
Abstract
Spinocerebellar ataxia type 31 (SCA31) is one of the most common forms of autosomal-dominant cerebellar ataxia in Japan. SCA31 has a strong founder effect, which is consistent with the fact that this disease is basically absent in other ethnicities. After searching the entire founder region of a 2-megabase (Mb), we finally identified a 2.5 to 3.8 kb-long complex penta-nucleotide repeat containing (TGGAA)n, (TAGAA)n, (TAAAA)n and (TAAAATAGAA)n as the only genetic change segregating SCA31 individuals from normal people. Furthermore, (TGGAA)n was isolated as the only repeat explaining the pathogenesis because other repeats were encountered in control Japanese. From the genomic point of view, the complex penta-nucleotide repeat lies in an intronic segment shared by two genes, BEAN1 (brain expressed, associated with Nedd4) and TK2 (thymidine kinase 2) transcribed in mutually opposite directions. While TK2 is ubiquitously expressed, BEAN1 is transcribed only in the brain. Thus, the complex repeat is bi-directionally transcribed exclusively in the brain, as two independent non-coding repeats. Furthermore, the complex repeat containing (UGGAA)n was found to form abnormal RNA structures, called RNA foci, in cerebellar Purkinje cell nuclei of SCA31 patients’ brains. Subsequent investigation by over-expressing (UGGAA)n in Drosophila revealed that the RNA containing (UGGAA)n exerts toxicity in a length- and expression level-dependent manner, whereas its toxicity could be dampened by (UGGAA)n-binding proteins, TDP-43, FUS and hnRNP A2/B1. It seems rational to formulate a treatment strategy through enhancing the role of RNA-binding proteins against (UGGAA)n-toxicity in SCA31.
Similar content being viewed by others

Identification of spinocerebellar ataxia type 31 (SCA31)
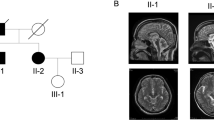
When we mapped the first group of families with pure cerebellar syndrome to human chromosome 19p, that later confirmed to be SCA6 [1], we found that there are a distinct group of families with SCA. This was the first description of SCA31 under the name of non-SCA6. Clinical features of SCA31 were summarized as, late-onset (average around 60 years old), slowly progressive ataxia. Extra-cerebellar features such as pyramidal tract signs, neuropathy or gaze limitations were not detected. In accord with this, magnetic resonance imaging showed cerebellar atrophy without brainstem involvement (Fig. 1).

T1-weighted magnetic resonance imaging of an SCA31 patient (female, 65 years old) sectioned in the sagittal pane (A: near midline, B: paramedian, C: lateral sections). At a near midline plane, the culmen shows prominent atrophy and the declive, folium, tuber vermis also appear atrophic (A). At the lateral planes (B, C), the cerebellar atrophy is obvious only the lobulus quadrangularis
We embarked a linkage analysis for these families, and three years later, we found their locus to a long arm of chromosome 16 [2]. Subsequent effort led us to finely map these families to chromosome 16q22.1 [3]. With a gradual accumulation of such families, we started to notice that affected individuals across different families share rare variants for a 2-megabase chromosomal region of 16q22.1. With a combination of these rare variants, we identified a haplotype that is not seen in control population. Therefore, SCA31 was considered to have a strong founder effect. For example, a single-nucleotide C-to-T change in the 5′ untranslated region of a gene, PLEKHG4 for a protein with a spectrin repeat and Rho guanine-nucleotide exchange-factor domain, also called puratrophin-1 [4], appeared to be segregated with the disease. However, subsequent identification of two affected subjects from different families without this single nucleotide exchange [5, 6], suggested that the change in PLEKHG4 was a polymorphism in a strong linkage disequilibrium (LD). The presence of strong founder effect made us to collect many families as much as we could, in order to search for a rare recombination within the locus. Thanks to many neurologists all over the country sending us their DNA samples. Continuing such efforts, we finally reached to seize a 900-kb critical region limited by two recombination events, first one as the C-to-T change in PLEKHG4 and the other end at rs11640843 [6]: all the affected individuals across different families had identical alleles within this 900-kb region, while alleles became discordant outside the 900-kb region. We thought that the cause of SCA31 must lie somewhere within this 900-kb.
Discovering SCA31 causative gene
In order to isolate the cause of SCA31, we had to check all the polymorphic markers that existed in the 900-kb critical region one by one. To do this, we took three different approaches: (1) Southern blot analysis for genetic rearrangement, (2) constructing BAC (bacterial artificial chromosome) tiling-path contig for the entire region followed by shot-gun sequencing for identifying smaller genetic changes, and (3) PCR-based Sanger sequencing of the entire 900-kb region. We embarked all the three approaches independently, and finally coincided with one genetic changes which all the affected members across different families harbored a 2.5 to 3.8 kb-long sequence not listed in public database [7]. Therefore, we thought this was an insertional mutation.
Cloning and sequencing of this aberrant sequence disclosed that this was a complex penta-nucleotide repeat containing (TGGAA)n, (TAGAA)n, (TAAAA)n and (TAAAATAGAA)n [7] (Table 1). In controls, a short, polymorphic (TAAAA)8–20 was seen. As we initially did not understand the implication of this change, we examined the relation between the length of this complex repeat and their age-of-onset for all available samples. Although the association was initially unclear, continuing this search by increasing new patients led us to find a weak inverse correlation between the two factors, supporting our discovery that the complex penta-nucleotide repeat containing (TGGAA)n, (TAGAA)n, (TAAAA)n and (TAAAATAGAA)n is indeed the mutation [7]. In addition, this insertion was not seen in a large set of control chromosomes: the vast majority (99.7%) of Japanese had a short TAAAA repeat of only 8 to 20 repeats. The only exception to this was noticed in three out of 1500 normal Japanese chromosomes: the three were, one chromosome with a long pure stretch of (TAAAA)n, and the other two chromosomes were complex repeats with (TAAAA)n, (TAGAA)n and (TAAAATAGAA)n. Thus, (TGGAA)n was never observed in controls. From these observations, we concluded that (TGGAA)n was the only repeat segregating with the phenotype, suggesting its importance in pathogenesis. The presence of TGGAA repeat in SCA31 patients was also confirmed independently by other researchers on different set of families [8].
The penta-nucleotide repeat responsible for SCA31 is transcribed in two mutually opposite directions
Public databases suggested that the complex penta-nucleotide repeat containing (TGGAA)n lay in a region between the two genes, BEAN1 (brain expressed, associated with Nedd4) and TK2 (thymidine kinase 2). As databases at that time suggested that two genes were transcribed in mutually opposite directions, and their genomic structure did not contain the complex penta-nucleotide repeat, we suspected that there could be additional exons downstream for both genes. In addition, it was suggested that BEAN1 drives brain-specific expression, while TK2 is expressed in all human tissues. We, therefore, underwent extensive 3′-RACE experiments for both genes on brain-extracted complimentary DNA (cDNA), and found that they indeed had multiple downstream exons that had not been deposited in databases. This implied that the 2.5 to 3.8 kb-long insertion is in an intronic region shared by the two genes, BEAN1 and TK2 [7]. Furthermore, it was assumed that the TGGAA repeat should be transcribed as UGGAA repeat and UUCCA repeat as independent RNA repeats in SCA31 brains.
The implication of founder effect in SCA31
As described, the SCA31 shows a strong founder effect. While SCA31 is a common ataxia in Japan, this disease has been reported from only a few countries such as Korea [9], Taiwan [10], and China [11, 12]. SCA31 was found in Brazilian SCA patients [13]. However, all these Brazilian patients were descendants of Japanese immigrants [13]. In addition, SCA31 in these four countries are extremely rare. In accord with this notion, SCA31 with (TGGAA)n was never found in the Caucasian SCA families (n = 320) in French and German cohorts nor in the 588 healthy control subjects [14]. From these observations, it is highly likely that SCA31 give rise from the founder chromosome where (TAAAA)n, (TAGAA)n, and (TAAAATAGAA)n are present.
Apart from this notion, we also found the fact that nearly 5.5% of the tested Caucasian cohorts harbored penta-nucleotide repeat expansions different from Japanese repeats. This raised an intriguing question why the SCA31 penta-nucleotide repeat locus is highly unstable depending on the ethnic backgrounds. The basic repeat is (TAAAA)n regardless of the ethnicity. In Caucasians, different repeats such as (TACAA)n, (GAAAA)n, (TGAAA)n, and (TAACA)n are frequently seen (Table 1). The most common (TACAA)n sometimes showed expansion up to 6.5 kb. Even in such individuals, no obvious clinical manifestations are seen. In addition, these Caucasian repeats were all pure repeats, while the SCA31 insertion consisted of three different repeats (TGGAA)n, (TAGAA)n, and (TAAAA)n. It seems possible that all the Caucasian repeats and the Japanese normal repeat (TAGAA)n, all arose through a single nucleotide transition. For example, (TACAA)n could arise from a A-to-C transition of the second A in the (TAAAA). If this view is correct, the (TGGAA)n could be regarded as the consequence of two transitions of A-to-G. If this is the case, some unknown genetic factor that predisposes to transitions may be present in SCA31 genome. Furthermore, such factors may also be present in other repeat expansion disorders.
Lessons learned from cultured cells and Drosophila model systems
As described, (TGGAA)n is expressed in both directions: TK2 gene drives (UUCCA)n in virtually ubiquitous tissues, while BEAN1 gene is expressed exclusively in the brain. As patients with SCA31 do not show any clear manifestation outside the brain [15, 16], we thought that BEAN1 gene expression fitted better than TK2 for explaining the pathogenesis. Therefore, we tested whether (UGGAA)n or (UAGAAUAAAA)n, the BEAN1-derived penta-nucleotide RNA repeats, in human cerebellum. In situ hybridization using a locked nucleic acid (LNA)-oligonucleotide (TTCCA)5 probe, Yusuke Niimi and our colleagues identified RNA foci within SCA31 Purkinje cells’ nuclei [17]. Similar RNA foci were also detected by probes against (UAGAAUAAAA)n21. We also tested whether (UGGAA)n is toxic in cultured cells by creating transient and stable expression cell systems. We found that cell toxicity and formation of RNA foci were both consistently observed upon expression of (UGGAA)n than by expressing (UAGAAUAAAA)n. These observations led us to conclude that (UGGAA)n could be toxic in cells.
Collaborating with Professor Yoshitaka Nagai, Taro Ishiguro and our colleagues created Drosophila models expressing an expanded (UGGAA)n repeat RNA [18]. We convinced that the expanded (UGGAA)n are toxic in Drosophila as well. In this model, the length of penta-nucleotide repeat had been contracted to 80–100 repeat (UGGAA)n compared to human SCA31 genome. Nevertheless, abundant RNA foci and remarkable eye degeneration were seen: some of the strongest lines were lethal, and some other lines with dramatic eye degenerations had a strong transgene expressions. On the other hand, lines with mild phenotypes had a low (UGGAA)n expression, in which the number of RNA foci was also small. The control repeat and a short UGGAA22 RNA, the latter happened to be generated by spontaneous contraction of the TGGAA repeats, had no significant effect.
Considering that one of the pathogeneses of noncoding repeat expansion disorders is mediated by RNA-binding proteins by binding to noncoding repeat responsible for human diseases [19], we thought that there could be (UGGAA)n binding proteins that may mediate some sort of disease mechanisms. Therefore, Sato N. and our colleagues screened for potential (UGGAA)n-binding proteins by in vitro RNA pull-down assay using nuclear fraction of PC12 cells, and found that TDP-43 (TAR DNA-binding protein, 43 kilodalton), FUS, and hnRNPs may bind to (UGGAA)n. After confirming that TDP-43 would bind to (UGGAA)n in western blot [18], we crossed (UGGAA)n expressing flies with a line that expresses human TDP-43, and found that the phenotype and RNA foci brought by (UGGAA)n were both alleviated. Similar effects were also seen for other (UGGAA)n binding proteins, FUS and hnRNPA2/B1. When the (UGGAA)n expressing flies were crossed with the fly, in which the endogenous protein homologous to TDP-43 was deleted, the (UGGAA)n toxicity was further enhanced. Further details of our findings could be seen in reference [18]. As these RNA binding proteins were thought to fix abnormal RNA structure of (UGGAA)n, we considered these proteins act as RNA chaperone.
From these and other related findings, we built a new idea that the RNA toxicity initiated by (UGGAA)n-toxicity and its counteracting effect by the (UGGAA)n-binding proteins as RNA chaperones are maintained in a well-balanced state when healthy, but once it tilts towards the RNA toxicity due to overexpression of (UGGAA)n, SCA31 emerges.
Conclusions
If our hypothesis is true for SCA31 human brains, overexpression of RNA binding proteins to an appropriate level may be rational, as we saw in Drosophila models.
However, the number of (UGGAA)n-containing RNA foci is low. Therefore, it seems that there are many other hidden factors behind SCA31 pathogenesis. Further studies are needed to discover mechanisms that (TGGAA)n in SCA31 patients’ genome leads to neurodegeneration.
References
Ishikawa K, Tanaka H, Saito M, Ohkoshi N, Fujita T, Yoshizawa K, et al. Japanese families with autosomal dominant pure cerebellar ataxia map to chromosome 19p13.1-p13.2 and are strongly associated with mild CAG expansions in the spinocerebellar ataxia type 6 gene in chromosome 19p13.1. Am J Hum Genet. 1997;61:336–46.
Nagaoka U, Takashima M, Ishikawa K, Yoshizawa K, Yoshizawa T, Ishikawa M, et al. A gene on SCA4 locus causes dominantly-inherited pure cerebellar ataxia. Neurology. 2000;54:1971–5.
Li M, Ishikawa K, Toru S, Tomimitsu H, Takashima M, Goto J, et al. Physical map and haplotype analysis of 16q-linked autosomal dominant cerebellar ataxia (ADCA) type III in Japan. J Hum Genet. 2003;48:111–8.
Ishikawa K, Toru S, Tsunemi T, Li M, Kobayashi K, Yokota T, et al. An autosomal dominant cerebellar ataxia linked to chromosome 16q22.1 is associated with a single-nucleotide substitution in the 5’ untranslated region of the gene encoding a protein with spectrin repeat and Rho guanine-nucleotide exchange-factor domain. Am J Hum Genet. 2005;77:280–96.
Ohata T, Yoshida K, Sakai H, Hamanoue H, Mizuguchi T, Shimizu Y, et al. A 16C>T substitution in the 5’UTR of the puratrophin-1 gene is prevalent in autosomal dominant cerebellar ataxia in Nagano. J Hum Genet. 2006;51:461–6.
Amino T, Ishikawa K, Toru S, Ishiguro T, Sato N, Tsunemi T, et al. Redefining the disease locus of 16q22.1-linked autosomal dominant cerebellar ataxia. J Hum Genet. 2007;52:643–9.
Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, et al. Spinocerebellar ataxia type 31 (SCA31) is associated with “inserted” pentanucleotide repeat including (TGGAA)n. Am J Hum Genet. 2009;68:355–67.
Sakai H, Yoshida K, Shimizu Y, Morita H, Ikeda S, Matsumoto N. Analysis of an insertion mutation in a cohort of 94 patients with spinocerebellar ataxia type 31 from Nagano, Japan. Neurogenetics. 2010;11:409–15.
Lee PH, Park HY, Jeong S-Y, Hong J-H, Kim HJ. 16q-linked autosomal dominant cerebellar ataxia in a Korean family. Eur J Neurol. 2007;14:e16–e17.
Lee YC, Liu CS, Lee TY, Lo YC, Lu YC, Soong BW. SCA31 is rare in the Chinese population on Taiwan. Neurobiol Aging. 2012;33:426.e23–4.
Ouyang Y, He Z, Li L, Qin X, Zhao Y, Yuan L. Spinocerebellar ataxia type 31 exists in Northeast China. J Neurol Sci. 2012;316:164–7.
Yang K, Zeng S, Liu Z, Shi S, Sun W, Yuan Y, et al. Analysis of spinocerebellar ataxia type 31 related mutations among patients from mainland China. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018;35:309–13.
Pedroso JL, Abrahao A, Ishikawa K, Raskin S, Sgobbi de Souza PV, Vieira de Rezende Pinto WB, et al. When should we test patients with familial ataxias for SCA31? A misdiagnosed condition outside Japan? J Neurol Sci. 2015;355:206–8.
Ishikawa K, Dürr A, Klopstock T, Müller S, De Toffol B, Vidailhet M, et al. Pentanucleotide repeats at the spinocerebellar ataxia type 31 (SCA31) locus in Caucasians. Neurology. 2011;77:1853–5.
Owada K, Ishikawa K, Toru S, Ishida G, Gomyoda M, Tao O, et al. A clinical, genetic, and pathologic study in a family with 16q-linked ADCA type III. Neurology. 2005;65:629–32.
Nakamura K, Yoshida K, Matsushima A, Shimizu Y, Sato S, Yahikozawa H, et al. Natural history of spinocerebellar Ataxia Type 31: a 4-year prospective study. Cerebellum. 2017;16:518–24.
Niimi Y, Takahashi M, Sugawara E, Umeda S, Obayashi M, Sato N, et al. Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology. 2013;33:600–11.
Ishiguro T, Sato N, Ueyama M, Fujikake N, Sellier C, Kanegami A, et al. Regulatory role of RNA Chaperone TDP-43 for RNA misfolding and repeat-associated translation in SCA31. Neuron. 2017;94:108–.e7.
Echeverria GV, Cooper TA. RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res. 2012;1462:100–11.
Acknowledgements
We thank all the researchers described in this review article. Particularly, I am in dept with Drs. T. Amino, N. Sato, T. Ishiguro, Y. Niimi, Professors Yoshitaka Nagai and Hidehiro Mizusawa for their devotion to this study. This manuscript was supported by from Grants-in-Aid from the Research Committee of Ataxia, Health Labour Sciences Research Grant, the Ministry of Health, Labour and Welfare, Japan (JPMH20FC1041), Grants-in-Aid for Scientific Research, the Japan Society for the Promotion of Science, Japan, the Grant from AMED, Japan (22ek0109577h0001) and Grants-in-Aid from the Research Committee of Ataxia, Health Labour Sciences Research Grant, the Ministry of Health, Labour and Welfare, Japan (JPMH20FC1041).
Author information
Authors and Affiliations
Contributions
This paper was written by Kinya Ishikawa.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ishikawa, K. Spinocerebellar ataxia type 31 (SCA31). J Hum Genet 68, 153–156 (2023). https://doi.org/10.1038/s10038-022-01091-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-022-01091-4
