is a novel adjuvant for DC-based vaccines, inducing maturation and activation of DCs through TLR4. The interaction between RPS3 and TLR4 was confirmed by a Blitz assay, and luciferase activity was found to be increased in HEK293 cells. Moreover, after treatment with RPS3, the maturation and activation of TLR4−/− DCs were reduced compared to those of TLR4+/+ DCs.
Therefore, DAMP proteins are released from dying cells or tumor cells after chemotherapy or radiotherapy, and they bind and activate immune cells through TLRs to convey danger signals by initiating inflammatory immune responses.
DAMP proteins act as mediators between immune cells and tumors through TLRs
DAMP proteins from dying or apoptotic cells can both activate immune cells to produce pro-inflammatory cytokines and stimulate tumor cells to be aggressive by promoting metastasis. These two cell subsets can be affected by TLRs reacting with DAMP proteins. Therefore, DAMP proteins can act as mediators between TLR-expressing immune cells and tumors.
Chemotherapy and radiotherapy induce tumor recurrence and metastasis even though most tumor cells are destroyed by apoptosis13,88,89. Moreover, tumor cells express functional TLRs that play important roles in their proliferation, apoptosis resistance, invasion, and metastasis90,91,92 (Table 3). Volk-Draper et al.93 reported that paclitaxel therapy promotes breast cancer metastasis via TLR4 signaling. The levels of inflammatory cytokines were increased in nab-PXL-treated TLR4+/+ tumors compared to untreated and nab-PXL-treated TLR4−/− tumors. In fact, various types of cancer involve overexpression of TLRs, with poor prognosis in patients. Cammarota et al.94 observed that TLR4 expression is a potential prognostic marker in colorectal cancer, and the expression of TLR4 was associated with adenocarcinoma in human samples and a murine model. Increased TLR4 expression in the stromal compartment was associated with a significantly increased risk of disease progression, and significant tumor relapse occurred earlier in colon cancer patients with very high levels of TLR4 than in those with lower expression levels.
TLRs are conventionally known to be involved in the host immune response; however, it has recently been shown that TLRs are also expressed in tumor cells and influence tumor progression95,96,97,98. Therefore, DAMP proteins released from tumor cells can directly induce tumors to be aggressive to promote migration, invasion, and metastasis through TLRs. Moreover, TLR signaling facilitates evasion of immune surveillance in the tumor microenvironment and contributes to resistance to anticancer drugs99,100,101. Kelly et al.91 and Dan et al.102 reported that activation of TLR4 signaling promotes the growth of ovarian cancer with resistance to chemotherapy and leads to significant increases in the levels of X-linked inhibitor of apoptosis and phosphorylated AKT. Jego et al.103 and He et al.104 described how tumor cell apoptosis is inhibited through TLR signaling in lymphoma and lung cancer. Merrell et al.105 reported that the invasion of the breast cancer cell line MDA-MB-231 was promoted by increased matrix metalloproteinase activity after activation by a TLR9 agonist. Thus, activation of TLRs expressed by tumor cells is associated with tumor aggressiveness. As components of the host immune system, chemo-/radiotherapy-induced DAMP proteins activate immune cells through TLRs to secrete pro-inflammatory cytokines, thereby clearing T-cell trafficking into the tumor microenvironment and ultimately clearing tumor cells. These pro-inflammatory cytokines induce autocrine secretion by immune cells in order to boost inflammation, which can result in autoimmune disease106. Moreover, these cytokines can induce paracrine secretion of secretory proteins by tumor cells, which can activate tumor cells through TLR signaling. Thus, DAMP proteins are associated with immune responses and tumor progression, and identification of DAMP proteins released from tumors is necessary to overcome autoimmune diseases or chronic inflammatory disorders such as atherosclerosis and arthritis107.
Inflammation-induced immunosuppressive populations in the tumor microenvironment
Innate immunity and adaptive immunity are involved in host defense against tumor development108. First, innate immune cells, such as macrophages and DCs, recognize tumor antigens and deliver them to T cells to activate adaptive immune responses109. During the defense against tumors, immune cells produce pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, and tumor-specific T cells produce perforin and granzyme B to clear tumor cells110. However, tumors usually escape the immune response by altering their microenvironment with immunosuppressive populations such as M2 macrophages, myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs)111,112,113. These immune cells produce suppressive cytokines (e.g., IL-10 and TGF-β) that render tumor cells aggressive and exhaust CD8+ T cells114. In fact, chemo-/radiotherapy-induced inflammation results in a suppressive tumor microenvironment that induces downregulation of host immune responses115. Moreover, DAMP proteins released from tumors can activate inhibitory immune cells that express TLRs. Song et al.116 reported that PAUF is a DAMP protein that is involved in pancreatic tumorigenesis and metastasis and enhances tumor-infiltrating MDSC functional activity via the TLR4-mediated signaling pathway. Moreover, Parker et al.117 noted that HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of MDSCs. Liu et al.118 reported that knockdown of HMGB1 induces a decrease in regulatory T cells in tumors and restores CD8+ T-cell function. In conclusion, chronic inflammation induces a tumor microenvironment comprising immunosuppressive populations, where DAMP proteins activate TLR signaling expressed by inhibitory immune cells that are polarized toward suppressive characteristics.
Concluding remarks
Tumor-derived DAMP proteins are functional in both immune cells and tumors. In immune cells, identification of DAMP proteins that bind to TLRs is important for inducing inflammatory responses. For example, DC-based vaccines require novel adjuvants that induce the maturation and activation of DCs to secrete inflammatory cytokines. In fact, DAMP proteins binding to TLR4 (e.g., HMGB1, S100, HSPs, API5, and RPS3) have been used as adjuvants for DC-based vaccines, affecting tumor prevention, treatment, and survival. In contrast, tumor progression and metastasis are associated with the inflammatory response based on the interaction between TLR signaling and DAMP proteins. Regulation of these DAMP proteins in tumors can reduce excessive inflammation to create an immunogenic tumor microenvironment. For example, neutralizing antibodies against specific DAMP proteins could be used for tumor treatment in combination with chemotherapy or radiotherapy to prevent immune escape. In conclusion, various DAMP proteins are released from tumor cells after apoptosis and are ligands of TLRs that initiate an immune response. As mediators between immune cells and tumors, DAMP proteins must be identified and applied for immunotherapy and cancer treatment.
References
Srikrishna, G. & Freeze, H. H. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 11, 615–628 (2009).
Schirrmacher, V. & Fournier, P. Danger signals in tumor cells: a risk factor for autoimmune disease? Expert Rev. Vaccines 9, 347–350 (2010).
Patidar, A. et al. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine 104, 114–123 (2018).
Zhang, J. et al. High mobility group box 1 promotes the epithelial-to-mesenchymal transition in prostate cancer PC3 cells via the RAGE/NF-κB signaling pathway. Int. J. Oncol. 53, 659–671 (2018).
Choi, C. H. et al. Elevated expression of pancreatic adenocarcinoma upregulated factor (PAUF) is associated with poor prognosis and chemoresistance in epithelial ovarian cancer. Sci. Rep. 8, 12161 (2018).
Kelly, M. G. et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 66, 3859–3868 (2006).
Park, H. D. et al. Pancreatic adenocarcinoma upregulated factor promotes metastasis by regulating TLR/CXCR4 activation. Oncogene 30, 201–211 (2011).
Erlandsson Harris, H. & Andersson, U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur. J. Immunol. 34, 1503–1512 (2004).
Krysko, D. V. et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164 (2011).
Kaczmarek, A., Vandenabeele, P. & Krysko, DmitriV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013).
Nace, G., Evankovich, J., Eid, R. & Tsung, A. Dendritic cells and damage-associated molecular patterns: endogenous danger signals linking innate and adaptive immunity. J. Innate Immun. 4, 6–15 (2012).
Breugom, A. J. et al. Adjuvant chemotherapy after preoperative (chemo)radiotherapy and surgery for patients with rectal cancer: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 16, 200–207 (2015).
Carelle, N. et al. Changing patient perceptions of the side effects of cancer chemotherapy. Cancer 95, 155–163 (2002).
Kepp, O. et al. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. 30, 61–69 (2011).
Ma, Y. et al. How to improve the immunogenicity of chemotherapy and radiotherapy. Cancer Metastasis Rev. 30, 71–82 (2011).
Im, K.-I. et al. Regulation of HMGB1 release protects chemoradiotherapy-associated mucositis. Mucosal Immunol. 12, 1070–1081 (2019).
Pandolfi, F., Altamura, S., Frosali, S. & Conti, P. Key role of DAMP in inflammation, cancer, and tissue repair. Clin. Therapeutics 38, 1017–1028 (2016).
Sims, G. P., Rowe, D. C., Rietdijk, S. T., Herbst, R. & Coyle, A. J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 28, 367–388 (2010).
Tang, D., Kang, R., Zeh, H. J. 3rd & Lotze, M. T. High-mobility group box 1 and cancer. Biochim Biophys. Acta 1799, 131–140 (2010).
Yang, H. & Tracey, K. J. Targeting HMGB1 in inflammation. Biochimica et. Biophysica Acta (BBA) - Gene Regulatory Mechanisms 1799, 149–156 (2010).
Wang, H., Yang, H. & Tracey, K. J. Extracellular role of HMGB1 in inflammation and sepsis. J. Intern. Med. 255, 320–331 (2004).
Tsung, A. et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201, 1135–1143 (2005).
Andersson, U. & Erlandsson-Harris, H. HMGB1 is a potent trigger of arthritis. J. Intern. Med. 255, 344–350 (2004).
Gao, H.-M. et al. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 31, 1081 (2011).
Davalos, A. R. et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629 (2013).
Tang, D. et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310 (2010).
Campana, L., Bosurgi, L. & Rovere-Querini, P. HMGB1: a two-headed signal regulating tumor progression and immunity. Curr. Opin. Immunol. 20, 518–523 (2008).
Chen, X. et al. Radiotherapy-induced cell death activates paracrine HMGB1-TLR2 signaling and accelerates pancreatic carcinoma metastasis. J. Exp. Clin. Cancer Res. 37, 77 (2018).
Rovere-Querini, P. & Castiglioni, A. Adjuvant role for cell death during chemo- and radiotherapy of cancer? Expert Rev. Clin. Immunol. 4, 27–32 (2008).
Napolitano, A. et al. HMGB1 and its hyperacetylated isoform are sensitive and specific serum biomarkers to detect asbestos exposure and to identify mesothelioma patients. Clin. Cancer Res. 22, 3087 (2016).
Naumnik, W., Nilklińska, W., Ossolińska, M. & Chyczewska, E. Serum levels of HMGB1, survivin, and VEGF in patients with advanced non-small cell lung cancer during chemotherapy. Folia histochemica et. cytobiologica 47, 703–709 (2009).
Tang, D., Kang, R., Zeh, H. J. 3rd & Lotze, M. T. High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal 14, 1315–1335 (2011).
Bresnick, A. R., Weber, D. J. & Zimmer, D. B. S100 proteins in cancer. Nat. Rev. Cancer 15, 96–109 (2015).
Harpio, R. & Einarsson, R. S100 proteins as cancer biomarkers with focus on S100B in malignant melanoma. Clin. Biochem. 37, 512–518 (2004).
Vrakas, C. N. et al. The measure of DAMPs and a role for S100A8 in recruiting suppressor cells in breast cancer lung metastasis. Immunological Investig. 44, 174–188 (2015).
Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 60, 540–551 (2003).
Pedrocchi, M., Schäfer, B. W., Mueller, H., Eppenberger, U. & Heizmann, C. W. Expression of Ca2+-binding proteins of the S100 family in malignant human breast-cancer cell lines and biopsy samples. Int. J. Cancer 57, 684–690 (1994).
Cross, S. S., Hamdy, F. C., Deloulme, J. C. & Rehman, I. Expression of S100 proteins in normal human tissues and common cancers using tissue microarrays: S100A6, S100A8, S100A9 and S100A11 are all overexpressed in common cancers. Histopathology 46, 256–269 (2005).
Fanelli, M. A., Cuello Carrión, F. D., Dekker, J., Schoemaker, J. & Ciocca, D. R. Serological detection of heat shock protein hsp27 in normal and breast cancer patients. Cancer Epidemiol. Biomark. amp;amp; Prev. 7, 791 (1998).
Fucikova, J. et al. Prognostic and predictive value of DAMPs and DAMP-associated processes in cancer. Front. Immunol. 6, 402 (2015).
Fujita, Y. et al. Proteomics-based identification of autoantibody against heat shock protein 70 as a diagnostic marker in esophageal squamous cell carcinoma. Cancer Lett. 263, 280–290 (2008).
Wu, F.-H. et al. Extracellular HSPA1A promotes the growth of hepatocarcinoma by augmenting tumor cell proliferation and apoptosis-resistance. Cancer Lett. 317, 157–164 (2012).
Ren, B. et al. The expression of DAMP proteins HSP70 and cancer-testis antigen SPAG9 in peripheral blood of patients with HCC and lung cancer. Cell Stress Chaperones 22, 237–244 (2017).
Dutta, S. K. et al. Serum HSP70: a novel biomarker for early detection of pancreatic cancer. Pancreas 41, 530–534 (2012).
Zimmermann, M. et al. Discrimination of clinical stages in non-small cell lung cancer patients by serum HSP27 and HSP70: a multi-institutional case–control study. Clin. Chim. Acta 413, 1115–1120 (2012).
Chen, J. S. et al. Secreted heat shock protein 90alpha induces colorectal cancer cell invasion through CD91/LRP-1 and NF-kappaB-mediated integrin alphaV expression. J. Biol. Chem. 285, 25458–25466 (2010).
Peterson, C. L. & Laniel, M.-A. Histones and histone modifications. Curr. Biol. 14, R546–R551 (2004).
Chen, R., Kang, R., Fan, X. G. & Tang, D. Release and activity of histone in diseases. Cell Death Dis. 5, e1370–e1370 (2014).
Patwa, T. H. et al. The identification of phosphoglycerate kinase-1 and histone H4 autoantibodies in pancreatic cancer patient serum using a natural protein microarray. Electrophoresis 30, 2215–2226 (2009).
Allam, R., Kumar, S. V. R., Darisipudi, M. N. & Anders, H.-J. Extracellular histones in tissue injury and inflammation. J. Mol. Med. 92, 465–472 (2014).
Kawai, C. et al. Circulating extracellular histones are clinically relevant mediators of multiple organ injury. Am. J. Pathol. 186, 829–843 (2016).
Thålin, C. et al. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE 13, e0191231 (2018).
Lee, Y. et al. PAUF functions in the metastasis of human pancreatic cancer cells and upregulates CXCR4 expression. Oncogene 29, 56–67 (2010).
Kim, S. A. et al. Pancreatic adenocarcinoma up-regulated factor (PAUF), a novel up-regulated secretory protein in pancreatic ductal adenocarcinoma. Cancer Sci. 100, 828–836 (2009).
Garcia-Jove Navarro, M. et al. Api5 contributes to E2F1 control of the G1/S cell cycle phase transition. PLoS ONE 8, e71443 (2013).
Morris, E. J. et al. Functional identification of Api5 as a suppressor of E2F-dependent apoptosis in vivo. PLoS Genet. 2, e196 (2006).
Kim, Y. S. et al. A novel function of API5 (apoptosis inhibitor 5), TLR4-dependent activation of antigen presenting cells. Oncoimmunology 7, e1472187–e1472187 (2018).
Cho, H. et al. Apoptosis inhibitor-5 overexpression is associated with tumor progression and poor prognosis in patients with cervical cancer. BMC Cancer 14, 545 (2014).
Basset, C. et al. Api5 a new cofactor of estrogen receptor alpha involved in breast cancer outcome. Oncotarget 8, 52511–52526 (2017).
Lindström, M. S. Emerging functions of ribosomal proteins in gene-specific transcription and translation. Biochem. Biophys. Res. Commun. 379, 167–170 (2009).
Graifer, D., Malygin, A., Zharkov, D. O. & Karpova, G. Eukaryotic ribosomal protein S3: A constituent of translational machinery and an extraribosomal player in various cellular processes. Biochimie 99, 8–18 (2014).
Mitterer, V. et al. Sequential domain assembly of ribosomal protein S3 drives 40S subunit maturation. Nat. Commun. 7, 10336 (2016).
Jang, C.-Y., Lee, J. Y. & Kim, J. RpS3, a DNA repair endonuclease and ribosomal protein, is involved in apoptosis. FEBS Lett. 560, 81–85 (2004).
Kim, Y., Kim, H. D. & Kim, J. Cytoplasmic ribosomal protein S3 (rpS3) plays a pivotal role in mitochondrial DNA damage surveillance. Biochimica et. Biophysica Acta (BBA) - Mol. Cell Res. 1833, 2943–2952 (2013).
Kim, J. et al. Implication of mammalian ribosomal protein S3 in the processing of DNA damage. J. Biol. Chem. 270, 13620–13629 (1995).
Kim, Y., Lee, M. S., Kim, H. D. & Kim, J. Ribosomal protein S3 (rpS3) secreted from various cancer cells is N-linked glycosylated. Oncotarget 7, 80350–80362 (2016).
Park, H. J. et al. A novel TLR4 binding protein, 40S ribosomal protein S3, has potential utility as an adjuvant in a dendritic cell-based vaccine. J. Immunother. Cancer 7, 60–60 (2019).
Piccinini, A. M. & Midwood, K. S. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010, 672395 (2010).
Gong, T., Liu, L., Jiang, W. & Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 20, 95–112 (2020).
Feldman, N., Rotter-Maskowitz, A. & Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 24, 29–39 (2015).
Patel, S. Danger-associated molecular patterns (DAMPs): the derivatives and triggers of inflammation. Curr. Allergy Asthma Rep. 18, 63 (2018).
Franklin, T. C., Xu, C. & Duman, R. S. Depression and sterile inflammation: essential role of danger associated molecular patterns. Brain, Behav., Immun. 72, 2–13 (2018).
Botos, I., Segal, DavidM. & Davies, DavidR. The structural biology of toll-like receptors. Structure 19, 447–459 (2011).
Kang, J. Y. & Lee, J.-O. Structural biology of the toll-like receptor family. Annu. Rev. Biochem. 80, 917–941 (2011).
Beutler, B. & Rehli, M. in Toll-Like Receptor Family Members and Their Ligands (eds Beutler, B. & Wagner, H.) 1–21 (Springer Berlin Heidelberg, Berlin, Heidelberg, 2002).
Kawai, T. & Akira, S. TLR signaling. Semin. Immunol. 19, 24–32 (2007).
Delneste, Y., Beauvillain, C. & Jeannin, P. Innate immunity: structure and function of TLRs. Med. Sci. 23, 67–73 (2007).
Kumar, H., Kawai, T. & Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388, 621–625 (2009).
Schröder, N. W. J. et al. Lipoteichoic Acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J. Biol. Chem. 278, 15587–15594 (2003).
Takeda, K. & Akira, S. TLR signaling pathways. Semin. Immunol. 16, 3–9 (2004).
Erridge, C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J. Leukoc. Biol. 87, 989–999 (2010).
Wang, X., Sun, R., Wei, H. & Tian, Z. High-mobility group box 1 (HMGB1)-toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: Interaction of γδ T cells with macrophages. Hepatology 57, 373–384 (2013).
Curtin, J. F. et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 6, e10–e10 (2009).
Yang, D. et al. Eosinophil-derived neurotoxin acts as an alarmin to activate the TLR2–MyD88 signal pathway in dendritic cells and enhances Th2 immune responses. J. Exp. Med. 205, 79–90 (2008).
Zhao, Y. et al. Helicobacter pylori heat-shock protein 60 induces interleukin-8 via a Toll-like receptor (TLR)2 and mitogen-activated protein (MAP) kinase pathway in human monocytes. J. Med. Microbiol. 56, 154–164 (2007).
Roelofs, M. F. et al. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J. Immunol. 176, 7021 (2006).
Kang, T. H. et al. Pancreatic adenocarcinoma upregulated factor serves as adjuvant by activating dendritic cells through stimulation of TLR4. Oncotarget 6, 27751–27762 (2015).
Panetta, J. C. A mathematical model of periodically pulsed chemotherapy: tumor recurrence and metastasis in a competitive environment. Bull. Math. Biol. 58, 425–447 (1996).
Geara, F. B. et al. Carcinoma of the nasopharynx treated by radiotherapy alone: determinants of distant metastasis and survival. Radiother. Oncol. 43, 53–61 (1997).
Huang, B., Zhao, J., Unkeless, J. C., Feng, Z. H. & Xiong, H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene 27, 218–224 (2008).
Kelly, M. G. et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res 66, 3859 (2006).
Korneev, K. V. et al. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine 89, 127–135 (2017).
Volk-Draper, L. et al. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res. 74, 5421–5434 (2014).
Cammarota, R. et al. The tumor microenvironment of colorectal cancer: stromal TLR-4 expression as a potential prognostic marker. J. Transl. Med. 8, 112 (2010).
Yu, L. & Chen, S. Toll-like receptors expressed in tumor cells: targets for therapy. Cancer Immunol. Immunother. 57, 1271–1278 (2008).
Shcheblyakov, D. V. et al. Toll-like receptors (TLRs): the role in tumor progression. Acta Nat. 2, 21–29 (2010).
Zeromski, J., Mozer-Lisewska, I. & Kaczmarek, M. Significance of toll-like receptors expression in tumor growth and spreading: a short review. Cancer Microenviron. 1, 37–42 (2008).
Dajon, M., Iribarren, K. & Cremer, I. Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology 222, 89–100 (2017).
Netea, M. G., Van der Meer, J. W. M. & Kullberg, B.-J. Toll-like receptors as an escape mechanism from the host defense. Trends Microbiol. 12, 484–488 (2004).
Urban-Wojciuk, Z. et al. The role of TLRs in anti-cancer immunity and tumor rejection. Front. Immunol. 10, 2388 (2019).
Chen, R., Alvero, A. B., Silasi, D.-A. & Mor, G. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am. J. Reprod. Immunol. 57, 93–107 (2007).
Dan, H. C. et al. Phosphatidylinositol-3-OH kinase/AKT and survivin pathways as critical targets for geranylgeranyltransferase I inhibitor-induced apoptosis. Oncogene 23, 706–715 (2004).
Jego, G., Bataille, R., Geffroy-Luseau, A., Descamps, G. & Pellat-Deceunynck, C. Pathogen-associated molecular patterns are growth and survival factors for human myeloma cells through Toll-like receptors. Leukemia 20, 1130–1137 (2006).
He, W. et al. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol. Immunol. 44, 2850–2859 (2007).
Merrell, M. A. et al. Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol. Cancer Res. 4, 437 (2006).
Kim, E. Y. & Moudgil, K. D. Regulation of autoimmune inflammation by pro-inflammatory cytokines. Immunol. Lett. 120, 1–5 (2008).
Foell, D., Wittkowski, H. & Roth, J. Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat. Clin. Pract. Rheumatol. 3, 382–390 (2007).
Vesely, M. D., Kershaw, M. H., Schreiber, R. D. & Smyth, M. J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 29, 235–271 (2011).
Medzhitov, R. & Janeway, C. Innate immunity. N. Engl. J. Med. 343, 338–344 (2000).
Bonilla, F. A. & Oettgen, H. C. Adaptive immunity. J. Allergy Clin. Immunol. 125, S33–S40 (2010).
Nishikawa, H. & Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 127, 759–767 (2010).
Mantovani, A., Sozzani, S., Locati, M., Allavena, P. & Sica, A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 (2002).
Finke, J. et al. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int. Immunopharmacol. 11, 856–861 (2011).
Sanjabi, S., Zenewicz, L. A., Kamanaka, M. & Flavell, R. A. Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 9, 447–453 (2009).
Salazar-Onfray, F., López, M. N. & Mendoza-Naranjo, A. Paradoxical effects of cytokines in tumor immune surveillance and tumor immune escape. Cytokine Growth Factor Rev. 18, 171–182 (2007).
Song, J. et al. Pancreatic adenocarcinoma up-regulated factor (PAUF) enhances the accumulation and functional activity of myeloid-derived suppressor cells (MDSCs) in pancreatic cancer. Oncotarget 7, 51840–51853 (2016).
Parker, K. H. et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 74, 5723–5733 (2014).
Liu, Z., Falo, L. D. Jr. & You, Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J. Immunol. 187, 118–125 (2011).
He, Y. et al. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res. 73, 629–639 (2013).
Schlueter, C. et al. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am. J. Pathol. 166, 1259–1263 (2005).
Jube, S. et al. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 72, 3290–3301 (2012).
Pusterla, T. et al. Receptor for advanced glycation endproducts (RAGE) is a key regulator of oval cell activation and inflammation-associated liver carcinogenesis in mice. Hepatology 58, 363–373 (2013).
Huebener, P. et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Invest. 125, 539–550 (2015).
Dumitriu, I. E. et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 174, 7506–7515 (2005).
Luo, Y. et al. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur. J. Cancer 49, 741–751 (2013).
Kang, R. et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 17, 666–676 (2010).
Zhou, J. et al. HMGB1 induction of clusterin creates a chemoresistant niche in human prostate tumor cells. Sci. Rep. 5, 15085 (2015).
Apetoh, L. et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 220, 47–59 (2007).
Liu, Y. et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J. Hepatol. 63, 114–121 (2015).
Chiba, S. et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 13, 832–842 (2012).
Hernandez, C., Huebener, P. & Schwabe, R. F. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35, 5931–5941 (2016).
Ghavami, S. et al. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J. Leukoc. Biol. 83, 1484–1492 (2008).
Ichikawa, M., Williams, R., Wang, L., Vogl, T. & Srikrishna, G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol. Cancer Res. 9, 133–148 (2011).
Gebhardt, C. et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 205, 275–285 (2008).
Bettum, I. J. et al. Metastasis-associated protein S100A4 induces a network of inflammatory cytokines that activate stromal cells to acquire pro-tumorigenic properties. Cancer Lett. 344, 28–39 (2014).
Sack, U. et al. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. J. Natl Cancer Inst. 103, 1018–1036 (2011).
Taha, E. A., Ono, K. & Eguchi, T. Roles of extracellular HSPs as biomarkers in immune surveillance and immune evasion. Int. J. Mol. Sci. 20, 4588 (2019).
Gu, T. et al. Impact of elevated circulating histones on systemic inflammation after radiofrequency ablation in lung cancer patients. BioMed. Res. Int. 2017, 6894832 (2017).
Kabelitz, D. Expression and function of Toll-like receptors in T lymphocytes. Curr. Opin. Immunol. 19, 39–45 (2007).
Rahman, A. H., Taylor, D. K. & Turka, L. A. The contribution of direct TLR signaling to T cell responses. Immunol. Res. 45, 25–36 (2009).
van Maren, W. W. C., Jacobs, J. F. M., de Vries, I. J. M., Nierkens, S. & Adema, G. J. Toll-like receptor signalling on Tregs: to suppress or not tosuppress? Immunology 124, 445–452 (2008).
Applequist, S. E., Wallin, R. P. A. & Ljunggren, H. G. Variable expression of Toll‐like receptor in murine innate and adaptive immune cell lines. Int. Immunol. 14, 1065–1074 (2002).
Kadowaki, N. et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194, 863–869 (2001).
Schreibelt, G. et al. Toll-like receptor expression and function in human dendritic cell subsets: implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol., Immunother. 59, 1573–1582 (2010).
Billack, B. Macrophage activation: role of toll-like receptors, nitric oxide, and nuclear factor kappa B. Am. J. Pharm. Educ. 70, 102–102 (2006).
Gallego, C., Golenbock, D., Gomez, M. A. & Saravia, N. G. Toll-like receptors participate in macrophage activation and intracellular control of Leishmania (Viannia) panamensis. Infect. Immun. 79, 2871–2879 (2011).
Schmitz, F., Mages, J., Heit, A., Lang, R. & Wagner, H. Transcriptional activation induced in macrophages by Toll-like receptor (TLR) ligands: from expression profiling to a model of TLR signaling. Eur. J. Immunol. 34, 2863–2873 (2004).
Prince, L. R., Whyte, M. K., Sabroe, I. & Parker, L. C. The role of TLRs in neutrophil activation. Curr. Opin. Pharm. 11, 397–403 (2011).
Hua, Z. & Hou, B. TLR signaling in B-cell development and activation. Cell. Mol. Immunol. 10, 103–106 (2013).
Muccioli, M. & Benencia, F. Toll-like receptors in ovarian cancer as targets for immunotherapies. Front. Immunol. 5, 341 (2014).
Zhou, M. et al. Toll-like receptor expression in normal ovary and ovarian tumors. Cancer Immunol., Immunother. 58, 1375–1385 (2009).
Husseinzadeh, N. & Davenport, S. M. Role of Toll-like receptors in cervical, endometrial and ovarian cancers: a review. Gynecologic Oncol. 135, 359–363 (2014).
Kim, W. Y. et al. Increased expression of Toll-like receptor 5 during progression of cervical neoplasia. Int. J. Gynecol. Cancer. 18, 300–305 (2008).
Nishimura, M. & Naito, S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol. Pharm. Bull. 28, 886–892 (2005).
Hasimu, A., Ge, L., Li, Q.-Z., Zhang, R.-P. & Guo, X. Expressions of Toll-like receptors 3, 4, 7, and 9 in cervical lesions and their correlation with HPV16 infection in Uighur women. Chin. J. Cancer 30, 344–350 (2011).
Ren, T. et al. Functional expression of TLR9 is associated to the metastatic potential of human lung cancer cell: functional active role of TLR9 on tumor metastasis. Cancer Biol. Ther. 6, 1704–1709 (2007).
Droemann, D. et al. Human lung cancer cells express functionally active Toll-like receptor 9. Respir. Res. 6, 1 (2005).
Fukata, M. et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 133, 1869–1881 (2007).
Fukata, M. & Abreu, M. T. TLR4 signalling in the intestine in health and disease. Biochemical Soc. Trans. 35, 1473–1478 (2007).
Pedersen, G., Andresen, L., Matthiessen, M. W., Rask-Madsen, J. & Brynskov, J. Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin. Exp. Immunol. 141, 298–306 (2005).
Damiano, V. et al. Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin. Cancer Res. 12, 577–583 (2006).
Rhee, S. H., Im, E. & Pothoulakis, C. Toll-like receptor 5 engagement modulates tumor development and growth in a mouse xenograft model of human colon cancer. Gastroenterology 135, 518–528 (2008).
Goto, Y. et al. Activation of Toll-like receptors 2, 3, and 4 on human melanoma cells induces inflammatory factors. Mol. Cancer Ther. 7, 3642–3653 (2008).
Salaun, B., Lebecque, S., Matikainen, S., Rimoldi, D. & Romero, P. Toll-like receptor 3 expressed by melanoma cells as a target for therapy? Clin. Cancer Res. 13, 4565–4574 (2007).
Xie, W. et al. Toll-like receptor 2 mediates invasion via activating NF-kappaB in MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 379, 1027–1032 (2009).
Curtin, J. F. et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 6, e10 (2009).
Hassan, F. et al. Intracellular expression of toll-like receptor 4 in neuroblastoma cells and their unresponsiveness to lipopolysaccharide. BMC Cancer 6, 281 (2006).
Ilvesaro, J. M. et al. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate 67, 774–781 (2007).
Kundu, S. D. et al. The toll-like receptor pathway: a novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate 68, 223–229 (2008).
Schmaußer, B., Andrulis, M., Endrich, S., Müller-Hermelink, H.-K. & Eck, M. Toll-like receptors TLR4, TLR5 and TLR9 on gastric carcinoma cells: An implication for interaction with Helicobacter pylori. Int. J. Med. Microbiol. 295, 179–185 (2005).
Chochi, K. et al. Helicobacter pylori augments growth of gastric cancers via the lipopolysaccharide-toll-like receptor 4 pathway whereas its lipopolysaccharide attenuates antitumor activities of human mononuclear cells. Clin. Cancer Res. 14, 2909 (2008).
Chang, Y. J., Wu, M. S., Lin, J. T. & Chen, C. C. Helicobacter pylori-Induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase-2 induction through TLR2/TLR9 and promoter regulation. J. Immunol. 175, 8242–8252 (2005).
Acknowledgements
This study was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (NRF-2016R1A5A2012284 and NRF-2018R1A2B6008455).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jang, GY., Lee, J.w., Kim, Y.S. et al. Interactions between tumor-derived proteins and Toll-like receptors. Exp Mol Med 52, 1926–1935 (2020). https://doi.org/10.1038/s12276-020-00540-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-00540-4
This article is cited by
-
B-1 derived anti-Thy-1 B cells in old aged mice develop lymphoma/leukemia with high expression of CD11b and Hamp2 that different from TCL1 transgenic mice
Immunity & Ageing (2024)
-
DAMP sensing and sterile inflammation: intracellular, intercellular and inter-organ pathways
Nature Reviews Immunology (2024)
-
HSP72 is a stimulus for activating toll-like receptor 2 in M2 macrophages leading to IL-6-mediated tumor malignancy
Molecular & Cellular Toxicology (2024)
-
TET1 is a Diagnostic and Prognostic Biomarker Associated with Immune Infiltration in Papillary Thyroid Cancer
Biochemical Genetics (2024)
-
Colon-targeted S100A8/A9-specific peptide systems ameliorate colitis and colitis-associated colorectal cancer in mouse models
Acta Pharmacologica Sinica (2023)
