
Abstract
In recent decades, the role of the peroxisome in physiology and disease conditions has become increasingly important. Together with the mitochondria and other cellular organelles, peroxisomes support key metabolic platforms for the oxidation of various fatty acids and regulate redox conditions. In addition, peroxisomes contribute to the biosynthesis of essential lipid molecules, such as bile acid, cholesterol, docosahexaenoic acid, and plasmalogen. Therefore, the quality control mechanisms that regulate peroxisome biogenesis and degradation are important for cellular homeostasis. Current evidence indicates that peroxisomal function is often reduced or dysregulated in various human disease conditions, such as neurodegenerative diseases. Here, we review the recent progress that has been made toward understanding the quality control systems that regulate peroxisomes and their pathological implications.
Similar content being viewed by others

Introduction
Peroxisomes contain hydrogen peroxide-generating oxidases and were first described by Christian De Duve in the 1960s1,2. Peroxisomes are tiny (0.2–1-μm-diameter), single-membrane-bound organelles, and hundreds of peroxisomes can exist in a single mammalian cell2. They are highly dynamic and ubiquitous organelles that communicate with other organelles, including mitochondria, the endoplasmic reticulum (ER), lysosome, and lipid droplets, through direct interactions3. Peroxisomes function as multipurpose organelles in both catabolic and anabolic pathways, providing different responses in various cells. Peroxisomes play particularly important roles in lipid metabolism, ether-phospholipid biosynthesis, and reactive oxygen species (ROS) metabolism3. In cooperation with mitochondria, peroxisomes play important roles in fatty acid oxidation (FAO) and fatty acid production. Moreover, peroxisomes function as signaling platforms for the innate immune response and inflammatory signaling4,5. Unsurprisingly, peroxisome dysregulation has been associated with various human disorders, such as metabolic diseases, cancer, and neurodegenerative diseases, as well as peroxisome biogenesis disorders (PBDs)2. Zellweger syndrome, one of the three PBDs, is a rare congenital disorder characterized by a reduction in the number of or absence of functional peroxisomes in the cells of an individual. Zellweger syndrome can manifest as a reduction in central nervous system myelin and postdevelopmental sensorineuronal degeneration6. To avoid repetition, we refer to our recently published article on PBDs and Zellweger syndrome7.
The quality and quantity of peroxisomes are regulated in response to changes in the environment to maintain optimal peroxisome numbers and function7,8. Peroxisome regulation consists of active processes that modulate peroxisome abundance, including peroxisome biogenesis and degradation (pexophagy). Peroxisomes can be generated through the growth and division of pre-existing peroxisomes or through de novo synthesis, which requires the fusion of two preperoxisomal vesicles, which are generated by the ER and mitochondria9. The growth and division of pre-existing peroxisomes are mediated by elongation factors and fission regulators10. These processes are tightly regulated by peroxisome biogenesis factors, known as peroxins (PEXs), and peroxisomal membrane proteins (PMPs)11. Selective autophagy of cellular organelles is an important process that maintains homeostasis during various internal and external stress responses. Pexophagy, which refers to the selective autophagic degradation of peroxisomes, can be activated to eliminate dysfunctional or superfluous peroxisomes3,7 and is triggered by both stress conditions, such as starvation and hypoxia, and peroxisomal dysfunction, to maintain peroxisome homeostasis12,13.
Peroxisomes are essential for cellular redox status and lipid metabolism; however, the physiologic and pathologic roles of peroxisomes remain poorly understood, especially compared with those of mitochondria. Therefore, in this review, we highlight the current understanding regarding the roles played by peroxisome quality control and lipid metabolic dysfunction in neurodegenerative diseases.
Peroxisome functions
Lipid metabolism
FAO is tightly regulated at several steps in the oxidation pathway to achieve a balanced energy production and expenditure system. The degradation of oxidized fatty acids occurs in peroxisomes and mitochondria. The β-oxidation of short-, medium-, and long-chain fatty acids predominantly occurs in the mitochondria under physiological conditions. However, the oxidation of specialized fatty acids occurs in peroxisomes, including very-long-chain fatty acids (VLCFAs, i.e., C22:0, C24:0, and C26:0), pristanic acid, and di- or tri-hydroxycholestanoic acids, which cannot be oxidized by mitochondria14. During this process, fatty acids undergo successive rounds of FAO, which involves 2-carbon chain-shortening processes. Peroxisomes are involved not only in catabolic processes but also in anabolic processes, including the synthesis of bile acid, docosahexaenoic acid (DHA), cholesterol, and ether phospholipids14,15. Ether lipids account for ~20% of all phospholipids in humans, and plasmalogens are particularly abundant in the heart and brain, where they form cell membranes and mediate signals16,17. Plasmalogen biosynthesis is initiated in the peroxisome by the enzymes glyceronephosphate O-acyltransferase (GNPAT) and alkylglycerone phosphate synthase and is completed in the ER. Fluorescence anisotropy of membrane-bound fluorophores, which indicates increased membrane lipid mobility, has been consistently demonstrated in plasmalogen-deficient cells16,18. Remarkably, plasmalogen-deficient cells are more sensitive to ROS and cell death than wild-type cells19. Several neurodegenerative disorders have been associated with reduced brain plasmalogen levels. Changes in peroxisomal function in oligodendrocytes may be the primary pathologic factor that results in demyelination, one of the common phenotypes of PBDs, such as Zellweger syndrome20. Therefore, the demyelination observed in PBD patients may be due to the depletion of plasmalogen, which is the major component of normal myelin membranes, as a result of VLCFA accumulation in membrane lipids21.
Peroxisomes, together with the ER, are also essential for DHA synthesis. The DHA synthesis rate in fibroblasts derived from Zellweger syndrome patients was found to be <5% of that in control fibroblasts22. In addition, DHA facilitates peroxisomal division by promoting the oligomerization of peroxisomal biogenesis factor 11 beta (PEX11β), resulting in the initiation of peroxisome elongation23. Bile acid intermediates are converted to taurine or glycine conjugates by bile acid-CoA:amino acid N-acyltransferase in peroxisomes15. Deficiencies in ATP binding cassette subfamily D member 3 (ABCD3), an ABC transporter found in the peroxisomal membrane, result in bile acid synthesis abnormalities. Analysis of Abcd3 knockout mice revealed reduced levels of mature C24 bile acid24. Cholesterol is an essential determinant of membrane fluidity, permeability, and organization in animal cells. PEX2 deficiency has been associated with ER stress-induced pathway activation, leading to the dysregulation of the endogenous sterol response mechanism and decreased cholesterol levels in the plasma and liver25. In addition, disruption of critical peroxisome genes, such as PEX1, results in cholesterol accumulation in the lysosome lumen26.
Redox homeostasis
Redox imbalances are strongly associated with human disease initiation and progression, including neurodegenerative diseases27,28. Peroxisomes have emerged as a central source of redox imbalance, affecting ROS generation and scavenging, owing to the similar functions of peroxisomes and mitochondria29. Notably, peroxisomes account for ~20% of total cellular oxygen consumption and up to 35% of total H2O2 generation in mammalian tissues30. In addition, peroxisomes are associated with the initiation of the cellular oxidative damage response. Deficiencies in peroxisomal antioxidant proteins, such as catalase, can perturb the mitochondrial redox potential31. Furthermore, local oxidative damage to peroxisomes eventually results in mitochondrial dysfunction and cell death5,28, implicating that peroxisomes act as upstream initiators of mitochondrial ROS signaling. Peroxisomes also contain several oxidases that can generate various ROS, such as superoxide radicals and hydroxyl radicals5,14. Antioxidants are essential for scavenging harmful ROS produced in the peroxisome to maintain the redox balance in cells. In addition to oxidases, peroxisomes also contain other antioxidant enzymes, such as catalase, superoxide dismutase 1 (SOD1), peroxiredoxin 5 (Prx5), S-transferase kappa, epoxide hydrolase, and glutathione peroxidase (GPx)14,32. Together, these antioxidant enzymes are responsible for inhibiting excessive ROS generation by peroxisomal oxidases14,32. Catalase is a heme-containing enzyme and represents the most abundant peroxisomal antioxidant14. Prx5 has a cytoprotective effect against H2O2- and lipid hydroperoxide-generated oxidative stress33. GPx reduces lipid hydroperoxides to their corresponding alcohols and reduces free H2O2 to form water14. Recently, several peroxisomal proteins, such as LonP2, insulin-degrading enzyme, and PEX11β, have been suggested to contribute to the maintenance of peroxisomal redox homeostasis, similar to the abovementioned antioxidants34,35,36.
Peroxisomal quality control
Peroxisomal quality and quantity are regulated in response to environmental changes to maintain the optimal numbers and functions of peroxisomes7. Both peroxisome biogenesis and pexophagy control the number of peroxisomes.
Peroxisome biogenesis
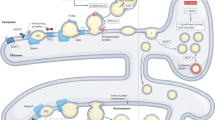
The number of peroxisomes can be controlled by the de novo biogenesis of peroxisomes through the fusion of mitochondria- and ER-derived preperoxisomal vesicles and the growth and division of pre-existing organelles (Fig. 1). These processes are complicated and are tightly regulated by more than 30 PEX proteins37,38. PEX proteins play important roles in many biological processes, such as targeting PMPs to peroxisomes, controlling peroxisomal size, and maintaining peroxisomal functions37,38,39.

The number of peroxisomes is regulated by de novo biogenesis and the growth and division of pre-existing organelles. First, peroxisomes can be formed by the maturation of preperoxisomal vesicles that emerge from the ER or mitochondria and contain peroxisomal membrane proteins, including PEX16, PEX3, and PEX14. Preperoxisomal vesicle fusion results in the generation of mature peroxisomes mediated by PEX19. Second, peroxisomes can be formed by the elongation and division of mature peroxisomes, which are cleaved by the proteins PEX11, Fis1, MFF, and DNM1L (top). Pexophagy is regulated by ubiquitination-dependent and ubiquitination-independent pathways. The ubiquitination of the cytosolic region of peroxisomes triggers their degradation by pexophagy. During oxidative stress, ATM interacts with and phosphorylates PEX5, which promotes PEX5 ubiquitination by PEX2. Ubiquitinated PEX5 is recognized by p62, which recruits the autophagosome. The peroxisomal AAA-type ATPase complex, PEX1, PEX6, and PEX26, prevents pexophagy by regulating the accumulation of ubiquitinated PEX5. During amino acid starvation conditions, PEX2 regulates the ubiquitination of PEX5 and PMP70, increasing pexophagy in an NBR1-dependent manner. USP30 prevents pexophagy by counteracting PEX2. In contrast, PEX14 directly interacts with LC3 under nutrient deprivation conditions (bottom).
Preperoxisomal vesicles emerge from a distinct subdomain of the ER that is enriched in PEX1611. Peroxisome biogenesis is regulated by the insertion of PEX16 into the ER membrane and the recruitment of another peroxin, PEX3, which serves as a docking factor for PEX19 on peroxisomes. PEX3 binds newly synthesized PMPs in the cytosol and delivers them to the peroxisome for insertion into the membrane40,41. Recently, PEX3 and PEX14 were reported to be selectively released into vesicular preperoxisomal structures. Mitochondria-derived vesicles containing PEX3 and PEX14 fuse with ER-derived vesicles; thus, newly generated peroxisomes can obtain membranes from diverse sources and expand their functional linkages to the mitochondria and the ER9.
Alternatively, peroxisomes can be formed through elongation and division processes. Mature peroxisomes are elongated and segregated by the cooperation between the PEX11 family and fission mitochondrial 1 (Fis1) and mitochondrial fission factor (MFF), which recruit the GTPase dynamin1-like (DNM1L) to cleave the peroxisomal membrane42. The peroxisome maturation process is facilitated by the import of matrix proteins by receptor proteins and peroxisome-targeting signals43. Peroxisomal matrix proteins are synthesized in the cytosol and transported to the peroxisome matrix. Two types of targeting signals direct most proteins to the peroxisome matrix. Most peroxisomal matrix proteins contain a C-terminal peroxisomal targeting signal (PTS1), although some contain the less common N-terminal PTS244,45. These signals are recognized by the soluble import receptors PEX5 and PEX746,47. PEX5 binds to the C-terminus of PTS1 and imports the target protein into the peroxisome48. PEX14 and PEX13 comprise the protein import machinery that forms the docking complex that binds PEX5-PTS1-containing proteins48. During the last step of the matrix protein import cycle, PEX5 is recycled back into the cytoplasm in a monoubiquitin-dependent manner for further rounds of import or is subjected to proteasome-mediated turnover of dysfunctional receptors49.
Peroxisomal proteins are influenced by several gene regulators at the transcriptional level, including peroxisome proliferator-activated receptors (PPARs), which represent the best-characterized nuclear receptors and are essential for regulating the transcriptional activation of peroxisomal proteins, especially peroxisomal beta-oxidation50. PPARs have been reported to act as autophagy regulators, even under feeding conditions51.
Peroxisomal degradation (pexophagy)
Autophagy is responsible for degrading cellular components and initiated by the nucleation of isolated membranes, which gradually elongate to form a mature autophagosome that captures cytosolic materials. Upon maturation, the outer membrane of the autophagosome fuses with the lysosome membrane, leading to the degradation of autophagosomal contents. Autophagy has long been considered a nonselective degradation process; however, recent research has indicated that autophagy selectively eliminates specific components, referred to as selective autophagy, including peroxisomes (pexophagy), mitochondria (mitophagy), lysosomes (lysophagy), and ER (reticulophagy)52. Autophagy is a sequential process that is primarily regulated by autophagy-related genes (ATGs) and various adaptor/receptor proteins, including sequestosome 1 (SQSTM1/p62), optineurin (OPTN), NBR1, NDP52, NIX, and FUNDC153.
Peroxisomal quality control is regulated by two distinct mechanisms. During peroxisomal degradation, 70–80% of excess peroxisomes are degraded through autophagy, whereas the remaining 20–30% of peroxisomes are degraded by other processes mediated by peroxisomal LonP2 and 15-lipoxygenase-154,55. Recent advances in the understanding of selective autophagy have suggested that the ubiquitination of membrane proteins found in specific organelles mediates the initiation of selective autophagy56,57,58. According to this notion, the ubiquitination of the cytosolic region of peroxisome-associated proteins triggers peroxisome degradation by pexophagy (Fig. 1, Table 1). Kim et al. showed that the ectopic expression of PEX3 or PMP34 fused to ubiquitin on the cytosolic face decreases the number of peroxisomes and that the ubiquitin signal on the peroxisomal membrane is recognized by p62 or NBR1, which targets peroxisomes to the autophagosome59. Moreover, the exogenous expression of NBR1 induces peroxisome clustering and targeting to lysosomes, promoting pexophagy60. NBR1 has a similar domain composition as p62, consisting of a PB1 domain at the N-terminus, a ZZ domain in the coiled coil, an LIR motif in the middle part of the molecule, an amphipathic alpha-helical J domain (JUBA) and a UBA domain at the C-terminus61,62. The PB1 domain of NBR1 mediates interactions with p62, and both the JUVA and UBA domains are involved in the localization of NBR1 on peroxisomes. Mutation studies have shown that the JUBA, UBA, and LIR domains of NBR1 contribute to pexophagy60. Among the PEX proteins, PEX5 closely regulates pexophagy. The inhibition of PEX5 recruitment by PEX14 depletion significantly reduces pexophagy60. During the PMP import cycle, PEX5 is regulated in a ubiquitination-dependent manner, whereas polyubiquitinated PEX5 is degraded by the proteasome system. Nordgren et al. showed that export-deficient monoubiquitinated PEX5, which maintains monoubiquitinated PEX5 at the membrane long enough to be recognized by the autophagic machinery, promotes peroxisomal removal63. In addition, Zhang et al. also reported that PEX5 binds to the protein ataxia-telangiectasia mutated (ATM). During oxidative stress, ATM directly phosphorylates PEX5 at Ser 141, which subsequently promotes PEX5 monoubiquitination at Lys 209. Ubiquitinated PEX5 is then recognized by p62, which recruits the autophagosome64. The peroxisomal AAA–ATPase complex consisting of PEX1, PEX6, and PEX26 prevents pexophagy and peroxisome biogenesis disorder development65. The loss of the ATPase associated with diverse cellular activities (AAA)–ATPase complex, which is required to cycle PEX5 for PMP import, results in the accumulation of ubiquitinated PEX5 on the peroxisomal membrane, triggering pexophagy65. Each of the three RING peroxins, namely, PEX2, PEX10, and PEX12, exhibits ubiquitin-protein isopeptide ligase activity. Members of the E2D (UbcH5) family act as specialized ubiquitin-conjugating enzymes that mediate the ubiquitination of PEX566. PEX5 and PMP70 are ubiquitinated by PEX2 during pexophagy triggered by amino acid starvation. PEX2 expression results in the gross ubiquitination of peroxisomes and pexophagy in an NBR1-dependent manner67. Conversely, the deubiquitinating enzyme USP30 prevents pexophagy by counteracting the activity of PEX2. USP30, which is known as a mitophagy regulator, can also be localized to peroxisomes68,69. USP30 overexpression prevents pexophagy during amino acid starvation by counteracting the PEX2-mediated ubiquitination of PEX5 and PMP70, whereas USP30 depletion results in pexophagy induction, even under basal conditions70. PEX14 and PEX13 comprise the protein import machinery that serves as a docking complex for PEX5. PEX14 has been suggested to directly interact with the LC3II autophagosomal protein71. During this process, PEX14 preferentially interacts with LC3 rather than PEX5 under nutrient-deprived conditions71. In addition to PEX14, PEX3 may target peroxisomes for pexophagy. In PEX3-overexpressing cells, peroxisomes are ubiquitinated and degraded via an NBR1-dependent process58. Taken together, these previous studies describe the regulatory mechanisms associated with ubiquitination-dependent pexophagy. However, the precise regulatory mechanisms that control the ubiquitination process require further investigation.
Several methods have using various model systems been proposed to study pexophagy. Pexophagy is a dynamic process that ends in the lysosome, which has the most acidic cellular microenvironment of any organelle. This property of lysosomes has led to the development of peroxisome-targeted forms of pH-dependent systems to monitor pexophagy. To observe and quantify pexophagy activity, Nazrko and coworkers utilized an mRFP-EGFP protein containing the PTS1 domain72. Similarly, Deosaran et al. used a tandem chimera of mCherry and EGFP fused to the peroxisome-membrane-targeting sequence of PEX2660. The GFP fluorescence of the fused protein is quickly quenched, whereas the mRFP fluorescence exhibits more stability under acidic conditions in the lysosome73. In addition to these tandem systems, we also developed a pexophagy assay model using a pH-sensitive pexo-dKeima generated by fusing the PTS1 sequence to the dKeima protein74. The dKeima protein is a pH-sensitive, dual-excitation, ratiometric fluorescent protein that exhibits lysosomal protease resistance. At the physiological pH of the peroxisome (pH 6.9–7.1), shorter-wavelength excitation predominates. At the end of pexophagy, pexo-Keima undergoes a gradual shift to longer-wavelength excitation within the acidic lysosomal environment (pH 4.5)74,75,76.
Peroxisome dysfunction in neurodegenerative diseases
The brain is a lipid-rich organ, with membrane lipids constituting 50–60% of the total solid brain matter77. Therefore, slight alterations in fatty acid composition may lead to considerable changes in neuronal function. Several inherited peroxisomal disorders have been associated with severe neurologic dysfunctions, including hypotonia, seizures, cerebellar ataxia, sensory impairment, and developmental deficits78. Recent studies have suggested that peroxisomal metabolic function is also disrupted in age-related neurological disorders, including Alzheimer’s disease (AD) and Parkinson’s disease (PD)28,32. Therefore, in this review, we focus on metabolic dysregulation associated with peroxisome dysfunction in AD and PD.
Peroxisome dysfunction in Alzheimer’s disease
AD is the most common neurological disorder that affects the elderly population and is clinically characterized by the progressive deterioration of cognition, behavior and functionality, leading to significant impairment of activities of daily living79. Primary histopathologic lesions associated with AD pathology indicate neuroinflammation and neuronal loss, which are accompanied by beta-amyloid (Aβ) plaques and neurofibrillary tangles80,81,82. The toxic properties of Aβ plaques are mediated by diverse mechanisms, including oxidative stress, inflammation, synaptic dysfunction, and excitotoxicity83. Tauopathy is another widely accepted component of AD pathology. When tau protein becomes highly phosphorylated, it aggregates, inhibiting microtubule function, impairing neuronal axonal transport, and thus leading to neuronal cytotoxicity84. Emerging evidence has suggested that in addition to Aβ and tau, inflammation may play a causal role in AD pathogenesis80. Serial studies of lipid metabolism have shown that lipid alterations can be detected during early AD progression85,86,87. Remarkably, a significant and selective decrease in plasmalogen can be observed in postmortem brain samples from AD patients88,89. Kuo et al. measured the levels of VLCFAs in cortical brain regions affected by AD and found that VLCFAs, such as C24:0 and C26:0, accumulate in all cortical areas except the parasubiculum87. In addition, increased VLCFA levels have been associated with the presence of neurofibrillary tangles87,89. Consistent with this finding, total plasmalogen concentrations have been found to be significantly decreased in the gyrus frontalis region of AD patients87,88.
Notably, cells from patients with Zellweger syndrome, a PBD, show lysosomal cholesterol accumulation26. Several epidemiologic studies have indicated that hypercholesterolemia is closely associated with AD pathology, although the exact mechanism through which cholesterol affects AD pathogenesis is largely unknown90. Plasma cholesterol levels are ~10% higher in AD patients than in normal controls, and several genes associated with hypercholesterolemia, such as ApoE4, increase the incidence of AD91,92. ApoE4, a strong genetic risk factor for late, sporadic AD onset, transports cholesterol, and other lipid components into neurons93. Cholesterol and oxysterol imbalances can cause alterations in cell membrane properties and increase intracellular cholesterol levels, enhancing the activities of beta-secretase 1 (BACE1) and increasing γ-secretase levels, which are associated with increased levels of Aβ generation94.
Similar to plasmalogen and cholesterol, DHA has also been identified as a causal factor in AD pathogenesis and progression95. DHA plays an important role in normal neurological development, especially in the brain and retina96. However, the DHA concentration has been shown to be reduced in the hippocampus in AD85, and the levels of DHA in the hippocampus, frontal cortex and temporal cortex are lower in AD patients than healthy individuals84,97. DHA inhibits AD pathogenesis by attenuating the Aβ burden, inhibiting tau phosphorylation and decreasing neuroinflammation98,99. Accordingly, various studies have suggested that DHA supplementation can effectively reduce key AD-associated risk factors. A DHA-enriched diet can increase cerebral blood volume and decrease vascular Aβ deposition, leading to selective changes in the phospholipid profiles of different brain regions in mouse models of AD100,101. In addition, DHA can suppress proinflammatory cytokine expression in neurons97,102. Neuroprotectin D1, a bioactive metabolite of DHA, inhibits neuroinflammation and toxicity103.
PPARs, which act as lipid sensors and peroxisomal gene activators, are associated with the transcriptional control of genes that regulate metabolism104. PPAR agonists, such as pioglitazone, can ameliorate AD-related pathology and improve cognition by decreasing Aβ production105,106. PPAR agonists also inhibit inflammatory gene expression and immune responses and inhibit the secretion of proinflammatory cytokines107,108. Icariin, a prenylated flavonol glycoside found in various medicinal herbs, attenuates M1 microglial activation and Aβ plaque formation in the hippocampus and prefrontal cortex by increasing PPARγ levels in an AD mouse model109,110. Neuroinflammation also plays a role in AD pathophysiology and is considered a promising target for AD treatment111.
Peroxisome dysfunction in Parkinson’s disease
PD is the second most common neurodegenerative disease after AD and is characterized by the selective loss of dopaminergic neurons in the substantia nigra and the underproduction of dopamine coupled with α-synuclein (SNCA) accumulation. In recent decades, studies have confirmed that various genetic factors, including DJ‐1, LRRK2, Parkin, PINK 1, SNCA, and VPS35, contribute to the complex pathogenesis of PD112,113,114.
Postmortem lipid composition analysis of lipid rafts from the frontal cortices of PD patients have indicated remarkable reductions in polyunsaturated fatty acid contents, including DHA and arachidonic acid, whereas saturated fatty acid levels are enhanced in the brains of PD patients compared with the brains of control subjects115. Another study that examined serum lipid profiles in PD patients showed lower levels of total cholesterol and triglycerides in PD patients than in control individuals116. Furthermore, several studies have indicated a relationship among the levels of peroxisomal lipids, such as cholesterol, the use of drugs that regulate cholesterol levels and PD development117,118. Ethanolamine plasmalogens are also diminished in the blood and brains of PD patients, and supplementation with the ethanolamine plasmalogen precursor PPI-1011 helps reverse striatal dopamine loss in a PD mouse model119,120. Thus, these lipids may be used as markers of PD severity. The neuroprotective effects of PPAR agonists have been assessed in several PD models as in AD models121. Pioglitazone and rosiglitazone, which were originally designed as PPARγ agonists, have been shown to block dopaminergic neurodegeneration and reduce astrocytic and microglial activation122. In addition, PPAR α/γ agonists, such as fenofibrate and MHY908, prevent neurotoxicity in a mouse model of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD123,124. The role played by oxidative stress in dopaminergic neuron degeneration has been extensively studied. Oxidative damage to lipids, proteins, and DNA occurs during PD, and the toxic products generated by oxidative damage can react with proteins, proteasome systems, and autophagy, impairing cell viability125,126,127,128.
Increased ROS production combined with defects in peroxisomal antioxidant mechanisms and the accumulation of lipid intermediates in the peroxisomal FAO system has been suggested to alter mitochondrial function and may contribute to PD pathogenesis. Marked mitochondrial abnormalities have been observed in several organs in PEX5-deficient mice129. In addition, deficiencies in peroxisome biogenesis associated with a mutation in PEX3 prevents the binding of SNCA to lipid droplets in lipid-loaded yeast130. Recently, our group also showed that HSPA9/mortalin depletion induces pexophagy by increasing peroxisomal ROS74. The overexpression of wild-type HSPA9 reverses peroxisome loss, whereas an HSPA9 mutant associated with PD fails to rescue HSPA9-depleted neuronal cells74.
Although peroxisome abundance and lipid metabolism play roles in several pathological neuronal conditions, it remains unclear whether these conditions represent secondary changes associated with general cellular dysfunction. Therefore, to better understand the roles played by peroxisomes in neurodegenerative diseases, further studies are warranted.
Conclusion and perspective
Peroxisomes are key metabolic organelles that have protective functions and wide-reaching impacts on human health and may contribute to a large number of globally important human diseases. Further systematic studies are necessary to determine whether peroxisome alterations/dysfunctions contribute to disease etiology. In addition, the functional correlations between disease pathogenesis and alterations in peroxisome physiology remain to be elucidated. Emerging research areas include the roles played by peroxisomes in cellular redox balance, metabolic balance, and pexophagy. Because peroxisomes are one of the most unexplored subcellular organelles in eukaryotic cells, the continued exploration of their functional significance is likely to reveal additional and useful information in the future.
References
De Duve, C. & Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 46, 323–357 (1966).
Braverman, N. E., D’Agostino, M. D. & Maclean, G. E. Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives. Dev. Disabil. Res. Rev. 17, 187–196 (2013).
Jo, D. S. & Cho, D. H. Peroxisomal dysfunction in neurodegenerative diseases. Arch. Pharm. Res. 42, 393–406 (2019).
Dixit, E. et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141, 668–681 (2010).
Fransen, M., Nordgren, M., Wang, B. & Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim. Biophys. Acta 1822, 1363–1373 (2012).
Zellweger, H., Maertens, P., Superneau, D. & Wertelecki, W. History of the cerebrohepatorenal syndrome of Zellweger and other peroxisomal disorders. South Med. J. 81, 357–364 (1988).
Cho, D. H., Kim, Y. S., Jo, D. S., Choe, S. K. & Jo, E. K. Pexophagy: molecular mechanisms and implications for health and diseases. Mol. Cells 41, 55–64 (2018).
Oku, M. & Sakai, Y. Peroxisomes as dynamic organelles: autophagic degradation. Febs J. 277, 3289–3294 (2010).
Sugiura, A., Mattie, S., Prudent, J. & McBride, H. M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 542, 251–254 (2017).
Williams, C. et al. The membrane remodeling protein Pex11p activates the GTPase Dnm1p during peroxisomal fission. Proc. Natl Acad. Sci. USA 112, 6377–6382 (2015).
Agrawal, G. & Subramani, S. De novo peroxisome biogenesis: evolving concepts and conundrums. Biochim. Biophys. Acta 1863, 892–901 (2016).
Walter, K. M. et al. Hif-2α promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 20, 882–897 (2014).
Eun, S. Y. et al. PEX5 regulates autophagy via the mTORC1-TFEB axis during starvation. Exp. Mol. Med. 50, 4 (2018).
Islinger, M., Voelkl, A., Fahimi, H. D. & Schrader, M. The peroxisome: an update on mysteries 2.0. Histochem. Cell Biol. 150, 443–471 (2018).
Ferdinandusse, S., Denis, S., Faust, P. L. & Wanders, R. J. Bile acids: the role of peroxisomes. J. Lipid Res. 50, 2139–2147 (2009).
Brites, P., Waterham, H. R. & Wanders, R. J. Functions and biosynthesis of plasmalogens in health and disease. Biochim. Biophys. Acta 1636, 219–231 (2004).
Braverman, N. E. & Moser, A. B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 1822, 1442–1452 (2012).
Hoefler, G., Paschke, E., Hoefler, S., Moser, A. B. & Moser, H. W. Photosensitized killing of cultured fibroblasts from patients with peroxisomal disorders due to pyrene fatty acid-mediated ultraviolet damage. J. Clin. Invest. 88, 1873–1879 (1991).
Zoeller, R. A. et al. Plasmalogens as endogenous antioxidants: somatic cell mutants reveal the importance of the vinyl ether. Biochem. J. 338(Pt 3), 769–776 (1999).
Crane, D. I. Revisiting the neuropathogenesis of Zellweger syndrome. Neurochem. Int. 69, 1–8 (2014).
Trompier, D. et al. Brain peroxisomes. Biochimie 98, 102–110 (2014).
Barry, D. S. & O’Keeffe, G. W. Peroxisomes: the neuropathological consequences of peroxisomal dysfunction in the developing brain. Int. J. Biochem. Cell Biol. 45, 2012–2015 (2013).
Itoyama, A. et al. Docosahexaenoic acid mediates peroxisomal elongation, a prerequisite for peroxisome division. J. Cell Sci. 125, 589–602 (2012).
Ferdinandusse, S. et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet. 24, 361–370 (2015).
Faust, P. L. & Kovacs, W. J. Cholesterol biosynthesis and ER stress in peroxisome deficiency. Biochimie 98, 75–85 (2014).
Chu, B. B. et al. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306 (2015).
Wanders, R. J. Metabolic functions of peroxisomes in health and disease. Biochimie 98, 36–44 (2014).
Cipolla, C. M. & Lodhi, I. J. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 28, 297–308 (2017).
Fransen, M., Lismont, C. & Walton, P. The peroxisome-mitochondria connection: how and why? Int. J. Mol. Sci. 18, 1126 (2017).
Boveris, A., Oshino, N. & Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 128, 617–630 (1972).
Hwang, I. et al. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 61, 728–738 (2012).
Deori, N. M., Kale, A., Maurya, P. K. & Nagotu, S. Peroxisomes: role in cellular ageing and age related disorders. Biogerontology 19, 303–324 (2018).
Walbrecq, G. et al. Antioxidant cytoprotection by peroxisomal peroxiredoxin-5. Free Radic. Biol. Med. 84, 215–226 (2015).
Pomatto, L. C., Raynes, R. & Davies, K. J. The peroxisomal Lon protease LonP2 in aging and disease: functions and comparisons with mitochondrial Lon protease LonP1. Biol. Rev. Camb. Philos. Soc. 92, 739–753 (2017).
Morita, M. et al. Insulin-degrading enzyme exists inside of rat liver peroxisomes and degrades oxidized proteins. Cell Struct. Funct. 25, 309–315 (2000).
Fox, M. A., Nieuwesteeg, M. A., Willson, J. A., Cepeda, M. & Damjanovski, S. Knockdown of Pex11β reveals its pivotal role in regulating peroxisomal genes, numbers, and ROS levels in Xenopus laevis A6 cells. Vitr. Cell Dev. Biol. Anim. 50, 340–349 (2014).
Mayerhofer, P. U. Targeting and insertion of peroxisomal membrane proteins: ER trafficking versus direct delivery to peroxisomes. Biochim. Biophys. Acta 1863, 870–880 (2016).
Schlüter, A. et al. The evolutionary origin of peroxisomes: an ER-peroxisome connection. Mol. Biol. Evol. 23, 838–845 (2006).
Giannopoulou, E. A., Emmanouilidis, L., Sattler, M., Dodt, G. & Wilmanns, M. Towards the molecular mechanism of the integration of peroxisomal membrane proteins. Biochim. Biophys. Acta 1863, 863–869 (2016).
Fang, Y., Morrell, J. C., Jones, J. M. & Gould, S. J. PEX3 functions as a PEX19 docking factor in the import of class I peroxisomal membrane proteins. J. Cell. Biol. 164, 863–875 (2004).
Jones, J. M., Morrell, J. C. & Gould, S. J. PEX19 is a predominantly cytosolic chaperone and import receptor for class 1 peroxisomal membrane proteins. J. Cell. Biol. 164, 57–67 (2004).
Koch, J. & Brocard, C. PEX11 proteins attract Mff and human Fis1 to coordinate peroxisomal fission. J. Cell. Sci. 125, 3813–3826 (2012).
Walton, P. A., Hill, P. E. & Subramani, S. Import of stably folded proteins into peroxisomes. Mol. Biol. Cell. 6, 675–683 (1995).
Gould, S. J., Krisans, S., Keller, G. A. & Subramani, S. Antibodies directed against the peroxisomal targeting signal of firefly luciferase recognize multiple mammalian peroxisomal proteins. J. Cell. Biol. 110, 27–34 (1990).
Swinkels, B. W., Gould, S. J., Bodnar, A. G., Rachubinski, R. A. & Subramani, S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. Embo. J. 10, 3255–3262 (1991).
Dammai, V. & Subramani, S. The human peroxisomal targeting signal receptor, Pex5p, is translocated into the peroxisomal matrix and recycled to the cytosol. Cell 105, 187–196 (2001).
Braverman, N. et al. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat. Genet. 15, 369–376 (1997).
Hua, R. & Kim, P. K. Multiple paths to peroxisomes: mechanism of peroxisome maintenance in mammals. Biochim. Biophys. Acta 1863, 881–891 (2016).
Ma, C., Agrawal, G. & Subramani, S. Peroxisome assembly: matrix and membrane protein biogenesis. J. Cell Biol. 193, 7–16 (2011).
Pawlak, M., Lefebvre, P. & Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 62, 720–733 (2015).
Lee, J. M. et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516, 112–115 (2014).
Kirkin, V. & Rogov, V. V. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol. Cell. 76, 268–285 (2019).
Cho, D. H., Kim, J. K. & Jo, E. K. Mitophagy and innate immunity in infection. Mol. Cells 43, 10–22 (2020).
van Leyen, K., Duvoisin, R. M., Engelhardt, H. & Wiedmann, M. A function for lipoxygenase in programmed organelle degradation. Nature 395, 392–395 (1998).
Iwata, J. et al. Excess peroxisomes are degraded by autophagic machinery in mammals. J. Biol. Chem. 281, 4035–4041 (2006).
Feng, L. et al. Ubiquitin ligase SYVN1/HRD1 facilitates degradation of the SERPINA1 Z variant/α-1-antitrypsin Z variant via SQSTM1/p62-dependent selective autophagy. Autophagy 13, 686–702 (2017).
Vives-Bauza, C. et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl Acad. Sci. USA 107, 378–383 (2010).
Yamashita, S., Abe, K., Tatemichi, Y. & Fujiki, Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 10, 1549–1564 (2014).
Kim, P. K., Hailey, D. W., Mullen, R. T. & Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl Acad. Sci. USA 105, 20567–20574 (2008).
Deosaran, E. et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 126, 939–952 (2013).
Mardakheh, F. K., Auciello, G., Dafforn, T. R., Rappoport, J. Z. & Heath, J. K. Nbr1 is a novel inhibitor of ligand-mediated receptor tyrosine kinase degradation. Mol. Cell. Biol. 30, 5672–5685 (2010).
Zientara-Rytter, K. & Subramani, S. Autophagic degradation of peroxisomes in mammals. Biochem. Soc. Trans. 44, 431–440 (2016).
Nordgren, M. et al. Export-deficient monoubiquitinated PEX5 triggers peroxisome removal in SV40 large T antigen-transformed mouse embryonic fibroblasts. Autophagy 11, 1326–1340 (2015).
Zhang, J. et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell. Biol. 17, 1259–1269 (2015).
Law, K. B. et al. The peroxisomal AAA ATPase complex prevents pexophagy and development of peroxisome biogenesis disorders. Autophagy 13, 868–884 (2017).
Grou, C. P. et al. Members of the E2D (UbcH5) family mediate the ubiquitination of the conserved cysteine of Pex5p, the peroxisomal import receptor. J. Biol. Chem. 283, 14190–14197 (2008).
Sargent, G. et al. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell. Biol. 214, 677–690 (2016).
Wang, Y. et al. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11, 595–606 (2015).
Marcassa E. et al. Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep. 19, e45595 (2018).
Riccio, V. et al. Deubiquitinating enzyme USP30 maintains basal peroxisome abundance by regulating pexophagy. J. Cell. Biol. 218, 798–807 (2019).
Jiang, L., Hara-Kuge, S., Yamashita, S. & Fujiki, Y. Peroxin Pex14p is the key component for coordinated autophagic degradation of mammalian peroxisomes by direct binding to LC3-II. Genes Cells 20, 36–49 (2015).
Nazarko, T. Y. et al. Peroxisomal Atg37 binds Atg30 or palmitoyl-CoA to regulate phagophore formation during pexophagy. J. Cell. Biol. 204, 541–557 (2014).
Kimura, S., Noda, T. & Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 (2007).
Jo D. S. et al. Loss of HSPA9 induces peroxisomal degradation by increasing pexophagy. Autophagy 1–15, https://doi.org/10.1080/15548627.2020.1712812 (2020).
Sun, N. et al. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 12, 1576–1587 (2017).
Dansen, T. B., Wirtz, K. W., Wanders, R. J. & Pap, E. H. Peroxisomes in human fibroblasts have a basic pH. Nat. Cell. Biol. 2, 51–53 (2000).
Bourre, J. M. Roles of unsaturated fatty acids (especially omega-3 fatty acids) in the brain at various ages and during ageing. J. Nutr. Health Aging 8, 163–174 (2004).
Waterham, H. R. & Ebberink, M. S. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta 1822, 1430–1441 (2012).
Graham, W. V., Bonito-Oliva, A. & Sakmar, T. P. Update on Alzheimer’s disease therapy and prevention strategies. Annu. Rev. Med. 68, 413–430 (2017).
Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015).
LaFerla, F. M., Green, K. N. & Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509 (2007).
Congdon, E. E. & Sigurdsson, E. M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 14, 399–415 (2018).
Carrillo-Mora, P., Luna, R. & Colín-Barenque, L. Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid. Med. Cell Longev. 2014, 795375 (2014).
Giacobini, E. & Gold, G. Alzheimer disease therapy-moving from amyloid-β to tau. Nat. Rev. Neurol. 9, 677–686 (2013).
Söderberg, M., Edlund, C., Kristensson, K. & Dallner, G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids 26, 421–425 (1991).
Astarita, G. et al. Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS ONE 5, e12538 (2010).
Kou, J. et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 122, 271–283 (2011).
Ginsberg, L., Rafique, S., Xuereb, J. H., Rapoport, S. I. & Gershfeld, N. L. Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Res. 698, 223–226 (1995).
Lizard, G., Rouaud, O., Demarquoy, J., Cherkaoui-Malki, M. & Iuliano, L. Potential roles of peroxisomes in Alzheimer’s disease and in dementia of the Alzheimer’s type. J. Alzheimers Dis. 29, 241–254 (2012).
Xue-Shan, Z. et al. Imbalanced cholesterol metabolism in Alzheimer’s disease. Clin. Chim. Acta 456, 107–114 (2016).
Xiao, Z. et al. Association studies of several cholesterol-related genes (ABCA1, CETP and LIPC) with serum lipids and risk of Alzheimer’s disease. Lipids Health Dis. 11, 163 (2012).
Popp, J. et al. Cerebral and extracerebral cholesterol metabolism and CSF markers of Alzheimer’s disease. Biochem. Pharmacol. 86, 37–42 (2013).
Frieden, C. & Garai, K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc. Natl Acad. Sci. USA 109, 8913–8918 (2012).
Grimm, M. O. et al. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 283, 11302–11311 (2008).
Tully, A. M. et al. Low serum cholesteryl ester-docosahexaenoic acid levels in Alzheimer’s disease: a case-control study. Br. J. Nutr. 89, 483–489 (2003).
Innis, S. M. Essential fatty acids in growth and development. Prog. Lipid Res. 30, 39–103 (1991).
Lukiw, W. J. et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 115, 2774–2783 (2005).
Pan, Y., Khalil, H. & Nicolazzo, J. A. The impact of docosahexaenoic acid on alzheimer’s disease: is there a role of the blood-brain barrier? Curr. Clin. Pharmacol. 10, 222–241 (2015).
Belkouch, M. et al. The pleiotropic effects of omega-3 docosahexaenoic acid on the hallmarks of Alzheimer’s disease. J. Nutr. Biochem. 38, 1–11 (2016).
Hooijmans, C. R. et al. DHA and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in APPswe/PS1dE9 mice. Neurobiol. Dis. 33, 482–498 (2009).
Bascoul-Colombo, C. et al. Dietary DHA supplementation causes selective changes in phospholipids from different brain regions in both wild type mice and the Tg2576 mouse model of Alzheimer’s disease. Biochim. Biophys. Acta 1861, 524–537 (2016).
van Gelder, B. M., Tijhuis, M., Kalmijn, S. & Kromhout, D. Fish consumption, n-3 fatty acids, and subsequent 5-y cognitive decline in elderly men: the Zutphen Elderly Study. Am. J. Clin. Nutr. 85, 1142–1147 (2007).
Zhao, Y. et al. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer’s disease models. PLoS ONE 6, e15816 (2011).
Dubois, V., Eeckhoute, J., Lefebvre, P. & Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Invest. 127, 1202–1214 (2017).
Chang, K. L., Wong, L. R., Pee, H. N., Yang, S. & Ho, P. C. Reverting metabolic dysfunction in cortex and cerebellum of APP/PS1 mice, a model for alzheimer’s disease by pioglitazone, a peroxisome proliferator-activated receptor gamma (PPARγ) Agonist. Mol. Neurobiol. 56, 7267–7283 (2019).
Lee, W. J. et al. Activation of peroxisome proliferator-activated receptor delta suppresses BACE1 expression by up-regulating SOCS1 in a JAK2/STAT1-dependent manner. J. Neurochem. 151, 370–385 (2019).
Landreth, G. Therapeutic use of agonists of the nuclear receptor PPARγ in Alzheimer’s disease. Curr. Alzheimer Res. 4, 159–164 (2007).
Marchetti, D. P. et al. Inflammatory profile in X-linked adrenoleukodystrophy patients: understanding disease progression. J. Cell Biochem. 119, 1223–1233 (2018).
Urano, T. & Tohda, C. Icariin improves memory impairment in Alzheimer’s disease model mice (5xFAD) and attenuates amyloid β-induced neurite atrophy. Phytother. Res. 24, 1658–1663 (2010).
Wang, Y. et al. Icariin attenuates M1 activation of microglia and Aβ plaque accumulation in the hippocampus and prefrontal cortex by up-regulating PPARγ in restraint/isolation-stressed APP/PS1 mice. Front. Neurosci. 13, 291 (2019).
Shadfar, S., Hwang, C. J., Lim, M. S., Choi, D. Y. & Hong, J. T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharm. Res. 38, 2106–2119 (2015).
Thenganatt, M. A. & Jankovic, J. Parkinson disease subtypes. JAMA Neurol. 71, 499–504 (2014).
Hernandez, D. G., Reed, X. & Singleton, A. B. Genetics in Parkinson disease: mendelian versus non-Mendelian inheritance. J. Neurochem. 139(Suppl 1), 59–74 (2016).
Chen-Plotkin, A. S. et al. Finding useful biomarkers for Parkinsonas disease. Sci. Transl. Med. 10, eaam6003 (2018).
Fabelo, N. et al. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 17, 1107–1118 (2011).
Guo, X. et al. The serum lipid profile of Parkinson’s disease patients: a study from China. Int. J. Neurosci. 125, 838–844 (2015).
Doria, M., Maugest, L., Moreau, T., Lizard, G. & Vejux, A. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 101, 393–400 (2016).
Gudala, K., Bansal, D. & Muthyala, H. Role of serum cholesterol in Parkinson’s disease: a meta-analysis of evidence. J. Parkinsons Dis. 3, 363–370 (2013).
Dragonas, C., Bertsch, T., Sieber, C. C. & Brosche, T. Plasmalogens as a marker of elevated systemic oxidative stress in Parkinson’s disease. Clin. Chem. Lab. Med. 47, 894–897 (2009).
Miville-Godbout, E. et al. Plasmalogen augmentation reverses striatal dopamine loss in MPTP mice. PLoS ONE 11, e0151020 (2016).
Chaturvedi, R. K. & Beal, M. F. PPAR: a therapeutic target in Parkinson’s disease. J. Neurochem. 106, 506–518 (2008).
Breidert, T. et al. Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. J. Neurochem. 82, 615–624 (2002).
Barbiero, J. K. et al. PPAR-α agonist fenofibrate protects against the damaging effects of MPTP in a rat model of Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 53, 35–44 (2014).
Lee, Y. et al. Neuroprotective effects of MHY908, a PPAR α/γ dual agonist, in a MPTP-induced Parkinson’s disease model. Brain Res. 1704, 47–58 (2019).
Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 53(Suppl 3), S26–S36 (2003).
Dias, V., Junn, E. & Mouradian, M. M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 3, 461–491 (2013).
Cerri, S. & Blandini, F. Role of autophagy in Parkinson’s disease. Curr. Med. Chem. 26, 3702–3718 (2019).
Hu, G., Jia, H., Zhao, L., Cho, D. H. & Fang, J. Small molecule fluorescent probes of protein vivinal dithiols. Chi. Chem. Lett. 30, 1704–1716 (2019).
Baumgart, E. et al. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am. J. Pathol. 159, 1477–1494 (2001).
Wang, S. et al. A peroxisome biogenesis deficiency prevents the binding of alpha-synuclein to lipid droplets in lipid-loaded yeast. Biochem. Biophys. Res. Commun. 438, 452–456 (2013).
Acknowledgements
This research was supported by the National Research Foundation of Korea funded by the Ministry of Science & ICT [2020R1A2C2003523 & 2017M3A9G7073521] and the Ministry of Education [2020R1I1A1A01073696].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jo, D.S., Park, N.Y. & Cho, DH. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp Mol Med 52, 1486–1495 (2020). https://doi.org/10.1038/s12276-020-00503-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-00503-9
