
Abstract
As knowledge of cell metabolism has advanced, glutamine has been considered an important amino acid that supplies carbon and nitrogen to fuel biosynthesis. A recent study provided a new perspective on mitochondrial glutamine metabolism, offering mechanistic insights into metabolic adaptation during tumor hypoxia, the emergence of drug resistance, and glutaminolysis-induced metabolic reprogramming and presenting metabolic strategies to target glutamine metabolism in cancer cells. In this review, we introduce the various biosynthetic and bioenergetic roles of glutamine based on the compartmentalization of glutamine metabolism to explain why cells exhibit metabolic reliance on glutamine. Additionally, we examined whether glutamine derivatives contribute to epigenetic regulation associated with tumorigenesis. In addition, in discussing glutamine transporters, we propose a metabolic target for therapeutic intervention in cancer.
Similar content being viewed by others


Introduction
After Otto Warburg discovered that cancer cells exhibit significantly elevated glucose consumption and lactate secretion even in the presence of oxygen1, studies on cell metabolism have accumulated. The major findings are that aerobic glycolysis is not a symptom of impaired mitochondrial function, and that glutamine supports mitochondrial oxidative metabolism when pyruvate derived from glucose is converted into lactate and secreted2,3,4. Glutamine, which is a nonessential amino acid (NEAA) due to the endogenous glutamine biosynthesis pathway, is currently considered essential in cancer cells because transformed cells consume glutamine at a rate exceeding that of glutamine biosynthesis5. Glutamine has a versatile role in cell metabolism, participating in tricarboxylic acid (TCA) cycle supplementation and the biosynthesis of nucleotides, glutathione (GSH), and other nonessential amino acids. Thus, glutamine deprivation suppresses cancer growth and even induces cell death in several cancers6,7. This metabolic dependency of transformed cells on glutamine constitutes the recently defined glutamine addiction8.
Since glutaminase 1 (GLS1), a key mitochondrial enzyme that catalyzes the deamidation of glutamine, was first discovered in the kidney in 19589, many enzymes involved in glutamine metabolism have been reported4. In addition, glutamine has been confirmed to be a major nutrient source for oxidative metabolism in some cancer cell lines10,11,12, and specific genetic interference with glutaminase (GLS) inhibits tumor cell growth13. Moreover, CB-839, the first glutaminase inhibitor, has entered several clinical trials14,15. Despite the importance of mitochondrial glutamine metabolism, the mitochondrial glutamine transporter, encoded by a transcript variant of the SLC1A5 gene, which encodes a well-known plasma membrane glutamine transporter, was only recently discovered16. Thus, glutamine metabolism is intriguingly linked with intricate cell metabolic processes via enzymes associated with mitochondrial glutaminolysis, cytosolic glutamine metabolism, and glutamine-derived metabolites that perform diverse cellular functions.
In this review, we first introduce metabolic pathways that enable glutamine to respond to diverse cellular needs and then discuss the metabolic link by which glutamine-derived metabolites may affect cellular metabolic processes, including NEAA synthesis, epigenetic modifications, and hypoxia adaptation. We next discuss recent advances in glutamine metabolism with particular emphasis on tumorigenesis. We aim to offer both the principles underlying cellular dependence on glutamine metabolism under various conditions and a discussion of future directions that are leading our efforts to investigate the role of glutamine in cellular metabolism.
Glutamine metabolic pathways
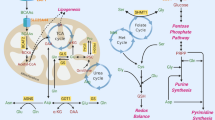
Glutamine is transported into cells through plasma membrane glutamine transporters such as SLC1A5, SLC38A1, and SLC38A217 and can then be used for the biosynthesis of hexosamine, nucleotides, and asparagine in the cytoplasm (Fig. 1). For mitochondrial glutaminolysis, cytosolic glutamine must be transported through the inner mitochondrial membrane via the SLC1A5 variant, a mitochondrial glutamine transporter16. Next, glutamine is converted into glutamate by GLSs, amidohydrolase enzymes that catalyze the conversion of glutamine into glutamate, releasing ammonium ions. GLSs have at least three isoforms, GLS1, GLS2, and GAC (a splicing isoform of GLS1), all of which were recently reported to be localized in mitochondria18,19,20. Mitochondrial glutamate generated via these catabolic pathways can be exported from mitochondria to the cytosol through the SLC25A18 and SLC25A22 transporters21, and cytosolic glutamate then participates in the biosynthesis of glutathione—a tripeptide comprising glutamate, cysteine, and glycine—and NEAAs (alanine, proline, aspartate, asparagine, and arginine) and is used as an exchange factor for importing extracellular cystine via SLC7A11. Mitochondrial glutamate is subsequently converted into alpha-ketoglutarate (α-KG) by glutamate dehydrogenase 1 (GLUD1 or GDH1) or by several mitochondrial aminotransferases, including glutamic-pyruvic transaminase 2 (GPT2) and glutamic-oxaloacetic transaminase 2 (GOT2). In addition, α-KG is exported from mitochondria through SLC25A11 to the cytosol21 and then participates in fatty acid biosynthesis and NADH generation22 (Fig. 1). Mitochondrial α-KG can then participate in the TCA cycle, supporting the oxidative phosphorylation (OXPHOS) pathway or the reductive carboxylation pathway23. In the oxidative phosphorylation pathway, metabolites of glutamine participate in the generation of an electron donor, such as NADH or FADH2, after synthesis of guanosine triphosphate (GTP) and adenosine triphosphate (ATP). In addition to pyruvate-derived acetyl-CoA, α-KG-derived metabolites (e.g., succinate and fumarate) generated via glutaminolysis are considered oncometabolites contributing to tumorigenesis23. Citrate, generated by reductive carboxylation of α-KG, is especially crucial for lipid synthesis under low-oxygen conditions24,25.

Glutamine enters through several plasma membrane glutamine transporters and is then utilized in the cytosol in processes such as the biosynthesis of nucleotides, asparagine, and UDP-GlcNAc. For glutaminolysis, glutamine is transported into the mitochondrial matrix through the SLC1A5 variant and subsequently converted to glutamate by GLS. Next, GLUD1 or several aminotransferases catalyze the deamidation of glutamate, producing α-KG. Glutamine-derived α-KG supplies metabolites for the TCA cycle and fuels the generation of 2-HG under conditions of IDH2 mutation or hypoxia. Citrate derived from glutamine via reductive carboxylation supports fatty acid synthesis under conditions of hypoxia or HIF-2α transcription factor stabilization. Glutamine-derived α-KG also activates the mTORC1 pathway. Α-KG and 2-HG affect epigenetic modification through α-KG-dependent dioxygenases. Gln glutamine, Glu glutamate, Asn asparagine, Cys cystine, Asp aspartate, αKG α-ketoglutarate, PRA 5-phosphoribosyl-1-amine, CP carbamoyl phosphate, GFAT glutamine-fructose-6-phosphate transaminase, ASNS asparagine synthetase, PPAT phosphoribosyl pyrophosphate amidotransferase, CPS carbamoyl phosphate synthetase, GLS glutaminase, GLUD glutamate dehydrogenase, GOT glutamic-oxaloacetic transaminase, GPT glutamic-pyruvate transaminase, IDH isocitrate dehydrogenase, 2-HG 2-hydroxyglutarate, Me methylation.
α-KG is considered an important cofactor for enzymes participating in epigenetic modification26. It is a substrate for α-ketoglutarate dehydrogenase (OGDH) in oxidative reactions generating succinyl-CoA and isocitrate dehydrogenase 1 (IDH1) or isocitrate dehydrogenase 2 (IDH2), which catalyze the reductive carboxylation reaction converting α-KG to isocitrate. Cancer cells in tumors with IDH1 or IDH2 mutations show oncogenic activity by converting glutamine-derived α-KG to 2-hydroxyglutarate (2-HG), which competitively inhibits α-KG-dependent histone and DNA modification enzymes27. Additionally, glutamine-derived aspartate plays a crucial role in hypoxic environments or environments with electron transport chain (ETC) impairment28. In addition, NADPH generation via glutamine metabolism in cancer cells supports redox homeostasis by maintaining the cytosolic NADPH pool used to restore oxidized glutathione29 (Fig. 1).
Nucleotides synthesized from glutamine
Cytosolic glutamine supports nucleotide biosynthesis, which is essential for rapidly proliferating cells30. The gamma-nitrogen of glutamine is used in five reactions in de novo nucleotide synthesis, and its bioavailability controls de novo biosynthesis of pyrimidines and purines (Fig. 2)5. In purine biosynthesis, two glutamines are used to generate inosine monophosphate (IMP), a precursor of both adenosine monophosphate (AMP) and guanosine monophosphate (GMP). Then, one glutamine molecule is needed for the conversion of IMP to GMP31. In pyrimidine biosynthesis, one glutamine molecule is consumed by a carbamoyl phosphate synthetase enzyme (CPS1 or CPS2, which are localized in the mitochondria and cytosol, respectively). One more glutamine molecule is used to synthesize cytidine triphosphate (CTP) from uridine triphosphate (UTP)31. Interestingly, the first step in de novo pyrimidine biosynthesis mediated by CPSs occurs mainly in mitochondria via CPS1 in K-Ras/LKB1-mutant lung cancer cells32 (Fig. 2). Although cytosolic CPS2 can produce a cytosolic pool of carbamoyl phosphate, CPS1 is a major rate-limiting enzyme in pyrimidine biosynthesis using nitrogen released via mitochondrial glutaminolysis32.

In purine biosynthesis, two glutamine molecules are consumed to synthesize AMP, and three glutamine molecules are used to synthesize GMP. Similarly, in pyrimidine biosynthesis, one glutamine molecule is consumed to synthesize UMP, and two glutamine molecules are spent to convert UTP into CTP. The initial step in de novo pyrimidine synthesis is the condensation reaction between glutamine and bicarbonate catalyzed by CPS to produce CP. In cells with an oncogenic mutational status, including K-Ras mutation, glutaminolysis sustains mitochondrial generation of CP by providing enough nitrogen fuel as ammonium ions, and mitochondrial CP then participates in cytosolic de novo pyrimidine synthesis. Glutamine-induced nucleotide biosynthesis is also enhanced by MYC or growth signals such as mTORC1 activation. PPAT phosphoribosyl pyrophosphate amidotransferase, PFAS phosphoribosylformylglycinamidine synthase, GMPS GMP synthetase, CPS carbamoyl phosphate synthetase, CTPS CTP synthetase, GLS glutaminase, PRPP 5-phosphoribosyl-1-pyrophosphate, PRA 5-phosphoribosyl-1-amine, FGAR N2-formyl-N1-(5-phospho-d-ribosyl)glycinamide, FGAM 2-(formamido)-N1-(5-phospho-d-ribosyl)acetamidine, IMP inosine monophosphate, SAMP adenylosuccinate, XMP xanthosine monophosphate, AMP adenosine monophosphate, GMP guanosine monophosphate, CP carbamoyl phosphate, UMP uridine monophosphate, UTP uridine triphosphate, CTP cytidine, Glu glutamine, Glu glutamate, αKG α-ketoglutarate.
In addition, glutamine can support nucleotide synthesis via other pathways. Aspartate, which is derived from the transamination of glutamine to form glutamate, participates in pyrimidine and purine biosynthesis28. Thus, exogenous aspartate can restore cell cycle arrest caused by glutamine deprivation33. Moreover, glutamine-induced activation of mTORC1 results in the phosphorylation of the enzyme complex called carbamoyl phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD), which catalyzes the condensation reaction converting glutamine-derived nitrogen into the pyrimidine precursor orotate34,35. Notably, increased expression of the transcription factor MYC, which is strongly associated with glutamine metabolism, induces the expression of several key enzymes in nucleotide biosynthesis, including phosphoribosyl pyrophosphate amidotransferase (PPAT)36. PPAT transfers glutamine-derived nitrogen to 5-phosphoribosyl pyrophosphate (PRPP), and this step is considered the initial step in purine biosynthesis37. In pancreatic cancer cells, oncogenic K-Ras maintains the nucleotide pool via the MAPK-dependent signaling pathway, leading to MYC upregulation, and the use of MEK inhibitors reduces the incorporation of glutamine-derived nitrogen into purine nucleotides38. Collectively, these studies describe a mechanism by which glutamine-derived nitrogen is essential for the rapid proliferation of cancer cells corresponding to an urgent need for nucleotide biosynthesis.
NEAAs synthesized from glutamine
Although glutamine has been considered an NEAA that is synthesized endogenously, most cancer cells cannot proliferate or survive in a medium that does not contain glutamine5. This inability is probably due to the function of glutamine metabolism, which provides both carbon and nitrogen for cellular biogenesis. Glutamine-derived carbon is an important substrate that supports the TCA cycle and the synthesis of glutathione. In addition, nitrogen derived from glutamine is required for the biosynthesis of molecules such as nucleotides, glucosamine, and NEAAs39. Notably, among NEAAs, the generation of glutamate and asparagine is directly dependent on glutamine (Fig. 3).

Intracellular glutamine is converted into diverse NEAAs and supports protein translation and amino acid signaling. Glutamine-derived glutamate plays a central role as a substrate for several aminotransferases producing aspartate, alanine, proline, arginine, serine, cysteine, and glycine. ASNS directly utilizes cytosolic glutamine to synthesize Asn, which plays a distinct role in glutamine-related metabolism. Collectively, glutamine-derived NEAAs suppress ATF4, which is a master transcriptional regulator stimulated under stress conditions. NEAAs nonessential amino acids, GLS glutaminase, GLUD glutamate dehydrogenase, GOT glutamic-oxaloacetic transaminase, GPT glutamic-pyruvate transaminase, PSAT phosphoserine aminotransferase, ATF activating transcription factor, ASNS asparagine synthetase, Gln glutamine, Glu glutamate, Pro proline, Asp aspartate, Ala alanine, Ser serine, Gly glycine, Cys cystine, Asn asparagine, Lys lysine, Thr threonine, Met methionine, aKG α-ketoglutarate, OAA oxaloacetate, Pyr pyruvate, PHP phosphohydroxypyruvate, PS phosphoserine.
Glutamate
Glutamate plays a central role in NEAA metabolism because it is crucial for the biosynthesis of alanine, aspartate, proline, and serine, which are in turn used for the biosynthesis of asparagine, arginine, cysteine, and glycine (Fig. 3). Glutamate is converted to α-KG both via GLUD1, generating glutamate-derived nitrogen as ammonia, and via aminotransferases, which transfer nitrogen from glutamate to α-KG to produce other NEAAs. Glutamate consumption by aminotransferases to generate NEAAs has also been indicated to be required for tumor growth in diverse cancer types29,40,41,42.
Although glutamate is the major downstream product of glutamine, glutamate supplementation during glutamine deprivation cannot rescue the impaired cell growth or mitochondrial respiration16,43,44,45, indicating that mitochondrial GLS-catalyzed cleavage of the gamma-nitrogen of glutamine is essential for glutaminolysis. A possible reason for this requirement is the charge difference between glutamine and glutamate. Glutamine is a neutral amino acid and thus does not induce a negative charge burden in the mitochondrial matrix, which is already more negatively charged than the cytosol. Glutamate, however, is a negatively charged amino acid, and most cancer cells export—instead of import—glutamate46. Glutamate efflux is more crucial when NRF2 is activated. In cells with NRF2 activation, most glutamate is secreted, and cystine is imported by the SLC7A11 (xCT) antiporter mechanism47 (Fig. 3).
Glutamate is also utilized to synthesize the antioxidant glutathione4. The first reaction in glutathione synthesis is the ligation of glutamate and cysteine catalyzed by glutamate-cysteine ligase (GCL). Next, glycine is added by glutathione synthetase (GSS). Additionally, glutamate can be converted to glycine through a transamination reaction catalyzed by phosphoserine aminotransferase (PSAT1) into phosphoserine (3-PS) and α-KG. Phosphoserine is subsequently converted to glycine via serine hydroxymethyltransferase (SHMT) (Fig. 3). In cancer cells, the use of glutamate-derived nitrogen for NEAA production may be favored in various types of cancer cells to preserve nitrogen for anabolic reactions48 and may prevent apoptosis induced by ATF4 activation upon glutamine deprivation6.
Asparagine
Asparagine can be synthesized de novo from glutamine via asparagine synthetase (ASNS). Interestingly, asparagine was reported to be able to rescue cancer cells from glutamine deprivation-induced apoptosis43. This finding is surprising because asparagine supplementation does not restore the levels of other NEAAs (alanine, proline, and glutamate) or any TCA cycle intermediates (α-KG, malate, and fumarate). Instead, asparagine supplementation enhances the expression of glutamine synthetase (GLUL) and increases intracellular glutamine usage via glutaminolysis, resulting in the recovery of global protein translation that is blocked by glutamine deprivation45. These studies suggest that most glutamine-dependent protein translation activities can still proceed under asparagine supplementation in a glutamine-deprived environment, although the exact mechanism is still unknown. Furthermore, studies performed in endothelial cells, Kaposi’s sarcoma-associated herpesvirus (KSHV)-transformed cancer cells and several normal fibroblast or epithelial cell lines reported a similar effect of asparagine on supporting cell survival and protein translation after glutamine deprivation44,49,50. Interestingly, high intracellular asparagine levels have recently been identified to be essential for breast cancer metastasis51. This study suggested that l-asparaginase treatment alone can reduce the incidence of breast cancer metastasis to the lung without affecting primary tumor growth. Although the clinical effect of l-asparaginase clearly indicates that asparagine is crucial for tumor survival and metastasis52, the importance of asparagine beyond protein synthesis and the mechanism by which asparagine supplementation enhances glutamine-associated metabolism are less well understood. Recently, asparagine has been reported to function as an exchange factor needed for the uptake of other amino acids that are required for mTORC1 activation53 and for enhanced nucleotide biosynthesis under mitochondrial electron chain transport system impairment54. Further investigation is needed to explain the considerable mechanistic importance of asparagine in cancer metabolism.
Redox control of glutamine
A low level of reactive oxygen species (ROS) activates tumorigenic growth signaling; however, when the level exceeds the cellular redox capacity, ROS can damage macromolecules such as proteins, lipids and nucleotides55. Recent studies suggest that cancer cells are under increased oxidative stress caused by oncogenic transformation, leading to metabolic alterations that result in ROS production56. Under these conditions, glutamine metabolism becomes essential for maintaining cellular redox homeostasis by harnessing enhanced ROS levels. The metabolic pathway by which glutamine mitigates ROS is the glutathione synthesis pathway57 (Fig. 3). Glutathione is a tripeptide (Glu–Cys–Gly) that deactivates peroxide-free radicals. Glutamine is considered the rate-limiting factor in glutathione synthesis58,59. Indeed, experiments using uniformly labeled 13C-glutamine showed that glutathione was enriched with five 13C atoms in glutathione, suggesting that glutamine is the major source of glutathione16,57,60. As shown in Fig. 3, glutamine is a direct fuel for the use of glutathione as a source of glutamate and is indirectly responsible for cystine uptake via the xCT antiporter system, which takes up cystine and simultaneously secretes glutamate61. Consistent with this observation, glutamine starvation has been associated with impaired uptake of cystine through xCT and decreased intracellular glutathione levels62. Furthermore, cells in several types of cancers are characterized by significant enhancement of glutathione biosynthesis, and this metabolic vulnerability has been targeted to sensitize these cancer cells to ROS-induced drugs63.
Glutathione can be recovered from its oxidized form, accompanied by the conversion of NADPH to NADP+. In pancreatic cancer cells, glutamine supports the production of NADPH via a noncanonical metabolic pathway29, and the mitochondrial glutamine transporter is strongly associated with glutaminolysis-induced NADPH generation16. In addition, IDH1-dependent reductive glutamine metabolism produces NADPH, which decreases mitochondrial ROS during anchorage-independent growth64. In summary, glutamine maintains cellular redox homeostasis by supplying fuels for glutathione synthesis and endowing reducing power in the form of NADPH for sustaining tumor growth.
Control of glutamine metabolism by hypoxia
Hypoxic conditions promote the uptake of glutamine by increasing the levels of glutamine transporters such as SLC1A5, the SLC1A5 variant, and SLC38A216,65 and switch the fate of glutamine from the oxidative pathway into the reductive carboxylation pathway24. This metabolic adaptation is critical because of the reduced entry of pyruvate into the TCA cycle by activated PDK1 and the increased lactate secretion in hypoxia66. Via this metabolic adaptation, cells can continually generate TCA metabolites, such as α-KG and citrate, which are converted to cytosolic acetyl-CoA for lipid biosynthesis (Fig. 4).

Hypoxia stabilizes HIF-α proteins such as HIF-1α and HIF-2α. HIF-1α enhances glucose uptake and increases the level of glycolytic enzymes. Under hypoxic conditions, most glucose-derived pyruvate is converted into lactate via LDHA and exported to the extracellular space through the lactate transporters SLC16A1 and SLC16A4. Under these conditions, HIF-2α-mediated glutaminolysis becomes essential to support the adaptation to hypoxia, altering the metabolic fate of glutamine via reductive carboxylation to generate citrate. Then, citrate participates in fatty acid synthesis in the cytosol, which is also activated by stabilized HIF-2α. Hypoxia-induced acidic pH also plays a crucial role in the production of L-2-HG by affecting the substrate affinities of LDHA and MDH. Next, L-2-HG can control DNA or histone methylation levels by regulating α-KG-dependent dioxygenases. HIF hypoxia-inducible factor, GLS glutaminase, GLUD glutamate dehydrogenase, IDH isocitrate dehydrogenase, MDH malate dehydrogenase, L-2HGDH L-2-hydroxyglutarate dehydrogenase, LDHA lactate dehydrogenase, TETs ten-eleven translocation enzymes, JHDMs JmjC domain-containing histone demethylases, Gln glutamine, Glu glutamate, α-KG α-ketoglutarate, L-2-HG L-2-hydroxyglutarate, Me methylation.
HIF-α is the most well-known transcription factor activated in hypoxia. HIF-1α is activated due to blockade of its degradation pathway mediated by low oxygen levels, thereby increasing the expression of target genes, including those encoding glycolytic enzymes and glucose transporters, and increasing lactate secretion67. Although HIF-2α has biochemical characteristics similar to those of HIF-1α, the metabolic role of HIF-2α in a low-oxygen environment is relatively unknown68. Recently, hypoxia-induced expression of the SLC1A5 variant was shown to be mediated by HIF-2α and to lead to metabolic reprogramming toward glutamine metabolism in pancreatic cancer cells16. Given that HIF-2α is an important transcription factor in cancer progression and leads to poor prognosis69,70, these findings suggest that targeting HIF-2α might be an effective therapeutic strategy by inhibiting glutamine metabolism in these notorious cancers. Furthermore, long-term exposure of cancer cells to acidic extracellular conditions induces metabolic reprogramming toward glutamine metabolism via HIF-2α activity71. In addition, extracellular lactate stabilizes HIF-2α, and HIF-2α then transactivates MYC, increasing the levels of glutamine transporters and GLS1, in turn resulting in increased glutamine catabolism72. These findings indicate that just as HIF-1α generally affects glucose metabolism in hypoxia, HIF-2α also plays a distinct role in glutamine metabolism to promote metabolic adaptation in hypoxia (Fig. 4).
Fatty acid synthesis is an anabolic process that uses cytosolic citrate to produce acetyl-CoA73. Glutamine acts as an alternative fuel for fatty acid synthesis, supplying citrate via mitochondrial reductive carboxylation, especially under hypoxic conditions74,75. In the context of constitutive HIF-2α stabilization75 or a defective mitochondrial electron transport chain76, glutamine-derived α-KG is reductively carboxylated through the consumption of NADPH by IDH2 to generate citrate. Next, mitochondrial citrate is transported across the inner mitochondrial membrane via a citrate carrier (CIC or SLC25A1) to support fatty acid synthesis for tumor progression in hypoxia73 (Fig. 4). This mechanism is very important in clear cell renal cell carcinoma (ccRCC) in which HIF-2α signaling is constitutively activated and intracellular lipid droplets are abundant. Fatty acid synthesis induced by HIF-2α is crucial for cell viability in ccRCC by sustaining endoplasmic reticulum (ER) homeostasis77. Furthermore, HIF-2α represses the transcription of carnitine palmitoyltransferase 1A (CPT1A), which is responsible for mitochondrial β-oxidation by transporting fatty acids and results in lipid deposition78. Indeed, recent studies have shown that HIF-2α can be targeted by selective inhibitors and have indicated that these molecules effectively suppress cancer cell growth and tumor angiogenesis characteristics in ccRCC79,80,81,82. Thus, HIF-2α-induced fatty acid synthesis using glutamine-derived citrate can be therapeutically targeted in several cancers, especially ccRCC.
In several cancers, glutamine metabolism is closely related to hypoxia-induced chemoresistance83. For example, glutamine depletion has been shown to abolish hypoxia-induced chemoresistance in cholangiocarcinoma. Impairing glutamine metabolism also induces sensitivity in gemcitabine-resistant pancreatic cancer cells16,84,85. This bolstered chemoresistance in cancer cells is partially supported by glutathione synthesis via glutaminolysis86. Given the importance of glucose and glutamine metabolism in pancreatic cancer cells, it is not surprising that gemcitabine resistance is closely associated with metabolic status, including cellular glucose and glutamine levels. Hypoxia increases the deoxycytidine triphosphate (dCTP) level through the pentose phosphate pathway (PPP) via glucose metabolism and results in resistance to gemcitabine, a dCTP analog87. Furthermore, redox modulation augmented by increased glutathione synthesis from glutamine was reported to be the mechanism of resistance to gemcitabine in pancreatic cancer cells16. Consistent with these findings, while NRF2 induces chemoresistance in KRAS-driven cancers, suppressing glutamine metabolism leads to weakened chemoresistance in these cancer cells85. These studies suggest that targeting glutamine metabolism can be an effective cancer treatment strategy when combined with conventional anticancer chemotherapy.
Under hypoxic conditions, L-2-hydroxyglutarate (L-2-HG) was proven to be generated by lactate dehydrogenase A (LDHA) and malate dehydrogenase (MDH)88,89. Under normal physiological conditions, LDHA catalyzes the conversion of pyruvate to lactate. However, under hypoxic conditions, LDHA can produce L-2-HG. The cellular metabolic alteration of increased L-2-HG levels contributes to the regulation of histone and DNA methylation levels by inhibiting epigenetic modification enzymes that use α-ketoacid as a cofactor. These events mitigate cellular reductive stress by suppressing key metabolic pathways, indicating a crucial role of L-2-HG. Acidic pH has also been reported to induce L-2-HG production via the promiscuous activity of LDHA and MDH enzymes. Acidic pH impairs the activity of the mitochondrial L-2-HG removal enzyme L-2-hydroxyglutarate dehydrogenase (L2HGDH) and enhances the protein stabilization of HIF-1α, leading to its escape from the degradation pathway90. In addition, L-2-HG accumulation in an acidic pH environment has been reported to result in HIF-1α stabilization in normoxia91 (Fig. 4).
Homozygous L2HGDH mutations in germline transmission cause a disease named 2-hydroxyglutaric aciduria (L-2-HGA)92. L-2-HGA is an autosomal recessive encephalopathy with an onset in childhood that causes developmental delays, epilepsy and cerebellar ataxia, the traditional clinical signs of this condition. Interestingly, patients with L-2-HGA are affected by tumors, including brain tumors93, bone tumors94, and nephroblastoma (Wilms tumor)95. Furthermore, increased L-2-HG levels caused by reduced expression of L2HGDH have been reported in renal cancer96. These studies indicate an oncogenic effect of L-2-HG and the association of L-2-HG with tumorigenesis under hypoxic conditions.
Control of epigenetic changes by glutamine
The metabolic state constitutes a fundamental component of chromatin modification and genome regulation97. As metabolites are the substrates used to generate chromatin modifications, including methylation and acetylation modifications of histones, a complicated interaction exists between metabolism and epigenetics. In particular, glutamine-derived α-KG has been implicated in regulating cellular histone and DNA methylation levels98.
α-KG, also named 2-oxoglutarate, is a cofactor for 2-oxoglutarate-dependent dioxygenases (2-OGDDs), which catalyze hydroxylation reactions on diverse substrates. The activities of 2-OGDDs are affected by the intracellular level of α-KG, succinate, fumarate, or 2-HG. These hydroxylation reactions also require Fe2+ as a cofactor, O2 as a cosubstrate and ascorbic acid (vitamin C) as a reductase, which restore the activity of 2-OGDD enzymes (Fig. 5a). Among 2-OGDDs, Jumonji C domain-containing histone demethylases and ten-eleven translocation (TET) family DNA demethylases are major enzymes that induce epigenetic modifications using glutamine-derived α-KG. In these reactions, α-KG is oxidized to succinate, and increasing levels of succinate can suppress the progression of α-KG-dependent histone or DNA demethylase reactions98.

a Several mutations in enzymes in the glutaminolysis pathway are responsible for the production of oncometabolites. Mutation of IDH1 and IDH2 produces R-2-HG from α-KG, which, when accumulated, leads to the inhibition of dioxygenases, in turn leading to the activation of TET and JHDM enzymes inside the nucleus. Mutation of SDH arrests the TCA cycle, resulting in an increase in the succinate concentration. A high concentration of succinate has an effect similar to the oncometabolite effect of R-2-HG. Additionally, impaired function of FH prevents further metabolism of fumarate, leading to its accumulation. FH impairment inhibits the function of Keap1 and PHD, which stimulates the transcription of protooncogenes. Gln glutamine, Glu glutamate, α-KG α-ketoglutarate, IDH isocitrate dehydrogenase, 2OGDH 2-oxoglutarate dehydrogenase, SDH succinate dehydrogenase, FH fumarate hydratase, R-2-HG R-2-hydroxyglutarate, Keap1 Kelch-like ECH-associated protein 1, PHD prolyl hydroxylase, TETs ten-eleven translocation enzymes, JHDMs JmjC domain-containing histone demethylases, Me methylation. b. Glutamine anaplerosis is a key mitochondrial metabolic pathway for cancer cell growth and survival. Influx of glutamine-derived α-KG into the TCA cycle replenishes the intermediates and consequently generates NADH, FADH2, and GTP. The generated GTP can be readily converted to an equal amount of ATP. Additionally, glutamate and α-KG produced via glutaminolysis participate in the malate-aspartate shuttle, promoting the transport of NADH from the cytosol into mitochondria. Elevated mitochondrial NADH and FADH2 levels collectively contribute to enhanced ATP production via OXPHOS through the ETC. Gln glutamine, Glu glutamate, Asp aspartate, αKG α-ketoglutarate, GOT1/2 glutamic-oxaloacetic transaminase 1/2, MDH1/2 malate dehydrogenase 1/2, OAA oxaloacetate, OGC 2-oxoglutarate carrier, AGC aspartate-glutamate carrier, ETC electron transport chain.
In cancer cells, mutations in succinate dehydrogenase subunit B (SDHB) cause susceptibility to familial pheochromocytoma99 and familial paraganglioma100 as well as gastrointestinal stromal tumors101. An increased ratio of succinate to α-KG in cancers resulting from impaired succinate dehydrogenase (SDH) activity is related to pervasive DNA hypermethylation, which contributes to the downregulation of key genes implicated in cell differentiation and cancer stages102. Moreover, the core region of solid tumors exhibits a deficiency of glutamine compared with other amino acids. This severe glutamine deprivation leads to dramatic histone hypermethylation due to decreased α-KG levels subsequent to decreased activity of Jumonji domain-containing histone demethylases and results in cancer cell dedifferentiation and resistance to BRAF inhibitors103.
In addition to its role in cancer cells, α-KG supports the self-renewal of naive murine embryonic stem cells (mESCs) by promoting histone and DNA demethylation104. In addition, at later stages of pluripotency, α-KG derived from glutamine can promote early differentiation, suggesting that the stage of cellular maturity can alter the effect of α-KG105. Furthermore, PSAT1 regulates changes in the level of glutamine-derived α-KG, which controls mESC pluripotency and differentiation106. These reports suggest that α-KG generated via glutaminolysis is closely related to the cellular decisions that characterize stem cells. In skeletal stem cells (SSCs), GLS and glutamine metabolism are required for the regulation of osteoblast and adipocyte specification and physiological bone formation107. In macrophage cells, α-KG produced via glutaminolysis promotes M2 activation via Jmjd3-dependent metabolic and epigenetic reprogramming108.
In T cell activation, glutamine deprivation has been shown to alter the activation of naive CD4+ T cells and result in their differentiation into forkhead box P3-positive (Foxp3+) regulatory T (Treg) cells, which have suppressor functions109. Recently, glutamine metabolism has been shown to be linked to white adipose tissue (WAT) inflammation in obesity110. The researchers discovered that glutamine metabolism is impaired in the obese state, leading to increased chromatin O-GlcNAcylation and activation of genes in proinflammatory pathways.
Collectively, glutamine-derived metabolites act as epigenetic modulators in a wide range of cell and tissue types, including various types of cancer cells, stem cells, immune cells, and even adipocytes. Considering that the SLC1A5 variant is an important regulator of the production of glutamine-derived α-KG16, confirming whether epigenetic regulation by glutamine-derived α-KG is affected by the SLC1A5 variant in cancer cells or stem cells is necessary (Fig. 5a).
Glutamine and its oncometabolites
The discovery of R-2-hydroxyglutarate (R-2-HG) accumulation in several tumors encouraged investigators to initially establish the term “oncometabolite”111. Genetic and metabolic studies have further shown that metabolites such as succinate and fumarate, which are generated under normal physiological conditions, are associated with tumorigenesis in several cancer types. Interestingly, these metabolites were often found to be associated with glutamine metabolism112. In particular, the production of these oncometabolites was affected by the level of glutamine-derived α-KG. Although additional studies are needed, ample experimental data support the recognition of R-2-HG, succinate, and fumarate as oncometabolites.
R-2-HG
Wild-type IDH1 and IDH2 catalyze the reaction by converting isocitrate and NADP+ into α-KG and CO2 with the concomitant generation of NADPH in the cytosol and mitochondrial matrix. However, mutant IDH enzymes convert α-KG into R-2-HG with the oxidation of NADPH into NADP+. Thus, various tumors, including glioma, secondary glioblastoma, and acute myeloid leukemia (AML), harboring heterozygous point mutations in the active sites of IDH1/2 show dramatic increases in the R-2-HG levels111,113,114,115. A high level of R-2-HG is sufficient to cause leukemia to arise from hematopoietic cells by maintaining their dedifferentiation and proliferation activities116. The role of R-2-HG as an oncometabolite has been implicated in epigenetic modifications through the inhibition of α-KG-dependent dioxygenases and demethylases, which has been assumed to be a driver of tumorigenesis117,118. In addition, dysregulated α-KG flux from normal reductive anabolism via the TCA cycle toward R-2-HG production has been associated with other metabolic flux impairments and disrupted redox balance119,120 (Fig. 5a).
Interestingly, the generation of R-2-HG from glutamine has been proven to occur rapidly in patient-derived chondrosarcoma cell lines harboring endogenous IDH mutations, indicating fundamental metabolic differences between cells that harbor IDH1/2 mutations and those that do not121. In this study, glutamine flux was directed toward the generation of R-2-HG in IDH1/2 mutant cells, and the kinetics of R-2-HG formation were proportionally of the same order of magnitude as those of glutamate or α-KG formation via glutaminolysis. Indeed, glutamine-derived R-2-HG accumulates and prevents the differentiation of myeloblasts, resulting in uncontrolled growth of blood cells122. After FDA approval of enasidenib, a first-in-class drug targeting cancer metabolism via inhibition of IDH2 activity, more studies were conducted with R-2-HG positioned as an oncometabolite. CB-839, a GLS inhibitor that blocks the conversion of glutamine into glutamate, reduced the production of R-2-HG in AML cell lines and patient tissues harboring IDH1/2 mutations123. As the importance of R-2-HG in boosting tumor initiation, proliferation and metastasis is emphasized, identifying whether metabolic enzymes or transporters associated with glutamine metabolism could be involved in the generation of R-2-HG is interesting.
Succinate
The normally functioning SDH enzyme is localized in the inner mitochondrial membrane and plays a role in the electron transport chain as well as the conversion of succinate into fumarate. In 2008, mutation of SDH was discovered in cancers such as paraganglioma and pheochromocytoma cells124. Later, similar observations were made in gastrointestinal tumors, neuroblastomas, renal tumors, thyroid tumors, and testicular tumors125,126. Several research groups have focused on the mechanism that underlying the features of tumorigenesis and cancer cell survival in the setting of SDH mutations. As succinate accumulates via the inhibition of the 2-OGDD enzyme, epigenetic modification acts in the process of cell transformation into a hypermethylated phenotype100. Several studies have shown that SDH-deficient cells exhibit increased tumorigenesis and that this increase is reversed by the addition of α-KG, supporting the idea that succinate accumulation contributes to tumorigenesis through epigenetic modification100. Succinate-specific effects are initiated by epigenetic alterations through the inhibition of KDMs and the TET family 5mC hydroxylases, which induce the translation of tumorigenic genes (Fig. 5a). The other mechanism by which succinate supports tumorigenesis acts through the inhibition of hypoxia-inducible factor prolyl hydroxylase (PHD). PHD activates the pseudohypoxic response by stabilizing HIF-1α, which is a well-known tumorigenesis enhancer, and as a transcription factor, maintains the metabolic reprogramming of cancer cells to support their survival127. In addition to the tumorigenic effects of succinate accumulation, SDH5 mutation is the key driver supporting the acquisition of epithelial–mesenchymal transition (EMT) characteristics. The results of a clinical study further confirmed this observation by showing that patients with nonmetastatic lung cancer harbored loss-of-function mutations in SDH5128. The study of succinate as an oncometabolite has only recently begun, and more research needs to be conducted to completely understand its tumorigenic properties.
Fumarate
Fumarate is another example of an oncometabolite produced by the action of fumarate hydratase on succinate. In 2001, mutation of fumarate hydratase leading to its inactivation was discovered in renal cell cancer129. Mutation of this enzyme leads to fumarate accumulation not only in skin cancer and uterine leiomyomas but also in breast, bladder, and Leydig cell tumors130. Further confirmation of fumarate as an oncometabolite was verified by experimental data showing that tumor cells lost their ability to invade and migrate when the function of fumarate hydratase was restored by an external expression vector131. In attempts to understand the cause of these effects, it was found that cells with high concentrations of fumarate display a phenotype of DNA hypermethylation. In addition, fumarate inhibits TET enzymes, which stimulate EMT, leading to cancer metastasis131,132. Similar to succinate, fumarate contributes to the inactivation of PHD, stabilizing HIF proteins to promote cell survival133 (Fig. 5a). In addition, accumulated fumarate can participate in different reactions of the addition of a succinate group to the thiol group of various proteins. For example, in hereditary leiomyomatosis and renal cell cancer (HLRCC), a high level of fumarate caused by genetic mutation of fumarate hydratase induces the succination of Kelch-like ECH-associated protein 1 (KEAP1) accompanied by the consumption of a fumarate molecule134,135. Endogenously, succinylated KEAP1 dissociates from the NRF2 protein to help cancer cells survive stress. High concentrations of fumarate bind to glutathione, augmenting ROS signaling and accumulation, as observed in not only in vitro models but also in vivo models136,137. Additionally, high levels of fumarate react with the cysteine group of mitochondrial aconitase-2 and iron-sulfur cluster binding protein-2, facilitating cellular metabolic adaptation to stresses138. The importance of fumarate hydratase mutation for cancer survival and growth is being studied in depth to completely understand the role of fumarate as a tumorigenic oncometabolite. This knowledge will aid in the complete comprehension of cancer metabolism.
Glutamine-derived energy production
The influx of α-KG into the TCA cycle and its subsequent oxidization generates two molecules of NADH and one molecule of FADH2 from the series of reactions catalyzed by OGDH, SDH, and MDH. Additionally, when succinyl-CoA is converted to succinate by succinate thiokinase, one molecule of GTP is generated, which can be readily converted to ATP by nucleoside-diphosphate kinase (NDPK). NADH and FADH2 produced via glutaminolysis are then fed into the electron transport chain to create the electrochemical gradient necessary for ATP production via oxidative phosphorylation139,140 (Fig. 5b). Correspondingly, in K-Ras mutant cells, the oxygen consumption rate and ATP generation are enhanced by glutamine, contributing to tumorigenesis55. Moreover, after the activation of K-Ras and Akt in transformed cells, 60% of the total FADH2 and NADH2 are synthesized from glutamine, while only 30% is derived from glucose140. Additionally, the level of the mitochondrial glutamine transporter controls the cellular ATP level stimulated by glutamine, suggesting that glutamine is an important energy source via mitochondrial glutaminolysis16. Collectively, these observations indicate that anaplerotic glutamine metabolism is highly responsible for energy generation in cancer cells.
Additionally, NADH can be generated by fatty acid oxidation (FAO) in the cytoplasm in tissues with high energy demand, such as cardiac muscle tissues, as well as in cancer cells141. Recent studies have suggested that in cancer cells with elevated cytosolic NADH levels, the malate-aspartate shuttle (MAS) actively takes up NADH to produce ATP in mitochondria through the electron transport chain142. The MAS comprises MDH1/2, GOT1/2, the malate-α-KG antiporter and the glutamate-aspartate antiporter, which exchanges mitochondrial α-KG for cytosolic malate that is synthesized from oxaloacetic acid (OAA) by cytosolic MDH (Fig. 5b). Glutamate and α-KG serve as important exchangers in the MAS, and since GLS1 knockdown significantly suppresses NADH and ATP production in cancer cells143, the supply of glutamate and α-KG for the induction of MAS activity is evidently critical for ATP production in cancer cells (Fig. 5b).
Glutamine metabolism upon cellular stresses
Glutamine is the most abundant amino acid in the blood. During cellular stress, such as nutrient starvation and catabolic stress after trauma, surgery, infection, sepsis, or cancer cachexia, blood glutamine levels are severely decreased144. Under these conditions, several studies have reported that glutamine supplementation can offer a therapeutic approach for these critical illnesses145,146,147. Glutamine has been considered an immunomodulatory amino acid in several disease states, yet the mechanisms underlying the therapeutic effects of glutamine supplementation in critical illness remain poorly understood. Conceivably, glutamine could exert its beneficial effects by producing glutathione for redox homeostasis, maintaining nitrogen balance, or other functions in immune cells2.
Consistent with the importance of glutamine in stressful situations, glutamine deprivation induces cellular stress. Upon glutamine starvation, p53 activity is induced and can help cancer cells adapt to nutrient starvation through diverse mechanisms148. Recently, SLC1A3, as a crucial effector of p53, has been shown to support cell survival and growth in the absence of glutamine149. Under DNA damage such as radiation, glutamine is conditionally essential to support the synthesis of nucleotides and redox homeostasis. It has recently been demonstrated that radioresistant cancer cells reprogram metabolic flux toward glutamine anabolism. Under these conditions, cancer cells highly express glutamine synthetase, facilitating cancer cell growth under radiation stress150. Moreover, evidence has shown that during the DNA damage response, normal cells show a decrease in glutaminolysis controlled by SIRT4 protein suppressing GLUD1. In the absence of SIRT4, a failure to undergo cell cycle arrest induced by DNA damage causes a delay in DNA repair and increased chromosomal instability, suggesting a tumor suppressor effect of SIRT4151.
Numerous studies have described the presence of alternative adaptive pathways upon the perturbation of glutamine metabolism. For instance, a recent study has shown that GLS1 inhibition induces an increase in mitochondrial glutamate-pyruvate transaminase 2 (GPT2) to assist in TCA cycle anaplerosis for sustaining cancer cell growth and survival152. Of note, GLS1 inhibition causes an elevation of the ROS level and induces GPT2 expression via ATF4, which again implies the importance of ATF4-mediated metabolic adaption during glutamine starvation.
Additionally, metabolic profiling has revealed that suppression of GLS1 induces a compensatory anaplerotic mechanism via pyruvate carboxylase (PC), which allows the release of a glutamine-independent supply of TCA intermediates by catalyzing the transformation of pyruvate to oxaloacetate153. This PC-mediated alternative anaplerosis is considered important in specific types of cancers, including liver cancers and glioblastoma, for maintaining biosynthesis and redox homeostasis154,155,156. Collectively, cancer glutamine metabolism shows extraordinary flexibility and is intertwined with diverse metabolic pathways.
Metabolic reprogramming induced by glutamine metabolism
Unsurprisingly, glutamine metabolism plays a critical role in tumor progression since it not only supports mitochondrial oxidative phosphorylation but also supplies metabolic intermediates for the TCA cycle, glutathione synthesis, and NEAA synthesis and simultaneously produces NADPH157,158,159. Recently, glutamine was shown to be a major fuel for mitochondrial oxygen consumption in pancreatic cancer cells; in addition, the expression of the SLC1A5 variant affected the levels of metabolites derived from glucose metabolism, including lactate and ribulose-5-phosphate, the intermediate metabolites in the PPP16. Intriguingly, this study regarding elevated glutamine metabolism in cancer cells also showed that glutaminolysis could in turn reinforce metabolic reprogramming, thus implying that glutamine metabolism plays a crucial role in tumorigenesis and tumor progression16 (Fig. 6a). Indeed, the process of adaptation to glutamine deprivation weakens the response to hypoxia, which normally strongly induces the expression of glycolytic enzymes83.

a Aerobic glycolysis is a hallmark of cancer metabolism. During this process, most glucose-derived pyruvate is secreted extracellularly as lactate, and glutamine becomes a conditionally essential amino acid. Glutaminolysis sustains mitochondrial function, supplying TCA cycle metabolites such as αKG and generating diverse biomolecules, including NEAAs, NADPH, and nucleotides. Increased glutamine flux into the mitochondrial matrix via the SLC1A5 variant can enhance glutaminolysis and lead to metabolic reprogramming toward enhanced aerobic glycolysis. b Glutamine-derived α-KG activates the mTORC1 signaling pathway, resulting in aerobic glycolysis and protein translation, which are crucial for tumor proliferation. c During glutaminolysis, ammonium ions are generated via a deamidation reaction catalyzed by glutaminase and glutamate dehydrogenase. Most ammonium ions are used as a nitrogen source for nucleotide biosynthesis and are disposed of via the urea cycle, but an excess of ammonium ions promotes autophagy. Augmented autophagy is associated with drug resistance by enhancing aerobic glycolysis and is involved in cancer cell survival, progression, and metastasis. Gln glutamine, Glu glutamate, α-KG a-ketoglutarate, PHD prolyl hydroxylase.
As previously described, glutamine is metabolized by mitochondrial enzymes into α-KG, which serves as an important intermediate in the TCA cycle for anaplerosis. Furthermore, enhanced production of α-KG causes other critical effects, such as stimulation of the signaling pathways that support cell growth. α-KG induces mTORC1 activation by enhancing GTP loading of the RagB protein in a PHD-dependent manner, thus promoting cell growth160,161. Accordingly, high mTORC1 activity in cancer cells promotes aerobic glycolysis and drives glucose addiction162,163 (Fig. 6b). In addition, mTORC1 activation via glutaminolysis suppresses autophagy and the DNA damage response164,165. Therefore, enhanced glutaminolysis might eventually contribute to the initiation and progression of cancer by stimulating cell growth via the mTORC1 pathway and enhancing aerobic glycolysis while disrupting the proper elimination of misfolded proteins, damaged DNA and organelles through the inhibition of autophagy and the DNA damage response166.
Enhanced glutaminolysis in cancer cells ensures a stable supply of glutamate and α-KG via sequential deamination processes inside mitochondria. Notably, ammonia is simultaneously generated as a byproduct of glutamine deamination. Hence, the facilitation of glutaminolysis leads to the accumulation of excess ammonia within cells, and a high concentration of ammonia is a potent inducer of autophagy167 (Fig. 6c). Although mTORC1 activation hinders autophagy, evidence has shown that autophagy can be upregulated in tumors with mTORC1 hyperactivation168. Therefore, glutaminolysis can suppress autophagy by activating the mTORC1 pathway but, on the other hand, can stimulate autophagy in the context of excess ammonia production. The fundamental need for ammonia-mediated induction of autophagy in cancer cells could be due to the cytoprotective functions of this event that allow cells to survive under extreme conditions166. Specifically, autophagy suppresses anoikis induced by the detachment of cancer cells from the extracellular matrix (ECM) and hence promotes metastasis169. Furthermore, autophagy has been shown to promote glycolysis in hepatocellular carcinoma (HCC) cells by upregulating monocarboxylate transporter 1 (MCT1), which plays an important role in the transport of lactic acid170. Therefore, autophagy supports cancer progression and chemoresistance by allowing tumor cells to overcome both environmental and intracellular stress signals, including nutrient deprivation and chemotherapeutic cytotoxicities167,171,172 (Fig. 6c).
However, the connection between glutamine and metabolic remodeling in cancer from the perspective of glucose metabolic flux, the mTORC1 pathway and autophagy has yet to be fully explored. This link might partially be explained by considering that the intimately entwined glucose and glutamine metabolic pathways cooperatively support the TCA cycle and that glutamine performs diverse functions for maintaining cellular homeostasis. Collectively, in-depth investigation of the role of glutaminolysis in tumor progression might hold the key for decoding cancer metabolic plasticity.
Crosstalk between glutamine metabolism and oncogenic signaling
The excessive proliferation exhibited by cancer cells demands a constant supply of fuels such as glucose and glutamine. Therefore, cancer cells orchestrate their metabolic pathways to coordinate their high demand for these nutrients. Metabolic reprogramming that promotes enhanced glutamine consumption in cancer cells is closely connected with dysregulation of oncogenes. Efforts have been undertaken to reveal the mechanism by which oncogenes modulate metabolic pathways that favor cancer cell growth173. Notably, cancer cells driven by oncogenic MYC, K-Ras, and PIK3CA require glutamine for their survival and display extensive anabolic utilization of glutamine29,174,175 (Fig. 7).

Oncogenes such as MYC, K-Ras, and PI3KCA modulate cancer metabolic reprogramming, favoring cancer cell growth and survival partially via the promotion of glutamine metabolism. Glutamine uptake is enhanced in MYC- and K-Ras-driven cells in which the expression of the glutamine transporter SLC1A5 is upregulated. Deamination of glutamine to form glutamate in mitochondria is enhanced by MYC-mediated upregulation of GLS1. Conversion of glutamate into α-KG is mediated by GLUD1 or aminotransferases such as GOT1/2 and GPT2. The expression of these enzymes is upregulated in cancer cells with MYC-driven, K-Ras-driven, and PI3KCA-driven signaling activation. Gln glutamine, Glu glutamate, Ala alanine, Asp aspartate, α-KG α-ketoglutarate, GLS1 glutaminase 1, GLUD1 glutamate dehydrogenase 1, GOT1/2 glutamic-oxaloacetic transaminase 1/2, GPT1/2 glutamic-pyruvate transaminase 1/2, MDH1 malate dehydrogenase 1, ME1 malic enzyme 1.
In cancer cells, genetic and epigenetic dysregulation of MYC expression and the loss of checkpoint components unleash the ability of MYC to promote cell growth, eventually leading to malignant transformation176. Oncogenic Myc stimulates mitochondrial glutaminolysis via transcriptional regulation of genes necessary for cellular glutamine catabolism177. MYC-driven cancer cells exhibit enhanced glutamine utilization accompanied by increased expression of key glutaminolysis enzymes, including GLS1/GLS2 and GLUD1178,179,180. Moreover, MYC upregulates the glutamine transporter SLC1A5 to facilitate glutamine uptake into cells177. MYC-dependent enhancement of mitochondrial glutaminolysis leads to the reprogramming of mitochondrial metabolism to accommodate the requirements for TCA cycle anaplerosis to sustain cellular viability and growth.
Similar to the situation in MYC-driven cancer cells, glutamine uptake is enhanced in K-Ras-driven cells via upregulation of SLC1A5181. Additionally, K-Ras-driven cells are characterized by increased expression of GOT1 and GOT2182,183. GOT1 and GOT2 catalyze the transamination reaction between oxaloacetate and glutamate to produce aspartate and α-KG. Significantly, enhanced transamination and aspartate synthesis in K-Ras-driven cancer cells are important in the promotion of nucleotide biosynthesis184 and maintenance of redox balance29.
Intriguingly, the glutamine-dependent checkpoint at late G1 phase in the cell cycle is dysregulated in K-Ras-driven cancer cells185. In normal cells, the cell cycle is tightly regulated by various checkpoints. Nutrient-dependent checkpoints regulate cell cycle passage through late G1 phase by sensing nutrient availability; glutamine is a particularly critical nutrient sensed in late G1 phase, and its deprivation causes cell cycle arrest at G1 phase186. Importantly, activation of K-Ras in cancer cells results in bypass of the late G1 glutamine-dependent checkpoint. Specifically, glutamine deprivation in K-Ras-driven cancer cells leads to growth arrest in S or G2/M phase instead of in G1 phase. Consistent with this observation, K-Ras sensitizes cells to glutamine deprivation, and K-Ras knockdown rescues cells from apoptosis induced by low glutamine levels187. Collectively, these findings indicate that enhanced glutamine metabolism and cell growth dysregulation are established in K-Ras-driven cancer cells to promote uncontrolled cell growth and to assist with glutamine acquisition and utilization for cell growth.
The PI3K signaling pathway is dysregulated in many tumors, and analyses have shown that PIK3CA is an oncogene that also contributes to tumor progression partially via metabolic reprogramming188. Oncogenic PIK3CA increases the dependency of cancer cells on glutamine by upregulating the expression of mitochondrial GPT2, which catalyzes the transamination reaction that converts glutamate and pyruvate into α-KG and alanine175. Thus, cells with PIK3CA mutations exhibit increased sensitivity to glutamine deprivation. Additionally, compared with wild-type cells, PIK3CA mutant colorectal cancer (CRC) cells exhibit elevated anaplerotic α-KG production and ATP generation from glutamine.
In addition to oncogenic regulators, there are some key upstream regulators of glutamine metabolism that are widely recognized for their pivotal role during tumorigenesis. mTORC1, which is well known for its function at the center of cancer metabolic reprogramming, promotes mitochondrial glutaminolysis via the migration of SIRT4-mediated inhibition of GLUD1189. Specifically, mTORC1 promotes proteasome-mediated destabilization of cAMP response element binding-2 (CREB2) to suppress transcription of SIRT4. Accordingly, loss of SIRT4 enhances glutamine-dependent proliferation and genomic instability, which simultaneously contribute to tumorigenesis151.
Furthermore, mTORC1 also acts as a downstream effector of glutamine. Glutamine itself, or after its conversion into α-KG, activates the mTORC1 pathway and participates in the growth signaling pathway. Evidence has shown that glutamine activates the mTORC1 pathway via Arf1 rather than via the Rag GTPase complex in MEFs190. According to another study, glutaminolysis increases the level of α-KG production, resulting in GTP loading of RagB and lysosomal translocation of the mTORC1 complex in human cancer cell lines160. It has been reported that cellular uptake of glutamine and its subsequent efflux in the presence of essential amino acids, including leucine, is the rate-determining step that activates mTORC1191. Moreover, glutamine also acts as a precursor for the synthesis of various NEAAs, including asparagine and arginine, implicated in mTORC1 activation39. Thus, cells have diverse mechanisms of mTORC1 activation for glutamine, and cancer cells efficiently utilize glutamine for mTORC1 pathway activation to drive unrestrained oncogenic growth.
Targeting glutamine metabolism and therapeutic implications
Although the essential role of glutamine metabolism in cancer cells has been well demonstrated in vitro, the extent to which glutamine supports tumor growth and survival in vivo remains elusive. It has been reported that K-Ras-driven mouse lung tumors preferentially utilize glucose more than glutamine to supply carbon to the TCA cycle via pyruvate carboxylase192. Furthermore, human glioblastoma cells do not rely much on circulating glutamine for proliferation but rather more on glutamate to synthesize glutamine via glutamine synthetase to fuel purine biosynthesis193. Nevertheless, the specific metabolic importance of glutamine in tumorigenesis and tumor growth has also been reported194,195,196, and these studies have led many researchers to target glutamine metabolism for the treatment of cancer8. Throughout the discovery of agents targeting glutaminolysis, none have yet been used clinically197. A recent attempt focused on the inhibition of GLSs. GLS overexpression has been observed in different tumor cells, and these enzymes are found to function in the metabolic reprogramming of glutamine addiction in cancer198. Chemical agents targeting GLSs have been studied, and CB-839, 968, and BPTES have been found to exhibit tumor-specific antiproliferative effects199. Among these agents, CB-839 is the only one to proceed to clinical trials; however, its selectivity toward GLS1 and failure to inhibit the compensatory effect of GLS2 require in-depth study14. A recent study discovered a prodrug (JHU083) of the glutamine antagonist DON, which was designed to selectively become activated inside a tumor. The researchers showed that blocking glutamine metabolism through JHU083 not only suppressed tumor cell metabolism but also mitigated the tumor microenvironment, which is hostile to the immune response due to its hypoxic, acidic, and nutrient-depleted conditions, unleashing the natural antitumor T cell response. They also confirmed that concurrent treatment with JHU083 and anti-PD-1 checkpoint inhibitor improved the antitumor effects compared with anti-PD-1 treatment alone, suggesting the presence of metabolic plasticity between cancer cells and effector T cells, which could be exploited as a metabolic checkpoint for cancer immunotherapy200.
The plasma membrane glutamine transporters SLC6A14, SLC7A11, and SLC38A1 have been targeted and found to be inhibited by erastin, α-Me-Trp, and MeAIB, respectively (Fig. 8). In addition, SLC1A5 was shown to have clinical importance, and it is considered the most critical plasma membrane glutamine transporter in cancer cells201. Many attempts have been made to explore the possibility that SLC1A5 suppression via small molecules might exert anticancer effects. As part of this effort, benzylserine and benzylcysteine were discovered in 2004 as the first substrate analog inhibitors of SLC1A5202. In an effort to improve the potency and efficacy of such inhibitors, some studies have discovered GPNA, which is widely used as a tool compound for suppressing SLC1A5203. Other studies have developed antibodies with high affinity for SLC1A5, which induce antibody-dependent cellular toxicity in gastric cancer models204. Recently, a potent inhibitor of SLC1A5, V-9302, has been reported to be effective in several cancer cell lines and in vivo tumor models205. However, other researchers have argued that controversial issues exist because GPNA also inhibits other glutamine transporters, such as SLC38A1, and V-9302 is effective even in SLC1A5 knockout models206,207. Hence, to date, no suitable compound has been identified to inhibit the plasma membrane glutamine transporter SLC1A5 with excellent sensitivity and specificity.

For the principal inhibition of glutaminolysis, attempts have been made to target the amino acid transporters related to these pathways. SLC6A14 and SLC38A1 are inhibited by α-Me-Trp and MeIAB, respectively. The most intensely researched topic is inhibitors of SLC1A5, a major glutamine transporter, which include substrate analog competitive inhibitors such as GPNA, benzylserine, and V-9302 and the inhibitory antibody MEDI7247. Although they exhibit low potency, inhibitors of SLC7A11 include erastin and SSZ. Inhibitors of glutaminolytic enzymes are agents that target GLS1, GOT2, and GLUD1. CB-839, an agent in its 2nd clinical trial, inhibits GLS1 similarly to BPTES and 968. AOA inhibits GOT2 activity, and EGCG, purpurin, and R162 inactivate GLUD1. However, the SLC1A5 variant, the sole glutamine transporter discovered to date, is expected to be a much more effective target for cancer therapeutics than previously studied glutaminolysis inhibitors. Cys cysteine, Glu glutamate, α-KG α-ketoglutarate, GLS glutaminase, GOT2 glutamic-oxaloacetic transaminase 2, GPT2 glutamic-pyruvate transaminase 2, GLUD1 glutamate dehydrogenase 1, α-Me-Trp alpha-methyl-tryptophan, MeAIB methylaminoisobutyric acid, GPNA L-γ-glutamyl-p-nitroanilide, SSZ sulfasalazine, DON 6-diazo-5-oxo-l-norleucine, AOA aminooxyacetate, EGCG epigallocatechin-3-gallate.
SLC1A5 might not be an appropriate target for suppressing glutamine uptake by cancer cells because it is not the only plasma membrane glutamine transporter, and its function would therefore be compensated by other redundant glutamine transporters such as SLC38A1 and SLC38A2. Thus, as the SLC1A5 variant is the only currently known glutamine transporter in the mitochondrial inner membrane16, targeting the SLC1A5 variant could be an effective strategy for selectively inhibiting glutamine metabolism in cancer cells (Fig. 8). Given the clinicopathological significance of SLC1A5201 and the observation that the level of the SLC1A5 variant is negatively correlated with prognosis in several cancer types16, targeting the SLC1A5 variant is a promising strategy to starve cancer cells and induce antitumor effects. Therefore, further studies on the development of selective inhibitors of the mitochondrial SLC1A5 variant are needed and should help to establish whether the level of the SLC1A5 variant is a predictive marker of glutamine dependency in cancer21.
Conclusion
Although Otto Warburg characterized cancer metabolism by its enhanced glucose consumption and loss of mitochondrial function, many studies have shown that mitochondrial function in cancer cells is still robust and even enhanced. Moreover, glutamine has been discovered to be required for the maintenance of active mitochondrial function in cancer cells. Glutamine has historically been one of the most intensely investigated nutrients in cancer metabolism and is involved in various aspects of biosynthesis and bioenergetics, including NEAA production, epigenetic gene control, adaptation to hypoxic conditions, ATP synthesis, cell signaling, and tumorigenesis. In this review, we offer an updated overview of glutamine metabolism and discuss the reason for glutamine dependency in cell metabolism.
Certain types of cancer, including renal cell carcinoma, hematologic malignancies, glioblastoma, pancreatic cancer, and those reported to depend on HIF-2α, seem to depend on glutamine; hence, targeting glutamine metabolism may show therapeutic effects in these cancers. Moreover, metabolite transporters have recently been shown to be involved in tumorigenesis; for example, low levels of mitochondrial pyruvate carriers initiate colon cancer development208. Conversely, suppression of the SLC1A5 variant, a mitochondrial glutamine transporter, is sufficient to inhibit tumor growth by impairing glutamine metabolism in pancreatic cancer cells16. As the importance of subcellular metabolite transporters in controlling tumor initiation is poorly understood, it would be interesting to determine whether overexpression or knockout of these transporters is involved in tumorigenesis, metastasis, and immune modulation.
In conclusion, metabolic reliance on glutamine arises via the intrinsic functional diversity of glutamine, supporting macromolecule biosynthesis and reinforcing the TCA cycle. In the context of tumorigenesis, glutamine-derived 2-HG alters the epigenetic landscape of chromosomes and induces oncogenic transformation. Further investigations to explain the mechanism underlying glutaminolysis-induced metabolic reprogramming are needed. These efforts are anticipated to reveal new metabolic vulnerabilities of cancer cells that can be targeted by therapeutic interventions.
References
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
DeBerardinis, R. J. & Cheng, T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324 (2010).
Hensley, C. T., Wasti, A. T. & DeBerardinis, R. J. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123, 3678–3684 (2013).
Altman, B. J., Stine, Z. E. & Dang, C. V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 749 (2016).
Zhang, J., Pavlova, N. N. & Thompson, C. B. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. Embo J. 36, 1302–1315 (2017).
Qing, G. et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 22, 631–644 (2012).
Chen, L. & Cui, H. Targeting glutamine induces apoptosis: a cancer therapy approach. Int. J. Mol. Sci. 16, 22830–22855 (2015).
Wise, D. R. & Thompson, C. B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 35, 427–433 (2010).
Klingman, J. D. & Handler, P. Partial purification and properties of renal glutaminase. J. Biol. Chem. 232, 369–380 (1958).
Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 122, 501–514 (1955).
Kovacevic, Z. & Morris, H. P. The role of glutamine in the oxidative metabolism of malignant cells. Cancer Res. 32, 326–333 (1972).
Reitzer, L. J., Wice, B. M. & Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 254, 2669–2676 (1979).
Lobo, C. et al. Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem. J. 348, 257–261 (2000).
Gross, M. I. et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13, 890–901 (2014).
Kizilbash, S. H. et al. The addition of CB-839 to temozolomide significantly reduces glioma aspartate and glutamate in an IDH mutated patient derived glioma xenograft model. Cancer Res. https://doi.org/10.1158/1538-7445.AM2019-3870 (2019).
Yoo, H. C. et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 31, 267–283 (2020).
Scalise, M., Pochini, L., Galluccio, M., Console, L. & Indiveri, C. Glutamine transport and mitochondrial metabolism in cancer cell growth. Front. Oncol. 7, 306 (2017).
Wang, J. B. et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 397–397 (2010).
Li, B. et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 39, 239–254 (2019).
Lukey, M. J. et al. Liver-type glutaminase GLS2 is a druggable metabolic node in luminal-subtype breast cancer. Cell Rep. 29, 76–88 e77 (2019).
Stine, Z. E. & Dang, C. V. Glutamine skipping the Q into mitochondria. Trends Mol. Med. 26, 6–7 (2020).
Mullen, A. R. et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 7, 1679–1690 (2014).
Yang, L. F., Venneti, S. & Nagrath, D. Glutaminolysis: a hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 19, 163–194 (2017).
Metallo, C. M. et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–U166 (2012).
Sun, R. C. & Denko, N. C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 19, 285–292 (2014).
Lu, V. & Teitell, M. A. Alpha-ketoglutarate: a “magic” metabolite in early germ cell development. Embo J. 38, e100615 (2019).
Chang, S., Yim, S. & Park, H. The cancer driver genes IDH1/2, JARID1C/KDM5C, and UTX/KDM6A: crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 51, 66 (2019).
Sullivan, L. B. et al. Supporting aspartate biosynthesis is an essential function of respiration in proliferating. Cell 162, 552–563 (2015).
Son, J. et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature https://doi.org/10.1038/nature12317 (2013).
Tong, X., Zhao, F. & Thompson, C. B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 19, 32–37 (2009).
Moffatt, B. A. & Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arabidopsis Book 1, e0018 (2002).
Kim, J. et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172 (2017).
Patel, D. et al. Aspartate rescues S-phase arrest caused by suppression of glutamine utilization in KRas-driven cancer cells. J. Biol. Chem. 291, 9322–9329 (2016).
Ben-Sahra, I., Howell, J. J., Asara, J. M. & Manning, B. D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 (2013).
Robitaille, A. M. et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320–1323 (2013).
Liu, Y. C. et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE 3, e2722 (2008).
Lane, A. N. & Fan, T. W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 43, 2466–2485 (2015).
Santana-Codina, N. et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 9, 4945 (2018).
Choi, B. H. & Coloff, J. L. The diverse functions of non-essential amino acids in cancer. Cancers 11, 675 (2019).
Thornburg, J. M. et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 10, 1–12 (2008).
Korangath, P. et al. Targeting glutamine metabolism in breast cancer with aminooxyacetate. Clin. Cancer Res. 21, 3263–3273 (2015).
Liu, B. Y. et al. Overexpression of phosphoserine aminotransferase 1 (PSAT1) predicts poor prognosis and associates with tumor progression in human esophageal squamous cell carcinoma. Cell. Physiol. Biochem. 39, 395–406 (2016).
Zhang, J. et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 56, 205–218 (2014).
Huang, H. et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. Embo J. 36, 2334–2352 (2017).
Pavlova, N. N. et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 27, 428 (2018).
LeBoeuf, S. E. et al. Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non-essential amino acids. Cell Metab. 31, 339 (2020).
Sayin, V. I. et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 6, e28083 (2017).
Coloff, J. L. et al. Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab. 23, 867–880 (2016).
Zhu, Y. et al. A critical role of glutamine and asparagine gamma-nitrogen in nucleotide biosynthesis in cancer cells hijacked by an oncogenic virus. Mbio 8, e01179–17 (2017).
Pant, A., Cao, S. & Yang, Z. L. Asparagine is a critical limiting metabolite for vaccinia virus protein synthesis during glutamine deprivation. J. Virol. 93, e01834–18 (2019).
Knott, S. R. V. et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature https://doi.org/10.1038/nature26162 (2018).
Hill, J. M. et al. L-asparaginase therapy for leukemia and other malignant neoplasms. Remission in human leukemia. Jama 202, 882–888 (1967).
Krall, A. S., Xu, S., Graeber, T. G., Braas, D. & Christofk, H. R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7, 11457 (2016).
Krall, A. S. et al. Asparagine signals mitochondrial respiration and can be targeted to impair tumour growth. bioRxiv, 2020.2003.2017.995670 (2020).
Weinberg, F. et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl Acad. Sci. USA 107, 8788–8793 (2010).
Gorrini, C., Harris, I. S. & Mak, T. W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 (2013).
Amores-Sanchez, M. I. & Medina, M. A. Glutamine, as a precursor of glutathione, and oxidative stress. Mol. Genet. Metab. 67, 100–105 (1999).
Welbourne, T. C. Ammonia production and glutamine incorporation into glutathione in the functioning rat kidney. Can. J. Biochem. 57, 233–237 (1979).
Sappington, D. R. et al. Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim. Biophys. Acta 1860, 836–843 (2016).
Tompkins, S. C. et al. Disrupting mitochondrial pyruvate uptake directs glutamine into the TCA cycle away from glutathione synthesis and impairs hepatocellular tumorigenesis. Cell Rep. 28, 2608–2619 (2019).
Muir, A. et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife https://doi.org/10.7554/eLife.27713 (2017).
Timmerman, L. A. et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465 (2013).
Okazaki, K., Papagiannakopoulos, T. & Motohashi, H. Metabolic features of cancer cells in NRF2 addiction status. Biophys. Rev. https://doi.org/10.1007/s12551-020-00659-8 (2020).
Jiang, L. et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258 (2016).
Morotti, M. et al. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc. Natl Acad. Sci. USA 116, 12452–12461 (2019).
Kim, J. W., Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 (2006).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705–713 (2008).
Goda, N. & Kanai, M. Hypoxia-inducible factors and their roles in energy metabolism. Int. J. Hematol. 95, 457–463 (2012).
Zhao, J., Du, F., Shen, G., Zheng, F. & Xu, B. The role of hypoxia-inducible factor-2 in digestive system cancers. Cell Death Dis. 6, e1600 (2015).
Li, Z. et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513 (2009).
Corbet, C. et al. The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 74, 5507–5519 (2014).
Perez-Escuredo, J. et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 15, 72–83 (2016).
Currie, E., Schulze, A., Zechner, R., Walther, T. C. & Farese, R. V. Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 18, 153–161 (2013).
Wise, D. R. et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl Acad. Sci. USA 108, 19611–19616 (2011).
Metallo, C. M. et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2011).
Mullen, A. R. et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 (2011).
Qiu, B. et al. HIF2alpha-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 5, 652–667 (2015).
Du, W. et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 8, 1769 (2017).
Scheuermann, T. H. et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat. Chem. Biol. 9, 271–276 (2013).
Wallace, E. M. et al. A small-molecule antagonist of HIF2alpha Is efficacious in preclinical models of renal cell carcinoma. Cancer Res. 76, 5491–5500 (2016).
Chen, W. et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539, 112–117 (2016).
Cho, H. et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 539, 107–111 (2016).
Wappler, J. et al. Glutamine deprivation counteracts hypoxia-induced chemoresistance. Neoplasia 22, 22–32 (2020).
Chen, R. et al. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci. Rep. 7, 7950 (2017).
Mukhopadhyay, S. et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-19-1363 (2020).
Ju, H. Q. et al. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol. Cancer Ther. 14, 788–798 (2015).
Shukla, S. K. et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 32, 392 (2017).
Intlekofer, A. M. et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 22, 304–311 (2015).
Oldham, W. M., Clish, C. B., Yang, Y. & Loscalzo, J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 22, 291–303 (2015).
Nadtochiy, S. M. et al. Acidic pH is a metabolic switch for 2-hydroxyglutarate generation and signaling. J. Biol. Chem. 291, 20188–20197 (2016).
Intlekofer, A. M. et al. L-2-hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 13, 494–500 (2017).
Kranendijk, M., Struys, E. A., Salomons, G. S., Van der Knaap, M. S. & Jakobs, C. Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 35, 571–587 (2012).
Aghili, M., Zahedi, F. & Rafiee, E. Hydroxyglutaric aciduria and malignant brain tumor: a case report and literature review. J. NeuroOncol. 91, 233–236 (2009).
Larnaout, A. et al. Osteoma of the calvaria in L-2-hydroxyglutaric aciduria. J. Inherit. Metab. Dis. 30, 980 (2007).
Rogers, R. E. et al. Wilms tumor in a child with L-2-hydroxyglutaric aciduria. Pediatr. Dev. Pathol. 13, 408–411 (2010).
Shim, E. H. et al. L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 4, 1290–1298 (2014).
Reid, M. A., Dai, Z. W. & Locasale, J. W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 19, 1298–1306 (2017).
Schvartzman, J. M., Thompson, C. B. & Finley, L. W. S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217, 2247–2259 (2018).
Astuti, D. et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 69, 640–640 (2001).
Letouze, E. et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23, 739–752 (2013).
Janeway, K. A. et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl Acad. Sci. USA 108, 314–318 (2011).
Killian, J. K. et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 3, 648–657 (2013).
Pan, M. et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 18, 1090–1101 (2016).
Carey, B. W., Finley, L. W., Cross, J. R., Allis, C. D. & Thompson, C. B. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 (2015).
TeSlaa, T. et al. alpha-Ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab. 24, 485–493 (2016).
Hwang, I. Y. et al. Psat1-dependent fluctuations in alpha-ketoglutarate affect the timing of ESC differentiation. Cell Metab. 24, 494–501 (2016).
Yu, Y. et al. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab. 29, 966–978 (2019).
Liu, P. S. et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18, 985 (2017).
Klysz, D. et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 8, ra97 (2015).
Petrus, P. et al. Glutamine links obesity to inflammation in human white adipose tissue. Cell Metab. 31, 375 (2020).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
Martinez-Reyes, I. & Chandel, N. S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102 (2020).
Ward, P. S. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 (2010).
Amary, M. F. et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 224, 334–343 (2011).
Borger, D. R. et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin. Cancer Res. 20, 1884–1890 (2014).
Losman, J. A. et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 (2013).
Figueroa, M. E. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012).
Ohka, F. et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumour Biol. 35, 5911–5920 (2014).
Tateishi, K. et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 28, 773–784 (2015).
Salamanca-Cardona, L. et al. In vivo imaging of glutamine metabolism to the oncometabolite 2-hydroxyglutarate in IDH1/2 mutant tumors. Cell Metab. 26, 830–841 e833 (2017).
Gonsalves, W. I. et al. Glutamine-derived 2-hydroxyglutarate is associated with disease progression in plasma cell malignancies. JCI Insight https://doi.org/10.1172/jci.insight.94543 (2018).
Matre, P. et al. Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget 7, 79722–79735 (2016).
Richter, S. et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 99, 3903–3911 (2014).
Bardella, C., Pollard, P. J. & Tomlinson, I. SDH mutations in cancer. Biochim Biophys. Acta 1807, 1432–1443 (2011).
Müller, S. N. U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 26, 268–270 (2000).
Selak, M. A. et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77–85 (2005).
Astuti, D. et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 69, 49–54 (2001).
Virpi Launonen, O. V. et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. PNAS 98, 3387–3392 (2001).
Tomlinson, I. P. et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410 (2002).
Sciacovelli, M. et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 (2016).
Laukka, T. et al. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 291, 4256–4265 (2016).
Isaacs, J. S. et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–153 (2005).
Kinch, L., Grishin, N. V. & Brugarolas, J. Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell 20, 418–420 (2011).
Adam, J. et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537 (2011).
Sullivan, L. B. et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236–248 (2013).
Ooi, A. et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20, 511–523 (2011).
Ternette, N. et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 3, 689–700 (2013).
Greenhouse, W. V. & Lehninger, A. L. Magnitude of malate-aspartate reduced nicotinamide adenine dinucleotide shuttle activity in intact respiring tumor cells. Cancer Res. 37, 4173–4181 (1977).
Fan, J. et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 9, 712 (2013).
Carracedo, A., Cantley, L. C. & Pandolfi, P. P. Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Cancer 13, 227–232 (2013).
Kang, J. H. et al. Aldehyde dehydrogenase inhibition combined with phenformin treatment reversed NSCLC through ATP depletion. Oncotarget 7, 49397–49410 (2016).
Lee, J. S. et al. Dual targeting of glutaminase 1 and thymidylate synthase elicits death synergistically in NSCLC. Cell Death Dis. 7, e2511 (2016).
Andrews, F. J. & Griffiths, R. D. Glutamine: essential for immune nutrition in the critically ill. Br. J. Nutr. 87, S3–S8 (2002).
Ferreira, C. et al. Glutamine supplementation improves the efficacy of miltefosine treatment for visceral leishmaniasis. PLoS Negl. Trop. Dis. 14, e0008125 (2020).
Gianotti, L., Alexander, J. W., Gennari, R., Pyles, T. & Babcock, G. F. Oral glutamine decreases bacterial translocation and improves survival in experimental gut-origin sepsis. J. Parenter. Enter. Nutr. 19, 69–74 (1995).
Bakalar, B. et al. Parenterally administered dipeptide alanyl-glutamine prevents worsening of insulin sensitivity in multiple-trauma patients. Crit. Care Med. 34, 381–386 (2006).
Reid, M. A. et al. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol. Cell 50, 200–211 (2013).
Tajan, M. et al. A role for p53 in the adaptation to glutamine starvation through the expression of SLC1A3. Cell Metab. 28, 721 (2018).
Fu, S. J. et al. Glutamine synthetase promotes radiation resistance via facilitating nucleotide metabolism and subsequent DNA damage repair. Cell Rep. 28, 1136 (2019).
Jeong, S. M. et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 23, 450–463 (2013).
Kim, M., Gwak, J., Hwang, S., Yang, S. & Jeong, S. M. Mitochondrial GPT2 plays a pivotal role in metabolic adaptation to the perturbation of mitochondrial glutamine metabolism. Oncogene 38, 4729–4738 (2019).
Cheng, T. et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl Acad. Sci. USA 108, 8674–8679 (2011).
Jitrapakdee, S., Vidal-Puig, A. & Wallace, J. C. Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cell. Mol. Life Sci. 63, 843–854 (2006).
Christen, S. et al. Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep. 17, 837–848 (2016).
Cappel, D. A. et al. Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining TCA cycle and redox metabolism in liver. Cell Metab. 29, 1291–1305 (2019).
Szeliga, M. & Obara-Michlewska, M. Glutamine in neoplastic cells: focus on the expression and roles of glutaminases. Neurochem. Int. 55, 71–75 (2009).
Eng, C. H. & Abraham, R. T. Glutaminolysis yields a metabolic by-product that stimulates autophagy. Autophagy 6, 968–970 (2010).
Rabinowitz, J. D. & White, E. Autophagy and metabolism. Science 330, 1344–1348 (2010).
Duran, R. V. & Hall, M. N. Glutaminolysis feeds mTORC1. Cell Cycle 11, 4107–4108 (2012).
Duran, R. V. et al. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 32, 4549–4556 (2013).
Poulain, L. et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 31, 2326–2335 (2017).
Dutchak, P. A. et al. Loss of a negative regulator of mTORC1 induces aerobic glycolysis and altered fiber composition in skeletal muscle. Cell Rep. 23, 1907–1914 (2018).
Sanli, T., Steinberg, G. R., Singh, G. & Tsakiridis, T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol. Ther. 15, 156–169 (2014).
Shen, C. & Houghton, P. J. The mTOR pathway negatively controls ATM by upregulating miRNAs. Proc. Natl Acad. Sci. USA 110, 11869–11874 (2013).
Villar, V. H., Merhi, F., Djavaheri-Mergny, M. & Duran, R. V. Glutaminolysis and autophagy in cancer. Autophagy 11, 1198–1208 (2015).
Eng, C. H., Yu, K., Lucas, J., White, E. & Abraham, R. T. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci. Signal. 3, ra31 (2010).
Medvetz, D., Priolo, C. & Henske, E. P. Therapeutic targeting of cellular metabolism in cells with hyperactive mTORC1: a paradigm shift. Mol. Cancer Res. 13, 3–8 (2015).
Debnath, J. Detachment-induced autophagy during anoikis and lumen formation in epithelial acini. Autophagy 4, 351–353 (2008).
Fan, Q. et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/beta-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 37, 9 (2018).
Soria, L. R. & Brunetti-Pierri, N. Ammonia and autophagy: An emerging relationship with implications for disorders with hyperammonemia. J. Inherit. Metab. Dis. 42, 1097–1104 (2019).
Marino, G. & Kroemer, G. Ammonia: a diffusible factor released by proliferating cells that induces autophagy. Sci. Signal. 3, pe19 (2010).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Dang, C. V. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 70, 859–862 (2010).
Hao, Y. et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 7, 11971 (2016).
Stine, Z. E., Walton, Z. E., Altman, B. J., Hsieh, A. L. & Dang, C. V. MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039 (2015).
Wise, D. R. et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl Acad. Sci. USA 105, 18782–18787 (2008).
Gao, P. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009).
Yuneva, M. O. et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 15, 157–170 (2012).
Xiao, D. et al. Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 6, 40655–40666 (2015).
Toda, K. et al. Clinical role of ASCT2 (SLC1A5) in KRAS-mutated colorectal cancer. Int. J. Mol. Sci. https://doi.org/10.3390/ijms18081632 (2017).
Bernfeld, E. & Foster, D. A. Glutamine as an essential amino acid for KRas-driven cancer cells. Trends Endocrinol. Metab. 30, 357–368 (2019).
Anglin, J. et al. Discovery and optimization of aspartate aminotransferase 1 inhibitors to target redox balance in pancreatic ductal adenocarcinoma. Bioorg. Med. Chem. Lett. 28, 2675–2678 (2018).
Patel, D. et al. Aspartate rescues S-phase arrest caused by suppression of glutamine utilization in KRas-driven cancer cells. J. Biol. Chem. 291, 9322–9329 (2016).
Saqcena, M. et al. Blocking anaplerotic entry of glutamine into the TCA cycle sensitizes K-Ras mutant cancer cells to cytotoxic drugs. Oncogene 34, 2672–2680 (2015).
Saqcena, M. et al. Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS ONE 8, e74157 (2013).
Gwinn, D. M. et al. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell 33, 91–107 (2018).
Mei, Z. B., Duan, C. Y., Li, C. B., Cui, L. & Ogino, S. Prognostic role of tumor PIK3CA mutation in colorectal cancer: a systematic review and meta-analysis. Ann. Oncol. 27, 1836–1848 (2016).
Csibi, A. et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153, 840–854 (2013).
Jewell, J. L. et al. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198 (2015).
Nicklin, P. et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534 (2009).
Davidson, S. M. et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016).
Tardito, S. et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 17, 1556–1568 (2015).
Le, A. et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 (2012).
Xiang, Y. et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Invest. 125, 2293–2306 (2015).
Shroff, E. H. et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl Acad. Sci. USA 112, 6539–6544 (2015).
jin, L., A., G. N. & Kang., S. Glutaminolysis as a target for cancer therapy. Oncogene 35, 3619–3625 (2016).
Lobo, C. et al. Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem. J. 348, 257–261 (2000).
Hoerner, C. R., Chen, V. J. & Fan, A. C. The ‘Achilles Heel’ of metabolism in renal cell carcinoma: glutaminase inhibition as a rational treatment strategy. Kidney Cancer 3, 15–29 (2019).
Leone, R. D. et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013 (2019).
Liu, Y. et al. The role of ASCT2 in cancer: a review. Eur. J. Pharm. 837, 81–87 (2018).
Grewer, C. & G., E. New inhibitors for the neutral amino acid transporter ASCT2 reveal its Na+-dependent anion leak. J. Physiol. 557, 747–759 (2004).
Esslinger, C. S., Cybulski, K. A. & Rhoderick, J. F. Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg. Med. Chem. 13, 1111–1118 (2005).
Osanai-Sasakawa, A. et al. An anti-ASCT2 monoclonal antibody suppresses gastric cancer growth by inducing oxidative stress and antibody dependent cellular toxicity in preclinical models. Am. J. Cancer Res. 8, 1499–1513 (2018).
Schulte, M. L. et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 24, 194–202 (2018).
Broer, A., Fairweather, S. & Broer, S. Disruption of amino acid homeostasis by novel ASCT2 inhibitors involves multiple targets. Front. Pharm. 9, 785 (2018).
Chiu, M. et al. GPNA inhibits the sodium-independent transport system L for neutral amino acids. Amino Acids 49, 1365–1372 (2017).
Bensard, C. L. et al. Regulation of tumor initiation by the mitochondrial pyruvate carrier. Cell Metab. 31, 284–300 (2020).
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (2018R1A6A1A03023718 and 2020R1I1A1A01067423) and by the NRF grant funded by the Korean government (MSIT) (2020M3E5E2040282). H.C.Y. was supported by the Graduate School of Yonsei University Research Scholarship Grants in 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoo, H.C., Yu, Y.C., Sung, Y. et al. Glutamine reliance in cell metabolism. Exp Mol Med 52, 1496–1516 (2020). https://doi.org/10.1038/s12276-020-00504-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-00504-8
This article is cited by
-
Ovarian aging: energy metabolism of oocytes
Journal of Ovarian Research (2024)
-
IDH2 regulates macrophage polarization and tumorigenesis by modulating mitochondrial metabolism in macrophages
Molecular Medicine (2024)
-
Spatial transcriptome and single-cell reveal the role of nucleotide metabolism in colorectal cancer progression and tumor microenvironment
Journal of Translational Medicine (2024)
-
Cancer metabolism and carcinogenesis
Experimental Hematology & Oncology (2024)
-
The solute carrier transporters (SLCs) family in nutrient metabolism and ferroptosis
Biomarker Research (2024)
