
Abstract
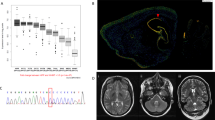
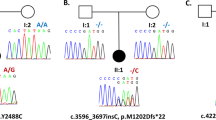
Paroxysmal kinesigenic dyskinesia (PKD) is a movement disorder characterized by episodic involuntary movement attacks triggered by sudden movements, acceleration, or intention to move. We ascertained two Japanese familial cases with PKD. The proband is a 22-year-old woman who had noted sudden brief (<30 s) of involuntary movements provoked by kinesigenic trigger such as starting to run, getting on a train, picking up a telephone receiver and so on at the age of 14. Interictal brain single photon emission computed tomography (SPECT) showed hyperperfusion in the left thalamus. A 46-year-old woman, the mother of the proband was also suffering from brief attacks triggered by starting to run in her high school days. On neurological examination, both showed no abnormality. Whole exome sequencing combined with rigorous filtering revealed two heterozygous nonsynonymous variants (NM_001447: c.8976G > C [p.Gln2992His] in FAT2 and NM_015678: c.8596C > T [p.Arg2866Trp] in NBEA). Real time quantitative PCR analysis of Nbea mRNA levels in the developing rat brain revealed peak at postnatal day 28 and decline at postnatal day 56. This result might match the most common clinical course of PKD from the point of view of the most common age at remission. NBEA has been reported to be responsible for neurodevelopmental disease accompanied by epilepsy. We concluded the variant in NBEA most likely to be responsible for our familial cases of PKD.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

Data availability
The NBEA variant data have been deposited to and openly available from ClinVar (Submission IDs: SUB7619122, https://www.ncbi.nlm.nih.gov/clinvar/variation/973380/). The FAT2 variant data have been deposited to and openly available from ClinVar (Submission IDs: SUB8292369, https://www.ncbi.nlm.nih.gov/clinvar/variation/992659/). The NUP98 variant data have been deposited to and openly available from ClinVar (Submission IDs: SUB8311015 https://www.ncbi.nlm.nih.gov/clinvar/variation/992658/). The data sets of this study are available from the corresponding author on reasonable request.
References
Silveira-Moriyama L, Kovac S, Kurian MA, Houlden H, Lees AJ, Walker MC, et al. Phenotypes, genotypes, and the management of paroxysmal movement disorders. Dev Med Child Neurol. 2018;60:559–65.
Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. 2004;63:2280–7.
Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. 2011;43:1252–5.
Wang HX, Li HF, Liu GL, Wen XD, Wu ZY. Mutation analysis of MR-1, SLC2A1, and CLCN1 in 28 PRRT2-negative paroxysmal kinesigenic dyskinesia patients. Chin Med J. 2016;129:1017–21.
Yin XM, Lin JH, Cao L, Zhang TM, Zeng S, Zhang KL, et al. Familial paroxysmal kinesigenic dyskinesia is associated with mutations in the KCNA1 gene. Hum Mol Genet. 2018;27:625–37.
Gardella E, Becker F, Møller RS, Schubert J, Lemke JR, Larsen LHG, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. 2016;79:428–36.
Méneret A, Grabli D, Depienne C, Gaudebout C, Picard F, Dürr A, et al. PRRT2 mutations: a major cause of paroxysmal kinesigenic dyskinesia in the European population. Neurology. 2012;79:170–4.
Becker F, Schubert J, Striano P, Anttonen AK, Liukkonen E, Gaily E, et al. PRRT2-related disorders: further PKD and ICCA cases and review of the literature. J Neurol. 2013;260:1234–44.
Lee YC, Lee MJ, Yu HY, Chen C, Hsu CH, Lin KP, et al. PRRT2 mutations in paroxysmal kinesigenic dyskinesia with infantile convulsions in a Taiwanese cohort. PLoS ONE. 2012;7:e38543.
Chen YP, Song W, Yang J, Zheng ZZ, Huang R, Chen K, et al. PRRT2 mutation screening in patients with paroxysmal kinesigenic dyskinesia from Southwest China. Eur J Neurol. 2014;21:174–6.
Pan G, Zhang L, Zhou S. Clinical features of patients with paroxysmal kinesigenic dyskinesia, mutations screening of PRRT2 and the effects of morning draughts of oxcarbazepine. BMC Pediatr. 2019;19:439.
Okumura A, Shimojima K, Kurahashi H, Numoto S, Shimada S, Ishii A, et al. PRRT2 mutations in Japanese patients with benign infantile epilepsy and paroxysmal kinesigenic dyskinesia. Seizure 2019;71:1–5.
Groffen AJA, Klapwijk T, van Rootselaar AF, Groen JL, Tijssen MAJ. Genetic and phenotypic heterogeneity in sporadic and familial forms of paroxysmal dyskinesia. J Neurol. 2013;260:93–9.
Bennett LB, Roach ES, Bowcock AM. A locus for paroxysmal kinesigenic dyskinesia maps to human chromosome 16. Neurology. 2000;54:125–30.
Mulhern MS, Stumpel C, Stong N, Brunner HG, Bier L, Lippa N, et al. NBEA: developmental disease gene with early generalized epilepsy phenotypes. Ann Neurol. 2018;84:788–95.
Wilson CB, McLaughlin LD, Nair A, Ebenezer PJ, Dange R, Francis J. Inflammation and oxidative stress are elevated in the brain, blood, and adrenal glands during the progression of post-traumatic stress disorder in a predator exposure animal model. PLoS ONE. 2013;8:e76146.
Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. 2015;138:3476–95.
Ko CH, Kong CK, Ngai WT, Ma KM. Ictal 99mTc ECD SPECT in paroxysmal kinesigenic choreoathetosis. Pediatr Neurol. 2001;24:225–7.
Joo EY, Hong SB, Tae WS, Kim JH, Han SJ, Seo DW, et al. Perfusion abnormality of the caudate nucleus in patients with paroxysmal kinesigenic choreoathetosis. Eur J Nucl Med Mol Imaging. 2005;32:1205–9.
Shirane S, Sasaki M, Kogure D, Matsuda H, Hashimoto T. Increased ictal perfusion of the thalamus in paroxysmal kinesigenic dyskinesia. J Neurol Neurosurg Psychiatry. 2001;71:408–10.
Zhou B, Chen Q, Gong Q, Tang H, Zhou D. The thalamic ultrastructual abnormalities in paroxysmal kinesigenic choreoathetosis: a diffusion tensor imaging study. J Neurol. 2010;257:405–9.
Nibbeling EAR, Duarri A, Verschuuren-Bemelmans CC, Fokkens MR, Karjalainen JM, Smeets CJLM, et al. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain. 2017;140:2860–78.
Nakayama M, Nakajima D, Nagase T, Nomura N, Seki N, Ohara O. Identification of high-molecular-weight proteins with multiple EGF-like motifs by motif-trap screening. Genomics. 1998;51:27–34.
Wang X, Herberg FW, Laue MM, Wüllner C, Hu B, Petrasch-Parwez E, et al. Neurobeachin: a protein kinase A-anchoring, beige/Chediak-Higashi protein homolog implicated in neuronal membrane traffic. J Neurosci. 2000;20:8551–65.
Zhou Y, Yuan Y, Liu Z, Zeng S, Chen Z, Shen L, et al. Genetic and clinical analyses of spinocerebellar ataxia type 8 in mainland China. J Neurol. 2019;266:2979–86.
Miller AC, Voelker LH, Shah AN, Moens CB. Neurobeachin is required postsynaptically for electrical and chemical synapse formation. Curr Biol. 2015;25:16–28.
Niesmann K, Breuer D, Brockhaus J, Born G, Wolff I, Reissner C, et al. Dendritic spine formation and synaptic function require neurobeachin. Nat Commun. 2011;2:557.
Medrihan L, Rohlmann A, Fairless R, Andrae J, Döring M, Missler M, et al. Neurobeachin, a protein implicated in membrane protein traffic and autism, is required for the formation and functioning of central synapses. J Physiol. 2009;587:5095–106.
Tuand K, Stijnen P, Volders K, Declercq J, Nuytens K, Meulemans S, et al. Nuclear localization of the autism candidate gene neurobeachin and functional interaction with the NOTCH1 intracellular domain indicate a role in regulating transcription. PLoS ONE. 2016;11:e0151954.
Gromova KV, Muhia M, Rothammer N, Gee CE, Thies E, Schaefer I, et al. Neurobeachin and the kinesin KIF21B are critical for endocytic recycling of NMDA receptors and regulate social behavior. Cell Rep. 2018;23:2705–17.
Smith M, Woodroffe A, Smith R, Holguin S, Martinez J, Filipek PA, et al. Molecular genetic delineation of a deletion of chromosome 13q12→q13 in a patient with autism and auditory processing deficits. Cytogenet Genome Res. 2002;98:233–9.
Castermans D, Wilquet V, Parthoens E, Huysmans C, Steyaert J, Swinnen L, et al. The neurobeachin gene is disrupted by a translocation in a patient with idiopathic autism. J Med Genet. 2003;40:352–6.
Nuytens K, Gantois I, Stijnen P, Iscru E, Laeremans A, Serneels L, et al. Haploinsufficiency of the autism candidate gene Neurobeachin induces autism-like behaviors and affects cellular and molecular processes of synaptic plasticity in mice. Neurobiol Dis. 2013;51:144–51.
Miura M, Ishiyama A, Nakagawa E, Sasaki M, Kurosawa K, Inoue K, et al. 13q13.3 microdeletion associated with apparently balanced translocation of 46,XX,t(7;13) suggests NBEA involvement. Brain Dev. 2020;42:581–86.
Méneret A, Gaudebout C, Riant F, Vidailhet M, Depienne C, Roze E. PRRT2 mutations and paroxysmal disorders. Eur J Neurol. 2013;20:872–8.
Acknowledgements
This work was supported by the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Miura, S., Shimojo, T., Morikawa, T. et al. Familial paroxysmal kinesigenic dyskinesia with a novel missense variant (Arg2866Trp) in NBEA. J Hum Genet 66, 805–811 (2021). https://doi.org/10.1038/s10038-021-00914-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-021-00914-0
