
Abstract
Monoclonal antibodies (mAbs) are one of the most widely used drug platforms for infectious diseases or cancer therapeutics because they selectively target pathogens, infectious cells, cancerous cells, and even immune cells. In this way, they mediate the elimination of target molecules and cells with fewer side effects than other therapeutic modalities. In particular, cancer therapeutic mAbs can recognize cell-surface proteins on target cells and then kill the targeted cells by multiple mechanisms that are dependent upon a fragment crystallizable (Fc) domain interacting with effector Fc gamma receptors, including antibody-dependent cell-mediated cytotoxicity and antibody-dependent cell-mediated phagocytosis. Extensive engineering efforts have been made toward tuning Fc functions by either reinforcing (e.g. for targeted therapy) or disabling (e.g. for immune checkpoint blockade therapy) effector functions and prolonging the serum half-lives of antibodies, as necessary. In this report, we review Fc engineering efforts to improve therapeutic potency, and propose future antibody engineering directions that can fulfill unmet medical needs.
Similar content being viewed by others

Introduction
Thirty-one new monoclonal antibodies (mAbs) have been approved by the Food and Drug Administration (FDA) in the USA and the European Medicines Agency in the EU since 2013, to constitute a total of 57 mAbs used in clinics by the end of 20171. Several financial reports revealed that the antibody market had exceeded 98 billion US dollars in sales by December 2017, with an annual growth rate of 18.3% and a predicted valuation of 137–200 billion US dollars by 20221. The antibody market is dominated by seven companies, with 87% of the total market sales out of 22 active companies led by Genentech, a member of the Roche group, with a 31% market share for 11 approved antibody molecules. In terms of disease indications, as of December 2017 15 out of 57 mAbs are approved for cancer therapy, the number one disease target, and 12 mAbs are used in hematology.
Among the five isotypes of immunoglobulins (Igs), which are IgG, IgA, IgM, IgD, and IgE, IgG comprises the majority, representing 60% of total serum Igs in humans. All FDA-approved therapeutic Igs belong to the IgG class2. The endogenous IgG molecule is composed of two identical fragment antigen binding (Fab) domains and one fragment crystallizable (Fc) domain that make it multivalent and multifunctional. The two Fab fragments each consist of a heterodimer of a light chain and the N-terminal part of the heavy chain, whereas the C-terminal half of the two heavy chains dimerizes to form the Fc fragment of the IgG antibody. The N-terminal domains of the Fab fragment are the variable domains (VL and VH) that are responsible for antigen recognition, whereas the C-terminal part of the heavy chains compose the Fc fragment that is responsible for humoral and cellular effector functions. The two Fabs and the Fc are connected by the hinge region, which facilitates the spatial alignment of the three moieties for binding to antigens and effector ligands. The native full-length mAb, recognizing one epitope, comprises 53 out of the total available mAbs in clinics as of December 20171.
Antibody Fc domains are responsible for function in antibodies and Fab domains are responsible for targeting. Thus, Fc engineering stands for engineering functions of antibodies, which are effector functions, such as antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), and controlling serum half-life. In this article, we discuss (i) Fc receptors, such as Fcγ receptors (FcγRs) and neonatal Fc receptor (FcRn); (ii) Fc engineering modulating binding capacity to FcγRs and FcRn; and (iii) other Fc engineering efforts, such as creating monovalency or bispecificity for improving therapeutic potency.
Fc receptors: Fcγ receptors (FcγRs) and neonatal Fc receptor (FcRn)
The Fcγ receptor IIIa
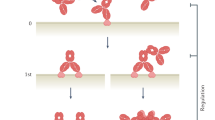
A family of receptors that recognize the Fc domain of IgG molecules is known as the FcγRs family. These receptors are expressed on the surfaces of immune effector cells, and upon being cross-linked by the IgG Fc domain, they induce downstream cellular processes, which affect innate and adaptive immunity. Among the FcγRs, which are highly homologous throughout mammalian species3, FcγRIIIa is a key surface receptor in terms of its contribution to ADCC activity (Fig. 1a). Typically, FcγRIIIa is found on the surfaces of natural killer (NK) cells, macrophages, monocytes, mast cells, eosinophils, and dendritic cells. However, it is the only FcγR expressed by NK cells4,5. FcγRIIIa is a transmembrane receptor containing two extracellular domains and a cytoplasmic tail, with medium affinity for IgG (KD = 2 × 10−7 M)6. In FcγRIIIa, an immunoreceptor tyrosine-based activation motif (ITAM) sequence is present in the intracellular region of the associated γ chain or ζ chain. The function of this receptor is dependent either on its γ-chain on monocytes and macrophages or on γ- or ζ-chains on NK cells. Humans express two FcγRIIIa allotypes that differ in a single amino acid at position 158; the residue can be either valine (V) or phenylalanine (F), whereby the isoform with V at position 158 has high affinity for the Fc domain of IgG1, and the one with F at position 158 has low affinity. This FcγRIIIa polymorphism has been shown to contribute to clinical responses of IgG1 therapeutic antibodies7,8,9,10. Engagement of the high-affinity FcγRIIIa-V158 by immune complexes (ICs) results in a stronger in vitro cytotoxic potency than that of FcγRIIIa-F15811. Regarding the allotype population, FcγRIIIa-V158 homozygotes represent only 10–20% of the population worldwide, whereas FcγRIIIa-F158 homozygotes constitute 40–50%12,13. Interestingly, expression of the high-affinity allotype of FcγRIIIa-V158 correlates with the improved clinical outcome in the treatment of lymphoma patients with rituximab7,10.

Human natural killer cells, which only express FcγRIIIa, activate antibody-dependent cellular cytotoxicity via FcγRIIIa (a). Macrophages are one of the main contributing lymphocytes for antibody-dependent cellular phagocytosis activity, which is known to be triggered by FcγRIIa intracellular signaling (b).
Fc-engineered antibodies with improved affinity to FcγRIIIa show higher therapeutic efficacy relative to native Fc because they are capable of priming and activating NK cells more efficiently14,15. However, several studies showed that although the depletion of NK cells or neutrophils did not significantly reduce tumor suppression activity, exhaustion of macrophages abrogated the therapeutic efficacy of anti-CD2016, anti-CD3017, or anti-CD40 antibodies18 in mouse models. These results highlight the importance of ADCP activity in therapeutic efficacy against cancer, because macrophages are one of the main contributing sources of leukocytes for tumor phagocytic activities (Fig. 1b)19. ADCP activity is known to be triggered by FcγRIIa intracellular signaling (Fig. 1b)20,21,22, which has been shown by glycoengineered antibodies to exhibit enhanced affinity toward FcγRIIa, resulting in increased ADCP activity21,23. However, the existence of other human Fc-glycoengineered antibodies that do not affect FcγRIIa binding24 but improve ADCP20 also indicates the possibility that FcγRIIIa contributes downstream signaling to phagocytic activity25.
Fcγ receptors II: FcγRIIa and FcγRIIb
FcγRII comprises a single-chain polypeptide α chain with an extracellular region of two immunoglobulin-like domains and a cytoplasmic domain. In humans, FcγRII is further classified into FcγRIIa and FcγRIIb. The activating FcγRIIa (KD = 2 × 10−7 M) contains an ITAM motif in the intracellular region of the α chain, whereas the inhibitory FcγRIIb (KD = 8 × 10−6 M) contains an immunoreceptor tyrosine-based inhibitory motif in the same region26. FcγRIIa initiates endocytosis, phagocytosis, ADCC, and inflammatory cytokine release. In contrast, FcγRIIb transduces inhibitory signals that downregulate immune functions triggered by the activating receptors. FcγRIIa has two allotypes that differ in a single amino acid at position 131. That residue can be either histidine (H) or arginine (R); the isoform with H is a high-affinity receptor, while that with R is a low-affinity receptor. Despite the high structural homology and protein sequence identity (96%) of FcγRIIa with FcγRIIb27, there are a number of ongoing efforts to create engineered antibodies with improved affinity for FcγRIIa to increase the therapeutic potential of the antibody28,29,30.
Several studies have established a correlation between Fc:FcγRs affinities and selectivity and the cytotoxic functions of immune effector cells engaged by ICs. For example, the increased binding toward activating FcγRs over inhibitory FcγRIIb has been shown to be important because it enhances the efficacy of trastuzumab or rituximab31. In other studies, the efficacies of various antibodies in the clinic have been shown to correlate with FcγRIIIa alleles (the high-affinity FcγRIIIa-V158 and the low-affinity FcγRIIIa-F158 alleles)7,10,11. Additionally, in vitro cytotoxicity was increased with improved affinities for FcγRs either by glycoengineering32 or by amino acid substitutions in the Fc domain33,34,35,36. In particular, Fc variants displaying high affinity for the FcγRIIa-R131 isoform and also high selectivity for FcγRIIa over FcγRIIb have been shown to mediate improved ADCP activity30. These results suggest that antibodies with increased binding affinity for activating FcγRs, but not for inhibitory FcγRIIb, elicit stronger ADCP and potentially represent more effective therapeutics for cancer treatment.
A human IgG recycling receptor: the neonatal Fc receptor (FcRn)
IgG is the most abundant antibody isotype largely because of its extremely long half-life of 7–23 days. The discovery and characterization of an intracellular trafficking receptor for IgG were first reported by Brambell and coworkers in 196637. This receptor is called the neonatal Fc receptor molecule (FcRn) and is a noncovalent heterodimer consisting of an MHC-class-I-like heavy chain and a β2-microglobin light chain. FcRn, which binds to IgG and albumin at different binding sites, is present at the cell surface and intracellularly within the ER.38,39 Simister and coworkers first described FcRn as a transporter of IgG within the intestinal epithelial cells of neonatal rats, enabling the transfer of passive immunity from a mother to her offspring40,41. Although rodent FcRn is upregulated in intestinal epithelial cells only during lactation, human FcRn is expressed throughout life in almost all other cell types, including endothelial cells42,43,44; kidney podocytes45; mammary epithelial cells46; pulmonary epithelial cells47; hepatocytes48; and hematopoietic cells, such as monocytes, macrophages, dendritic cells49, and B cells50.
Mice deficient in either FcRn or β2-microglobin have severely reduced serum IgG levels because the rates of catabolism of these antibodies are increased. In these mice, IgGs have a circulation half-life of approximately 1.4 days, whereas wild-type mice have a circulation half-life of 9 days38,51,52. In humans suffering from familial hypercatabolic hypoproteinemia, a mutation in the β2-microglobin gene results in expression of a nonfunctional protein and five-fold increased catabolism of IgG relative to that in healthy individuals53,54. Bone marrow chimeric mouse studies have shown that both wild-type mice chimerized with Fcgrt−/− bone marrow and Fcgrt−/− mice chimerized with wild-type bone marrow have approximately 50% serum IgG levels relative to those of wild-type mice55.
FcRn only binds to its ligands at acidic pH (≤6.5) but not at neutral pH (≥7.0)56,57,58. At physiological pH, histidine residues of the Fc domain of IgG, which interacts with FcRn, become deprotonated, thereby enabling ligand dissociation. In contrast, at more acidic pH, the protonated histidine residues stabilize the IgG:FcRn complex59. Thus, the strict pH-dependent ligand-binding property of FcRn seems to have evolved in the endo-lysosomal system to function as a mediator of IgG trafficking and pharmacological parameters, including stability, biodistribution, and immunogenicity, in humans.
Engineering effector functions of antibodies
The effector functions of antibodies, whether they are produced by our own immune systems or administered therapeutically, rely on the interactions of their Fc domains with FcγRs and complement components, such as C1, to clear pathogens or destroy tumor cells60,61,62. The effector functions of antibodies are: (i) ADCC and ADCP via the recruitment of FcγRs63 and (ii) complement-dependent cytotoxicity (CDC) via the recruitment of C1q. The known human FcγRs are FcγRI (CD64), FcγRIIa (CD32a), FcγRIIb (CD32b), FcγRIIc (CD32c), FcγRIIIa (CD16a), and FcγRIIIb (CD16b), and these receptors are expressed at different levels on the surface of various immune cells.63 The cytotoxic potential of mAbs results from improved affinity between their Fc domains to the activating FcγR relative to the inhibitory FcγRIIb31,64.
NK cells are well-known cytotoxic lymphocytes that kill tumor cells and constitute a major component of the innate immune system. Clinically, NK cells have been implicated as important mediators of the antitumor activity of trastuzumab in breast cancer patients. Patients with responsive tumors tend to have increased numbers of tumor-associated NK cells65. ICs are recognized by FcγRIIIa on NK cells, resulting in the activation of cytotoxic processes and potent ADCC66. Human NK cells, which only express FcγRIIIa, activate ADCC via FcγRIIIa (Fig. 1a). The efficacy of some cancer therapeutic antibodies has been correlated with a widespread FcγRIIIa polymorphism: improved clinical outcomes are observed in patients expressing the high-affinity isoform V158 rather than the low-affinity isoform F1587,10,11.
Optimization of the interactions between antibodies and FcγRs has emerged as a promising approach for enhancing the activity of therapeutic antibodies for the treatment of both cancer and autoimmune disease67. Thus, engineering IgGs with improved FcγRs affinity has been investigated extensively in recent years. Researchers at Xencor (CA, USA) capitalized on a structure-based computational design algorithm for engineering various therapeutic effector functions of clinically applicable antibodies. They isolated a series of Fc mutants with improved effector functions mediated by enhanced binding affinity, including increased FcγRIIIa binding over FcγRIIb for ADCC68 and increased FcγRIIa binding for ADCP28. Remarkably, a S239D/I332E/A330L (EU numbering) IgG mutant showed a > 300-fold increase in FcγRIIIa binding affinity relative to that of the wild-type IgG. This antibody also showed 58-fold increased binding to FcγRIIIa-F158. However, this antibody also bound to the inhibitory receptor FcγRIIb with much higher affinity68. For example, margetuximab, developed by MacroGenics, had adapted the same Fab target (Her2) as transtuzumab but had been Fc-engineered to maximize immune effector function by elevating relative affinity to activating Fcγ receptor, FcγRIIIa over inhibitory Fcγ receptor, FcγRIIb, and it recently showed a 24% risk reduction in patients relative to that of trastuzumab in a phase 3 clinical trial in 536 breast cancer patients69. Other significant efforts have been directed to the engineering of antibodies with improved affinity for FcγRIIIa and enhanced effector function by amino acid mutations36,70,71,72,73,74 or glycan modifications75,76,77,78,79,80,81. Hatori and coworkers also engineered an antibody Fc variant (P238D/L328E) that showed selectively enhanced FcγRIIb binding over both FcγRIIa-R131 and FcγRIIa-H13182. Recently, several engineered variants have shown promise in preclinical testing and are moving into clinical trials83,84,85. These reports indicate the clinical significance of engineered Fcs with improved affinity to FcγRs.
Although antibodies with potentiated tumor-destroying functions of Fcs are desirable for tumor targeting, those with benign immune-silenced Fc’s are advantageous over native Fcs when ADCC, ADCP, and/or CDC would be detrimental, such as when antibodies function as (i) systemic neutralizers of cytokines, (ii) blockers of cell-surface antigens on immune cells, or (iii) bispecific engagers of effector immune cells in proximity to target diseased cells86,87,88. Because it is best not to stimulate unwanted immune cell depletion by FcγRs cross-linking, it is critical to completely silence Fc effector function because of the variation in activation thresholds by FcγRs among patients. One way to reduce the effector function is to remove the N-linked glycan by substituting an asparagine residue at 297 position with other residues89,90,91,92. Another strategy is the incorporation of amino acid mutations into residues that are responsible for FcγRs or C1q binding93. For example, Orthoclone OKT3®, the first marketed therapeutic antibody, contains two mutations, L234A and L235A, in the Fc domain to minimize toxicity by cytokines94. Moving the Fab region to other IgG subclasses, such as IgG2 or IgG4, which engages less tightly to FcγRs could be another option64. Moreover, exchanging amino acid residues among IgG subclasses resulted in more Fc options with reduced complement and Fcγ receptor binding, such as IgG2/495, IgG2 m496, and “TM” (L234F, L235E, and P331S)97. Researchers at Janssen Research and Development capitalized on multiple methodologies to abolish FcγRs and C1q affinity to generate IgG2σ with undetectable effector functions mediated by FcγRs and C1q98. Extensive efforts to engineer Fc to lack effector function are continuously being executed using different approaches, such as proline sandwich99, for safety considerations because effector activation thresholds are variable among diseases and populations.
Engineering serum half-life of antibodies
Compared with other serum proteins in blood circulation, IgG antibodies have the advantage of prolonged serum stability (21 days). FcRn, which exists in endosomes of circulating monocytes or endothelial cells, is a key molecule responsible for controlling serum IgG levels in mammals through its pH-dependent binding characteristics100. The mechanism underlying how IgG antibodies can be recycled or transcytosed through endothelium has been established101,102 (Fig. 2).

When IgGs are internalized by an endothelial cell or a circulating monocyte, they initially encounter FcRn within the early acidified endosome. This binding protects IgG from being sorted into a lysosome, in which serum proteins without FcRn binding are degraded; instead, IgGs are recycled back into the extracellular space44. At neutral pH on the cell surface, IgGs immediately dissociate from FcRn and return to the circulation.
At pH 6.0, FcRn binds to the CH2–CH3 hinge regions of the Fc region of IgG with micromolar affinity103,104. However, it does not measurably bind to IgG at physiological pH. FcRn binds to IgG in acidic endosomal compartments after serum IgG is internalized via pinocytosis by endothelial cells and circulating monocytes (Fig. 2). FcRn then recycles the IgG back into circulation at physiological pH, thus extending its serum half-life, whereas serum proteins that do not bind to FcRn are directed for lysosomal degradation. In contrast to monomeric IgGs that are recycled into serum by FcRn, polymeric IgGs, such as ICs surrounded by multiple IgG molecules bound to antigens, have been shown to be efficiently directed into lysosomes by an unknown mechanism of action55. By discriminating IgGs, whether they are monomeric or polymeric, FcRn determines the fate of IgG, either to protect it or direct it to be degraded for antigen presentation purposes. In addition to the enhanced degradation of ICs relative to monomeric IgGs, FcRn has also been shown to contribute to cross-presenting degraded antigens by regulating the sorting of intracellular IgG ICs, thereby cross-priming CD8+ cytotoxic T cell responses59.
Extensive studies in non-human primates105,106 and in human FcRn transgenic mice107,108 have shown that IgG Fc engineering for higher FcRn binding is of great importance for enhancing the half-lives of therapeutic antibodies. Fc engineering for higher FcRn binding at endosomal pH has also been extensively studied. A M252Y/S254T/T256E (YTE) variant isolated by Medimmune using phage display109 demonstrated 10-fold greater affinity to human FcRn than that of wild-type IgG1 at pH 6.0 and efficient release at pH 7.4. IgG antibodies containing YTE mutations showed 3.8-fold improvement in pharmacokinetics (half-life) in human FcRn transgenic mice and 2.5-fold improvement in cynomolgus monkeys105,110,111. However, YTE exhibited decreased ADCC activity. The Medimmune YTE had been adapted to anti-RSV antibody Fc, which received fast track designation by the FDA for prevention of respiratory disease under the name of MEDI8897, and is currently undergoing phase IIb clinical evaluation111. Another corporate group, Xencor, developed an engineered variant, Xtend (M428L/N434S), with 11-fold increased FcRn binding at pH 6.0 relative to that of wild-type IgG1. Xtend showed 4.2-fold improved serum half-life in transgenic mice and 3.2-fold improved serum half-life in non-human primates. Xtend Fc was tested in xenograft mouse models that express human FcRn as either an anti-VEGF or anti-EGFR IgG1 antibody and led to extended serum half-life as well as reduced tumor burden relative to those of wild-type IgG1108. Xtend has been adapted to ravulizumab (ALXN1210), which has been approved by US FDA on December 2018 for treatment of paroxysmal nocturnal hemoglobinuria/hemolytic-uremic syndrome112, or VRC01LS, which is under clinical evaluation for the prevention of human immunodeficiency virus113. These studies indicate that prolonging the serum stability of engineered IgG antibodies in the blood circulation enables extended therapeutic or protective activity, which circumvents the need for frequent administration of antibody drugs.
In some cases, the extended half-life property of Fc is desirable when combined with “benign blockers” with abolished effector functions, which are described in the section outlining engineering effector functions of antibodies. Borrok et al. at Medimmune characterized the molecular stability of a combination of two variants: “TM” (L234F, L235E, and P331S), which lacks immune receptor binding97 and “YTE” (M252Y, S254T, and T256E), which has an extended serum half-life105. Furthermore, they dissected the contributions of individual mutations of TM-YTE, which in turn hamper thermostability, and then discovered a novel combination FQQ-YTE (L234F, L235Q, K322Q, M252T, S254T, and T256E), with improved molecular and serum stability, which capitalized on rational protein engineering design114.
Engineering valency or bispecificity of antibodies
Fc engineering efforts for better therapeutic efficacy, other than for modulating affinity profiles to Fc receptors, are also being extensively pursued. One example is an engineered Fc that does not form a homodimer but remains as a soluble monomer, mFc, with half size, displaying (i) high affinity for FcγRI, (ii) no detectable binding to FcγRIIIa, and (iii) similar pH-dependent FcRn binding115,116. This engineering was focused on downsizing the molecular weight of Fc for therapeutic simplicity. However, mFc could nonspecifically bind to several viral or cancer-related proteins, such as gp140 of HIV-1, EDII of Zika virus, mesothelin, 5T4, PD-L1, OX40, and TIM3. Nonspecific bindings have been excluded by phage-display library screening, and the critical role of T366R and L368H on building monomeric status has been identified117.
Fc engineering for heterodimeric Fc is another example of obtaining bispecific properties for antigen binding to circumvent homodimer formation. The bispecific antibodies recognize two different antigens, so that they not only neutralize soluble pathogenic or endogenous proteins, but also co-ligate discrete antigens on the same cells or engage unconnected cells to derive therapeutically favorable status. Until now, three strategic approaches have been adapted to achieve heterodimeric structure in the Fc domain: (i) steric complementarity design, which are generally called knobs-into-holes; (ii) charge-to-charge swap; and (iii) isotype strand swap118,119,120. Two bispecific mAbs are currently on the market: emicizumab (Hemlibra®) and blinatumomab (Blincyto®). Emicizumab, developed by Chugai Pharmaceutical, is an IgG4 knobs-into-holes bispecific for coagulation factors IX and X to mimic coagulation factor VIII (FVIII) activity and is more favorable than FVIII in terms of pharmacokinetics (21 days for emicizumab vs. 0.5 day for FVIII)121. The Fc-engineered bispecific IgG4 antibody, emicizumab has been approved by the FDA for prophylaxis to prevent or reduce the frequency of bleeding episodes in patients with hemophilia A on November 2017122,123,124. This Fc engineering for bispecificity enabled co-ligation of two different antigens and opened up new therapeutic opportunities, maintaining all of the beneficial properties of mAbs.
The other marketed bispecific antibody is blinatumomab, a fusion of the two single-chain Fvs targeting CD3 on T lymphocytes and CD19 on tumor cells, which was approved by the FDA in December 2014. Despite their significant efficacy, the current bispecific T-cell engager (BiTE) programs have serum half-lives of <1 day, and therefore require continuous intravenous infusion for 28 consecutive days per cycle. Hence, Amgen is currently advancing new versions of BiTE molecules by fusing the Fc domain. However, the fact that catumaxomab, a bispecific mAb for CD3, and EpCAM have been withdrawn due to toxicity issues, presumably because of FcγR or C1q binding of Fc125,126,127,128, indicates that for T-cell engagers, FcγR-related effector functions are detrimental with respect to clinical aspects. Therefore, simple Fc fusion for half-life extension purposes may lead to side effects, such as toxicity and/or shortened efficacy, at least in case of BiTE. Either Fc engineering for immune silencing or further protein engineering are also essential for immune cell-engaging antibodies.
Conclusion
Extensive research and development capitalizing on Fc engineering techniques to modulate effector functions of antibodies have been conducted by researchers in both academia and industry129. Despite these efforts, the exact function of Fc remains to be validated; for example, the function of FcγRIIIa on NK cells is the key contributor for ADCC activity but that on other lymphocytes, such as monocytes and macrophages, is not yet clear. Especially, Fc function in protection of infectious diseases, such as antiviral activity, has received increasing attention130,131, which suggests that it is responsible for inducing humoral and cellular responses that provide protective immunity, such as vaccine-like effects, in addition to neutralizing the activity of IgG antibodies. Fc engineers have also developed a few Fc variants that have stricter pH-dependent FcRn binding, such as YTE and Xtend, which are used in clinical settings as anti-infectious agents or against autoimmune diseases132. The recent approvals of bispecific antibodies, developed by using sophisticated Fc engineering that produced maximal ratio of heterodimeric Fcs relative to homodimeric Fcs, indicate that bispecific antibodies have high potential for therapeutic applications and that there is much room for improvement to meet unmet medical needs, such as in pharmacokinetics or tissue infiltration, while maintaining low toxicity. Despite insufficient knowledge of the functions of Fc receptors because of the diverse profiles of the receptors on discrete leukocytes, Fc engineering works for (i) improving effector functions by selective FcγR affinity in tumor targeting purposes, (ii) maximizing serum half-life by pH-selective FcRn affinity for various disease categories such as cancer, infectious diseases or autoimmune therapy, (iii) molecular downsizing to half relative to that of native antibodies by blocking homodimerization, and (iv) conferring bispecificity by maximizing the efficiency of Fc heterodimerization. These are powerful strategies to broaden the therapeutic applications of antibodies. Additionally, ICs comprising antibodies that have had their Fc functions optimized for adaptive immunity are potential candidates for future cancer vaccines and therapy133,134.
References
Grilo, A. L. & Mantalaris, A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 37, 9–16 (2019).
Brezski, R. J. & Georgiou, G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr. Opin. Immunol. 40, 62–69 (2016).
Bournazos, S. In Current Topics in Microbiology and Immunology (Springer, Berlin, Heidelberg, 2019).
Takai, T. Fc receptors and their role in immune regulation and autoimmunity. J. Clin. Immunol. 25, 1–18 (2005).
Hogarth, P. M. & Pietersz, G. A. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat. Rev. Drug Discov. 11, 311–331 (2012).
Okazaki, A. et al. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcgammaRIIIa. J. Mol. Biol. 336, 1239–1249 (2004).
Weng, W. K. & Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 21, 3940–3947 (2003).
Bibeau, F. et al. Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 27, 1122–1129 (2009).
Canete, J. D. et al. Influence of variants of Fc gamma receptors IIA and IIIA on the American College of Rheumatology and European League Against Rheumatism responses to anti-tumour necrosis factor alpha therapy in rheumatoid arthritis. Ann. Rheum. Dis. 68, 1547–1552 (2009).
Cartron, G. et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99, 754–758 (2002).
Dall’Ozzo, S. et al. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 64, 4664–4669 (2004).
Kyogoku, C. et al. Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. 46, 1242–1254 (2002).
Sullivan, K. E. et al. Analysis of polymorphisms affecting immune complex handling in systemic lupus erythematosus. Rheumatology 42, 446–452 (2003).
Hatjiharissi, E. et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood 110, 2561–2564 (2007).
Capuano, C. et al. Obinutuzumab-mediated high-affinity ligation of FcgammaRIIIA/CD16 primes NK cells for IFNgamma production. Oncoimmunology 6, e1290037 (2017).
Horikawa, M., Minard-Colin, V., Matsushita, T. & Tedder, T. F. Regulatory B cell production of IL-10 inhibits lymphoma depletion during CD20 immunotherapy in mice. J. Clin. Invest. 121, 4268–4280 (2011).
Oflazoglu, E. et al. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood 110, 4370–4372 (2007).
Oflazoglu, E. et al. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Brit. J. Cancer 100, 113–117 (2009).
Gul, N. et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Invest. 124, 812–823 (2014).
Dahal, L. N. et al. STING activation reverses lymphoma-mediated resistance to antibody immunotherapy. Cancer Res. 77, 3619–3631 (2017).
Nagelkerke, S. Q. et al. Red pulp macrophages in the human spleen are a distinct cell population with a unique expression of Fc-gamma receptors. Blood Adv. 2, 941–953 (2018).
Martinez, F. O., Sica, A., Mantovani, A. & Locati, M. Macrophage activation and polarization. Front. Biosci. 13, 453–461 (2008).
Minard-Colin, V. et al. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood 112, 1205–1213 (2008).
Golay, J. et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 122, 3482–3491 (2013).
Kang, T. H. et al. An engineered human Fc variant with exquisite selectivity for FcgammaRIIIaV158 reveals that ligation of FcgammaRIIIa mediates potent antibody dependent cellular phagocytosis with GM-CSF-differentiated macrophages. Front. Immunol. 10, 562 (2019).
Bruhns, P. et al. Specificity and affinity of human Fc {gamma} receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716 (2009).
Rankin, C. T. et al. CD32B, the human inhibitory Fc-gamma receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood 108, 2384–2391 (2006).
Richards, J. O. et al. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol. Cancer Ther. 7, 2517–2527 (2008).
Shang, L. et al. Selective antibody intervention of Toll-like receptor 4 activation through Fc gamma receptor tethering. J. Biol. Chem. 289, 15309–15318 (2014).
Jung, S. T. et al. Effective phagocytosis of low Her2 tumor cell lines with engineered, aglycosylated IgG displaying high FcgammaRIIa affinity and selectivity. ACS Chem. Biol. 8, 368–375 (2013).
Clynes, R. A., Towers, T. L., Presta, L. G. & Ravetch, J. V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 6, 443–446 (2000).
Shields, R. L. et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 277, 26733–26740 (2002).
Duncan, A. R., Woof, J. M., Partridge, L. J., Burton, D. R. & Winter, G. Localization of the binding site for the human high-affinity Fc receptor on IgG. Nature 332, 563–564 (1988).
Sarmay, G., Lund, J., Rozsnyay, Z., Gergely, J. & Jefferis, R. Mapping and comparison of the interaction sites on the Fc region of IgG responsible for triggering antibody dependent cellular cytotoxicity (ADCC) through different types of human Fc gamma receptor. Mol. Immunol. 29, 633–639 (1992).
Redpath, S., Michaelsen, T. E., Sandlie, I. & Clark, M. R. The influence of the hinge region length in binding of human IgG to human Fcgamma receptors. Hum. Immunol. 59, 720–727 (1998).
Shields, R. L. et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 276, 6591–6604 (2001).
Brambell, F. W. The transmission of immunity from mother to young and the catabolism of immunoglobulins. Lancet 2, 1087–1093 (1966).
Chaudhury, C. et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 197, 315–322 (2003).
Chaudhury, C., Brooks, C. L., Carter, D. C., Robinson, J. M. & Anderson, C. L. Albumin binding to FcRn: distinct from the FcRn-IgG interaction. Biochemistry 45, 4983–4990 (2006).
Simister, N. E. & Rees, A. R. Isolation and characterization of an Fc receptor from neonatal rat small intestine. Eur. J. Immunol. 15, 733–738 (1985).
Simister, N. E. & Mostov, K. E. An Fc receptor structurally related to MHC class I antigens. Nature 337, 184–187 (1989).
Borvak, J. et al. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int. Immunol. 10, 1289–1298 (1998).
Antohe, F., Radulescu, L., Gafencu, A., Ghetie, V. & Simionescu, M. Expression of functionally active FcRn and the differentiated bidirectional transport of IgG in human placental endothelial cells. Hum. Immunol. 62, 93–105 (2001).
Ober, R. J., Martinez, C., Vaccaro, C., Zhou, J. & Ward, E. S. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J. Immunol. 172, 2021–2029 (2004).
Akilesh, S. et al. Podocytes use FcRn to clear IgG from the glomerular basement membrane. Proc. Natl Acad. Sci. USA 105, 967–972 (2008).
Cianga, P., Cianga, C., Cozma, L., Ward, E. S. & Carasevici, E. The MHC class I related Fc receptor, FcRn, is expressed in the epithelial cells of the human mammary gland. Hum. Immunol. 64, 1152–1159 (2003).
Sakagami, M. et al. Expression and transport functionality of FcRn within rat alveolar epithelium: a study in primary cell culture and in the isolated perfused lung. Pharm. Res. 23, 270–279 (2006).
Blumberg, R. S. et al. A major histocompatibility complex class I-related Fc receptor for IgG on rat hepatocytes. J. Clin. Invest. 95, 2397–2402 (1995).
Zhu, X. et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J. Immunol. 166, 3266–3276 (2001).
Mi, W. et al. Targeting the neonatal fc receptor for antigen delivery using engineered fc fragments. J. Immunol. 181, 7550–7561 (2008).
Roopenian, D. C. et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J. Immunol. 170, 3528–3533 (2003).
Kim, J. et al. Beta 2-microglobulin deficient mice catabolize IgG more rapidly than FcRn- alpha-chain deficient mice. Exp. Biol. Med. 233, 603–609 (2008).
Waldmann, T. A. & Terry, W. D. Familial hypercatabolic hypoproteinemia. A disorder of endogenous catabolism of albumin and immunoglobulin. J. Clin. Invest. 86, 2093–2098 (1990).
Wani, M. A. et al. Familial hypercatabolic hypoproteinemia caused by deficiency of the neonatal Fc receptor, FcRn, due to a mutant beta2-microglobulin gene. Proc. Natl Acad. Sci. USA 103, 5084–5089 (2006).
Qiao, S. W. et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proc. Natl Acad. Sci. USA 105, 9337–9342 (2008).
Vaughn, D. E. & Bjorkman, P. J. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure 6, 63–73 (1998).
Raghavan, M., Gastinel, L. N. & Bjorkman, P. J. The class I major histocompatibility complex related Fc receptor shows pH-dependent stability differences correlating with immunoglobulin binding and release. Biochemistry 32, 8654–8660 (1993).
Rodewald, R. pH-dependent binding of immunoglobulins to intestinal cells of the neonatal rat. J. Cell Biol. 71, 666–669 (1976).
Baker, K. et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc. Natl Acad. Sci. USA 108, 9927–9932 (2011).
Nimmerjahn, F. & Ravetch, J. V. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 19, 239–245 (2007).
Nimmerjahn, F. & Ravetch, J. V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 (2008).
Cohen-Solal, J. F., Cassard, L., Fridman, W. H. & Sautes-Fridman, C. Fc gamma receptors. Immunol. Lett. 92, 199–205 (2004).
Jefferis, R. & Lund, J. Interaction sites on human IgG-Fc for FcgammaR: current models. Immunol. Lett. 82, 57–65 (2002).
Nimmerjahn, F. & Ravetch, J. V. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 310, 1510–1512 (2005).
Arnould, L. et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 94, 259–267 (2006).
Giodini, A., Rahner, C. & Cresswell, P. Receptor-mediated phagocytosis elicits cross-presentation in nonprofessional antigen-presenting cells. Proc. Natl Acad. Sci. USA 106, 3324–3329 (2009).
Mimoto, F., Kuramochi, T., Katada, H., Igawa, T. & Hattori, K. Fc engineering to improve the function of therapeutic antibodies. Curr. Pharm. Biotechnol. 17, 1298–1314 (2016).
Lazar, G. A. et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl Acad. Sci. USA 103, 4005–4010 (2006).
Taylor, N. P. MacroGenics’ margetuximab beats Herceptin in phase 3. FierceBiotech https://www.fiercebiotech.com/biotech/macrogenics-margetuximab-beats-herceptin-phase-3 (2019).
Oganesyan, V., Damschroder, M. M., Leach, W., Wu, H. & Dall’Acqua, W. F. Structural characterization of a mutated, ADCC-enhanced human Fc fragment. Mol. Immunol. 45, 1872–1882 (2008).
Stavenhagen, J. B. et al. Enhancing the potency of therapeutic monoclonal antibodies via Fc optimization. Adv. Enzym. Regul. 48, 152–164 (2008).
Saxena, A. & Wu, D. Advances in therapeutic Fc engineering—modulation of IgG-associated effector functions and serum half-life. Front. Immunol. 7, 580 (2016).
Ashoor, D. N., Ben Khalaf, N., Bourguiba-Hachemi, S., Marzouq, M. H. & Fathallah, M. D. Engineering of the upper hinge region of human IgG1 Fc enhances the binding affinity to FcgammaIIIa (CD16a) receptor isoform. Protein Eng. Des. Sel. 31, 205–212 (2018).
Zhang, D., Goldberg, M. V. & Chiu, M. L. Fc engineering approaches to enhance the agonism and effector functions of an Anti-OX40 antibody. J. Biol. Chem. 291, 27134–27146 (2016).
Davies, J. et al. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. Biotechnol. Bioeng. 74, 288–294 (2001).
Mossner, E. et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 115, 4393–4402 (2010).
Umana, P., Jean-Mairet, J., Moudry, R., Amstutz, H. & Bailey, J. E. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 17, 176–180 (1999).
Li, W., Zhu, Z., Chen, W., Feng, Y. & Dimitrov, D. S. Crystallizable fragment glycoengineering for therapeutic antibodies development. Front. Immunol. 8, 1554 (2017).
Li, T. et al. Modulating IgG effector function by Fc glycan engineering. Proc. Natl Acad. Sci. USA 114, 3485–3490 (2017).
Yu, X., Marshall, M. J. E., Cragg, M. S. & Crispin, M. Improving antibody-based cancer therapeutics through glycan engineering. BioDrugs 31, 151–166 (2017).
Lee, H. S. & Im, W. Effects of N-Glycan composition on structure and dynamics of IgG1 Fc and their implications for antibody engineering. Sci. Rep. 7, 12659 (2017).
Mimoto, F. et al. Engineered antibody Fc variant with selectively enhanced FcgammaRIIb binding over both FcgammaRIIa(R131) and FcgammaRIIa(H131). Protein Eng. Des. Sel. 26, 589–598 (2013).
Nordstrom, J. L. et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcgamma receptor binding properties. Breast Cancer Res. 13, R123 (2011).
Horton, H. M. et al. Antibody-mediated coengagement of FcgammaRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J. Immunol. 186, 4223–4233 (2011).
Salles, A., Morchhauser, F. & Thieblemont, C. Promising efficacy with the new anti-CD20 antibody GA101 in heavily pre-treated patients-first results from a phase II study in patients with relapsed/refractory indolent NHL (INHL). Haematologica 95, 229 (2010).
Strohl, W. R. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 20, 685–691 (2009).
Pollreisz, A. et al. Intravenous immunoglobulins induce CD32-mediated platelet aggregation in vitro. Br. J. Dermatol. 159, 578–584 (2008).
Kontermann, R. E. & Brinkmann, U. Bispecific antibodies. Drug Discov. Today 20, 838–847 (2015).
Jung, S. T. et al. Aglycosylated IgG variants expressed in bacteria that selectively bind FcgammaRI potentiate tumor cell killing by monocyte-dendritic cells. Proc. Natl Acad. Sci. USA 107, 604–609 (2010).
Hristodorov, D., Fischer, R. & Linden, L. With or without sugar? (A)glycosylation of therapeutic antibodies. Mol. Biotechnol. 54, 1056–1068 (2013).
Jacobsen, F. W. et al. Engineering an IgG scaffold lacking effector function with optimized developability. J. Biol. Chem. 292, 1865–1875 (2017).
Borrok, M. J., Jung, S. T., Kang, T. H., Monzingo, A. F. & Georgiou, G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem. Biol. 7, 1596–1602 (2012).
Chappel, M. S. et al. Identification of the Fc gamma receptor class I binding site in human IgG through the use of recombinant IgG1/IgG2 hybrid and point-mutated antibodies. Proc. Natl Acad. Sci. USA 88, 9036–9040 (1991).
Alegre, M. L. et al. A non-activating “humanized” anti-CD3 monoclonal antibody retains immunosuppressive properties in vivo. Transplantation 57, 1537–1543 (1994).
Mueller, J. P. et al. Humanized porcine VCAM-specific monoclonal antibodies with chimeric IgG2/G4 constant regions block human leukocyte binding to porcine endothelial cells. Mol. Immunol. 34, 441–452 (1997).
An, Z. et al. IgG2m4, an engineered antibody isotype with reduced Fc function. MAbs 1, 572–579 (2009).
Oganesyan, V., Gao, C., Shirinian, L., Wu, H. & Dall’Acqua, W. F. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr. D. Biol. Crystallogr. 64, 700–704 (2008).
Vafa, O. et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 65, 114–126 (2014).
Schlothauer, T. et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 29, 457–466 (2016).
Ghetie, V. & Ward, E. S. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu. Rev. Immunol. 18, 739–766 (2000).
Challa, D. K., Velmurugan, R., Ober, R. J. & Sally Ward, E. FcRn: from molecular interactions to regulation of IgG pharmacokinetics and functions. Curr. Top. Microbiol. Immunol. 382, 249–272 (2014).
D’Hooghe, L., Chalmers, A. D., Heywood, S. & Whitley, P. Cell surface dynamics and cellular distribution of endogenous FcRn. PLoS One 12, e0182695 (2017).
Presta, L. G. Molecular engineering and design of therapeutic antibodies. Curr. Opin. Immunol. 20, 460–470 (2008).
Roopenian, D. C. & Akilesh, S. FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. 7, 715–725 (2007).
Dall’Acqua, W. F., Kiener, P. A. & Wu, H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 281, 23514–23524 (2006).
Hinton, P. R. et al. Engineered human IgG antibodies with longer serum half-lives in primates. J. Biol. Chem. 279, 6213–6216 (2004).
Yeung, Y. A. et al. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J. Immunol. 182, 7663–7671 (2009).
Zalevsky, J. et al. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 28, 157–159 (2010).
Dall’Acqua, W. F. et al. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J. Immunol. 169, 5171–5180 (2002).
Oganesyan, V. et al. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol. Immunol. 46, 1750–1755 (2009).
Domachowske, J. B. et al. Safety, tolerability and pharmacokinetics of MEDI8897, an extended half-life single-dose respiratory syncytial virus prefusion f-targeting monoclonal antibody administered as a single dose to healthy preterm infants. Pediatr. Infect. Dis. J. 37, 886–892 (2018).
Roth, A. et al. Ravulizumab (ALXN1210) in patients with paroxysmal nocturnal hemoglobinuria: results of 2 phase 1b/2 studies. Blood Adv. 2, 2176–2185 (2018).
Gaudinski, M. R. et al. Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: A Phase 1 open-label clinical trial in healthy adults. PLoS Med. 15, e1002493 (2018).
Borrok, M. J. et al. An “Fc-silenced” IgG1 format with extended half-life designed for improved stability. J. Pharm. Sci. 106, 1008–1017 (2017).
Ying, T., Chen, W., Gong, R., Feng, Y. & Dimitrov, D. S. Soluble monomeric IgG1 Fc. J. Biol. Chem. 287, 19399–19408 (2012).
Ying, T., Feng, Y., Wang, Y., Chen, W. & Dimitrov, D. S. Monomeric IgG1 Fc molecules displaying unique Fc receptor interactions that are exploitable to treat inflammation-mediated diseases. MAbs 6, 1201–1210 (2014).
Wang, C. et al. Engineered soluble monomeric IgG1 Fc with significantly decreased non-specific binding. Front. Immunol. 8, 1545 (2017).
Ha, J. H., Kim, J. E. & Kim, Y. S. Corrigendum: immunoglobulin Fc heterodimer platform technology: from design to applications in therapeutic antibodies and proteins. Front. Immunol. 8, 1582 (2017).
Liu, H., Saxena, A., Sidhu, S. S. & Wu, D. Fc engineering for developing therapeutic bispecific antibodies and novel scaffolds. Front. Immunol. 8, 38 (2017).
Ko, S. & Jung, S. T. Engineering antibodies for dual specificity and enhanced potency. Biotechnol. Bioproc. Eng. 20, 201–210 (2015).
Kitazawa, T. et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat. Med. 18, 1570–1574 (2012).
Oldenburg, J. et al. Emicizumab prophylaxis in hemophilia A with inhibitors. New Engl. J. Med. 377, 809–818 (2017).
Mahlangu, J. et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. New Engl. J. Med. 379, 811–822 (2018).
Oldenburg, J. et al. The effect of emicizumab prophylaxis on health-related outcomes in persons with haemophilia A with inhibitors: HAVEN 1 Study. Haemophilia 25, 33–44 (2019).
Knodler, M. et al. Randomised phase II trial to investigate catumaxomab (anti-EpCAM x anti-CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. Brti. J. Cancer 119, 296–302 (2018).
Borlak, J., Langer, F., Spanel, R., Schondorfer, G. & Dittrich, C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcgamma receptors. Oncotarget 7, 28059–28074 (2016).
Kurbacher, C. M. et al. Outpatient intraperitoneal catumaxomab therapy for malignant ascites related to advanced gynecologic neoplasms. Oncologist 20, 1333–1341 (2015).
Seeber, A. et al. Detection of soluble EpCAM (sEpCAM) in malignant ascites predicts poor overall survival in patients treated with catumaxomab. Oncotarget 6, 25017–25023 (2015).
Wang, X., Mathieu, M. & Brezski, R. J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 9, 63–73 (2018).
Pelegrin, M., Naranjo-Gomez, M. & Piechaczyk, M. Antiviral monoclonal antibodies: can they be more than simple neutralizing agents? Trends Microbiol. 23, 653–665 (2015).
Gunn, B. M. et al. A role for Fc function in therapeutic monoclonal antibody-mediated protection against ebola virus. Cell Host Microbe 24, 221–233 e225 (2018).
Ward, E. S. & Ober, R. J. Targeting FcRn to generate antibody-based therapeutics. Trends Pharmacol. Sci. 39, 892–904 (2018).
Bournazos, S. & Ravetch, J. V. Fcgamma receptor function and the design of vaccination strategies. Immunity 47, 224–233 (2017).
Wen, Y. M., Mu, L. & Shi, Y. Immunoregulatory functions of immune complexes in vaccine and therapy. EMBO Mol. Med. 8, 1120–1133 (2016).
Acknowledgements
This work was supported by grants from the Basic Science Research Programs (2019R1F1A1059834 and 2019R1A4A1029000), the Bio & Medical Technology Development Program (2017M3A9C8060541), and the Pioneer Research Center Program (2014M3C1A3051460) through the National Research Foundation of Korea funded by the Ministry of Science and ICT.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, T.H., Jung, S.T. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med 51, 1–9 (2019). https://doi.org/10.1038/s12276-019-0345-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-019-0345-9
This article is cited by
-
Dosing of Convalescent Plasma and Hyperimmune Anti-SARS-CoV-2 Immunoglobulins: A Phase I/II Dose-Finding Study
Clinical Pharmacokinetics (2024)
-
Unlocking the Power of Complement-Dependent Cytotoxicity: Engineering Strategies for the Development of Potent Therapeutic Antibodies for Cancer Treatments
BioDrugs (2023)
-
Expression of mammalian proteins for diagnostics and therapeutics: a review
Molecular Biology Reports (2022)
-
Therapeutic Monoclonal Antibody Therapies in Chronic Autoimmune Demyelinating Neuropathies
Neurotherapeutics (2022)
-
A critical overview of current progress for COVID-19: development of vaccines, antiviral drugs, and therapeutic antibodies
Journal of Biomedical Science (2022)
