
Abstract
Genome-wide association studies have identified >10,000 genetic variants associated with various phenotypes and diseases. Although the majority are common variants, rare variants with >0.1% of minor allele frequency have been investigated by imputation and using disease-specific custom SNP arrays. Rare variants sequencing analysis mainly revealed have played unique roles in the genetics of complex diseases in humans due to their distinctive features, in contrast to common variants. Unique roles are hypothesis-free evidence for gene causality, a precise target of functional analysis for understanding disease mechanisms, a new favorable target for drug development, and a genetic marker with high disease risk for personalized medicine. As whole-genome sequencing continues to identify more rare variants, the roles associated with rare variants will also increase. However, a better estimation of the functional impact of rare variants across whole genome is needed to enhance their contribution to improvements in human health.
Similar content being viewed by others


Introduction
Over the last 15 years, genome-wide association studies (GWAS) have identified >10,000 (common in most cases) genetic variants associated with various diseases and phenotypes [1, 2]. Although the causal variants and genes to directly increase or decrease different disease risks and phenotypes remain unknown in most GWAS loci, positional (and functional) candidate genes in GWAS loci, as well as integrated analysis with other functional datasets, have elucidated novel biological pathways involved in a target disease. Polygenic risk scores per individual successfully identified high-risk individuals in a part of complex diseases [3]. In these GWAS, rare variants with lower minor allele frequency (MAF) (e.g., <1%) have been examined using a customized SNP array and imputation. Customized SNP arrays can be used to focus on the rare variants in the genes of interest for different types of diseases, namely Immunochip array for major autoimmune and inflammatory diseases [4], Metabochip array for metabolic, cardiovascular, and anthropometric traits [5], and iCOGS array for hormone-related cancers [6]. Imputation using reference panels has been used to infer the genotypes of rare variants not directly genotyped by SNP arrays [7]. However, it is not able to analyze all rare variants. Custom arrays have been used to focus on rare variants in genes of interest, which are previously identified in European populations [4]. Imputation has a limited accuracy, especially for rarer variants with <0.1% of MAF. This accuracy depends on the size of the reference panel and genetic background and is known to be lower in non-European populations [8].
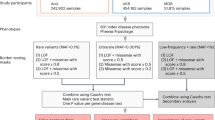
In order to analyze the contribution of rare variants to complex diseases, all rare variants will need to be identified by sequencing individuals. Next-generation sequencing (NGS) now allows for whole-genome sequencing (WGS) to be performed for under 1000 dollars [9] and WGS studies have reported on tens of thousands samples [10]. The importance of rare variants is increasing. The role of rare variants in the genetics of complex diseases in humans is not a simple extension of that of common variants—that is, simply for their association with diseases and phenotypes in WGS-based association studies. Rare variants have distinctive features, including lower linkage disequilibrium with flanking variants, a higher impact of some rare variants on gene function and expression, and a larger population specificity, with which rare variants play unique roles in the genetics of complex diseases. In this review, we discuss the roles played by rare variants in the genetics of complex diseases (Fig. 1), including especially inflammatory bowel diseases (IBD) consisting of Crohn’s disease and ulcerative colitis [11], and hereditary cancers the authors have contributed to.

Unique roles of rare variants in the genetic of complex diseases. Rare variants have unique roles which are different from common variants characterized by lower impact on gene function, and higher linkage disequilibrium with flanking variants. In this manuscript, four unique roles are discussed, and they contribute to different parts in the genetics of complex diseases in humans, which ultimately leads to the improvement of human health
It is worth noting that the threshold of MAF in rare variants has not yet been clearly defined. As shown in Table 1, this threshold varies between 0.1% and 5% in previous studies. The terms “low-frequency variants” and “less common variants” are used to indicate variants whose frequency lies between common variants and rare variants. However, since the unique roles of rare variants described in this review are slightly changed according to the threshold of MAF but generally applicable to any threshold of MAF, they will be defined where necessary.
Unique roles of rare variants
Hypothesis-free evidence for gene causality
The identification of causal genes responsible for disease onset is one of the goals of biological research. Various types of evidence are used to denote gene causality. Among them, hypothesis-free evidence is obtained by a hypothesis-free approach in which researchers do not start with a certain functional hypothesis [12]. GWAS is considered as a hypothesis-free approach because GWAS systematically analyzes SNPs across genome without a prior functional hypothesis. This type of evidence is considered robust because it does not depend on the accuracy of a prior functional hypothesis. Rare variants could provide hypothesis-free evidence for gene causality in complex diseases.
In 2001, two groups [13, 14] identified three common variants (p.Arg702Trp, p.Gly908Cys, and p.Leu1007ProfsTer2) in NOD2 that independently increased the risk of Crohn’s diseases [9]. In addition, the French group sequenced the coding regions of NOD2 in 457 patients with Crohn’s disease and 103 unaffected individuals to identify rare nonsynoymous variants. They found that patients had more rare nonsynonymous variants (17%) than unaffected individuals (5%). These results suggest that not only three common variants, but also rare variants, contribute to an increased risk of Crohn’s disease because linkage disequilibrium between common variants and rare variants is low. Since the identification of enrichment of rare variants is conducted without a prior functional hypothesis, rare variants could provide hypothesis-free evidence for gene causality independent of common variants.
This finding gained further attention around 2008, when GWAS became more widely reported. Several early GWAS in 2006 and 2007 [15,16,17,18] typically identified a few SNPs showing genome-wide significant association. Given a low-expected odds ratio (<1.2), large sample sizes were needed to improve the identification of SNPs with genome-wide associations. Three groups combined their GWAS data as a meta-analysis with adjustment for differences in SNP arrays by imputation [19] to identify as many as 32 loci (21 additional loci in this meta-analysis) associated with Crohn’s disease. Meta-analysis for other complex diseases also identified more loci around the same time, including 30 loci for polygenic dyslipidemia [20] and 10 loci for colorectal cancer [21]. Although meta-analysis dramatically increased the number of loci identified, the causal variants and genes remained largely unknown. To identify likely causal genes, various fine-mapping strategies were used, including expression quantitative trait locus (eQTL), functional annotation of genetic variants, PubMed text mining, protein–protein interactions, pathway prioritization protocol, and Bayesian methods to identify credible sets [22]. Among these, the use of Bayesian methods for the identification of credible sets [23] and the identification of rare variant enrichment follow the advantage of the hypothesis-free approach of GWAS.
Two groups from Belgium [24] and the USA [25] tried to identify rare variant enrichment in positional candidate genes (63 and 56 genes, respectively) in IBD using NGS. IL23R showed that controls had more rare variants than Crohn’s disease patients [24]. This enrichment was also observed in ulcerative colitis patients, consistent with GWAS results [18]. In addition, single rare coding variants in CARD9, IL18RAP, CUL2, C1orf106, PTPN22, and MUC19 showed additional associations [25]. As described in Table 1, the enrichment of rare variants was also investigated in other complex diseases. Whole exome and genome sequencing have also been used for IBD and other complex diseases. Luo et al. identified a novel rare missense variant in ADCY7 associated with ulcerative colitis [26]. In addition, IBD genes implicated by causal coding or eQTL variants showed the enrichment of rare variants in Crohn’s disease [26]. In the era of WGS, the enrichment of rare variants has been widely investigated (Table 1).
A statistical method about rare variants was also evolved. A higher statistical power was obtained in the above analysis method if all rare nonsynonymous variants were functional with the same magnitude and direction of impact on gene function. However, nonsynoymous variants can be nonfunctional, while others may have opposite effects in terms of function. Researchers also wanted to include various covariates such as sex and age into statistics analysis. As results, various statistical analyses have been proposed to maintain or increase statistical power in these different scenarios, which were grouped into four categories: burden tests, variance-component tests, combined tests, and other tests [27]. Details were reviewed by Povysil [28].
Statistical analysis is conducted in functional units because variants in the same functional unit are expected to show similar functions for the same target. In most cases, the coding regions of one gene have been used as a functional unit, since a target is the same and the annotation of variants, including synonymous, nonsynonymous, and loss-of-function variants, could be reliably determined. Multiple genes have also be investigated in the form of functional units, including prior knowledge associated gene sets [29, 30], sets of candidate genes selected by eQTL [26, 31], and known pathways [32], although some are not hypothesis-free approaches. Gene ontology is also used to characterize genes with variants [33]. Other researchers have focused on specific regions within a single gene. Specific known domains [32] and regions with different missense tolerance ratios [34] are used to identify functionally important parts. In this way, the role of hypothesis-free evidence for gene causality is being evolved.
Precise target of functional analysis for understanding disease mechanisms
GWAS and other hypothesis-free genetic analysis methods are expected to identify unknown mechanisms that cannot be identified using knowledge-based research. The identification of causal variants is plausible for further functional analysis. However, this is difficult, especially for common variants, since they are generally found in high linkage disequilibrium with flanking variants. In addition, it is common to have multiple causal variants, including rare variants in one GWAS locus. Therefore, it is challenging to identify a causal variant according to the association pattern. For example, the IRGM locus was previously identified in a GWAS on Crohn’s disease, wherein a 20-kb common deletion 2 kbp upstream of IRGM was considered to be a likely causal variant due to the fact that it showed perfect linkage disequilibrium with the highest signal and an eQTL effect on IRGM [35]. Three years later, a common synonymous variant (c.313C>T) was also considered as a potential causal variant. This variant also showed perfect linkage disequilibrium with the highest signal, but had been disregarded due to lacking an amino acid change. Brest et al. [36] found that microRNA (miR-196) was overexpressed in the inflammatory intestinal epithelia of patients with Crohn’s disease. c.313C>T was located within the seed region of miR-196 with different IRGM expression under the control of miR196.
Similar difficulty was observed in the FTO locus of individuals with obesity. While a top variant identified by GWAS was assumed to regulate FTO by a series of functional tests in 2009–2010 [37, 38], in 2014 it was found to interact with the promoters of IRX3 located several hundred kilobases away and obesity-associated variants were associated with IRX3 expression [39]. Indeed, Irx3-deficient mice showed a reduction in body weight of 25–30% primarily through the loss of fat mass and increase in basal metabolic rate with browning of white adipose tissue. Functional analyses are indispensable for the identification of the mechanisms of variants in disease onset. However, it remains difficult for researchers to validate mechanisms revealed by functional analysis since they are unable to test all possible mechanisms.
A potential strategy to decrease the possibility of this type of misinterpretation is to focus on loss-of-function (LoF) variants, such as nonsense, frame-shift, and splice-site variants, since the ambiguity about the direction and the magnitude of impact on gene function is limited. In particular, rare LoF variants showing significant association with diseases are ideal. CARD9 in IBD is one such example. While a common GWAS signal in this locus is considered to change the expression level of CARD9 [31], IVS11+1C>G presumably skipping exon 11 was also identified using target sequencing in candidate genes. The frequencies in cases and controls were 0.20% and 0.64%, respectively (P < 1×10−16; odds ratio = 0.29) [25]. A subsequent study with this rare LoF variant showed that ubiquitin ligase TRIM62 regulates CARD9-medicated anti-fungal immunity and intestinal inflammation [40]. Other examples about functional analysis with rare variants associated with diseases are provided in Table 2.
If researchers were to directly analyze the participants, it would be possible to deeply understand biological mechanisms behind genetic associations. Generally, studies have identified variants associated with phenotypes in previously collected individuals. When participants are recruited to a study, the volume of phenotype analysis is limited due to financial and practical constraints for the optimal number of participants that ensures the best statistical power. However, when a specific potentially causal variant is being studied, recruitment is limited to participants with the genotypes of interest to allow researchers for deep phenotyping, which is called Recall-by-Genotype [41].
One example of the Recall-by-Genotype is TYK2. TYK2 is differentially associated with common autoimmune diseases, including Crohn’s disease, ulcerative colitis, ankylosing spondylitis, multiple sclerosis, and psoriasis [42]. Dendrou et al. tried to resolve genotype-to-phenotype differences in autoimmunity in this locus with a potentially rare causal variant. They performed fine-mapping to identify rs34536443 (p.Pro1104Ala). Among their analyses, they recalled individuals from the Oxford BioBank for blood donation in a balanced, age- and sex-matched fashion based on preselected heterozygotes and homozygotes of a minor allele. In their study, peripheral blood mononuclear cells were separated into CD3+ T cells, CD4+ T cells, CD8+ T cells, CD19+ B cells, and CD14+ monocytes, followed by cytokine stimulations of peripheral blood mononuclear cells, which are unlikely to be collected without Recall-by-Genotype. The authors found that a potentially causal variant (rs34536443) had a demonstrable impact on TYK2 function, leading to impaired type I IFN, IL-12, and IL-23 signaling. This indicates that studies analyzing disease mechanisms have a greater chance of elucidating the causal mechanism of disease if they study a specific causal variant in subjects with genotypes of interest.
New favorable target for drug development
Accumulating information on the associations between genes and diseases is also important for drug development. Drug candidates with genetically supported targets are more likely to be successful in Phase II and III clinical trials [43]. Genetic associations can also be used to identify new favorable drug targets. In particular, rare variants that worked protectively against disease have been used to identify new drug targets because their allele mimics the effect of modulating drug target genes [44, 45]. A notable example is PCSK9. In black individuals, there are two rare nonsense variants (p.Tyr142Ter and p.Cys679Ter) in PCSK9, with 2.6% of individuals having at least one nonsense variant [46]. In 2006, a study on a cohort of black individuals found that nonsense mutations in PCSK9 resulted in a reduction in the mean LDL cholesterol and the risk of coronary heart disease of 28% and 88%, respectively [46]. In 2017, a monoclonal antibody inhibiting PCSK9, evolocumab, reduced the LDL cholesterol levels in individuals receiving statin therapy and the risk of cardiovascular events in a randomized, double-blind, placebo-controlled trial [47]. This drug was subsequently approved and launched.
Along with the development of genetic analysis techniques, methods used for the identification of rare variants that provide protection against disease have evolved. Currently, researchers use biobanks to target 18,228 LoF and 135 phenotypes at one time to systematically identify 27 associations, showing the value of collecting population-scale genomic data [48]. Another idea involves focusing on consanguineous unions, as they are more likely to result in offspring carrying homozygous LoF. In the Pakistan Risk of Myocardial Infarction Study, researchers sequenced the protein-coding regions in 10,503 adult participants to identify 49,138 rare LoF variants. They systematically identified their impact on >200 biochemical and disease traits, especially in homozygotes of LoF [49]. Another group sequenced the exomes of 3222 British adults of Pakistani heritage and identified 1111 rare-variant homozygous genotypes with LoF in 781 genes, although no significant relationship between gene knockouts and clinical consultation or prescription rate was found [50]. While such large-scale screening is promising for identifying new targets for drug development, more traditional strategies, including series of studies performing gene identification, functional analysis, and mechanisms identification, have also resulted in drug development, such as IL23R for Risankizumab [51] (Table 3).
Genetic marker with high disease risk for personalized medicine
Most odds ratios of common variants identified by GWAS are <1.2 in complex diseases. Therefore, most single variants associated with complex disease are not clinically useful, but some complex diseases are caused by a single pathogenic variants in some genes, including BRCA1 and BRCA2 in hereditary breast cancer and ovarian cancer syndrome [52], MLH1, MSH2, MSH6, and PMS2 in Lynch syndrome [53], and LDLR and PCSK9 in atherosclerotic cardiovascular disease [54]. In an individual with a pathogenic variant of BRCA1, the cumulative risk for breast cancer and ovarian cancer at 80 years of age is 72% and 44%, respectively [55]. These individuals are expected to have a longer median progression-free survival and a lower risk of disease progression or death by treatment with a PARP inhibitor [56]. Interventions such as risk-reducing bilateral mastectomy, salpingo-oophorectomy, and breast magnetic resonance imaging for early detection could be applied to carriers of pathogenic variants [57]. The same benefits could be obtained by their relatives. Therefore, rare variants have great potential for use as biomarkers in personalized medicine.
This seems a typical example of well-established personalized medicine. However, rare variants have many challenges in order for them to work well. In addition, there are also more potentials in this field. The greatest challenge in this field is the annotation of variants. Recently, a multiple gene panel for the analysis of several genes in one genetic test was used in-clinic to identify genetic variants in a patient. For breast cancer screening, 11 genes, including BRCA1 and BRCA2, are recommended for genetic testing in the National Comprehensive Cancer Network guidelines [57]. Typically, ~10 genetic variants are identified on average in one individual [58] but a subject wants to know whether she has a pathogenic variant that increases risk among the variants detected; this is not easy. If the variant is a LoF variant, in most cases it would be a pathogenic variant. However, this is not always true. The insertion of 4-bp frameshift (p.Lys1358fs, rs55740729) in MSH6 is known to be a benign variant in Lynch syndrome. Indeed, this variant was equally observed in colorectal cancer patients (2.13%) and controls (2.09%) in a Japanese cohort [59]. In contrast, synonymous variants are likely benign because they do not undergo changes in their amino acid sequences. However, p.Gln1395Gln in BRCA1 is registered as pathogenic in ClinVar [60] since it alters splicing [61]. Nonsynonymous variants are less straightforward because it is difficult to estimate the direction and magnitude of the impact of each nonsynonymous variant on gene function. To resolve this, guidelines have been developed that allow determining the clinical interpretation of variants. The guidelines by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology have 16 criteria for pathogenicity and 12 criteria for benign. These criteria have to be checked for each variant to determine the appropriate clinical interpretation [57]. Despite this guideline providing a consensus in the community of genetic research, inconsistencies in clinical interpretation have been found between different laboratories [62], and recommendations for the modification of the criteria regarding LoF variants have been discussed [63].
Another difficulty is the differences in pathogenic variants between populations. Most pathogenic variants are singleton variants. For example, 75.8% of 244 pathogenic variants in 11 genes found in breast cancer in Japanese individuals were singleton [58] but there are founder mutations shared in patients. Since founder mutations are specific to racial, ethnic, or geographic groups [64], they change the importance of genes in such groups. For example, three founder mutations in BRCA1/2 have a combined prevalence of as high as 2–3% in American Ashkenazi Jews [65], while this prevalence is estimated to be 0.1–0.5% in other populations based on Exome Aggregation Consortium data [66]. Other genes were also influenced by population-specific rare variants. In European populations, c.1100delC in CHEK2, one of the 11 genes recommended for genetic testing, is common to breast cancer patients in the UK (1.2–1.3%), Netherlands (2.5–3.8%), Finland (2.1–2.9%), Germany (0.33–1.1%), and Australia (0.68%) [67]. On the other hand, this founder mutation has not been observed in Japanese individuals, resulting in the number of patients with pathogenic variants in CHEK2 in European populations (1.12% of breast cancer patients) to be threefold higher than in the Japanese population (0.38%) [58]. As another example, NBN was recommended for genetic testing as a founder mutation (c.657del5, p.Lys219Asnfs, rs587776650) in NBN associated with breast cancer [68] and prostate cancer [69] in European populations. However, this variant was not found in Japanese breast or prostate cancer patients, and there were no associations between other pathogenic variants in NBN and both cancers in Japanese cohorts [58, 70]. Therefore, NBN does not need to be included in Japanese cohorts. The inverse may also be true: Japanese-specific founder mutations in one unknown gene may contribute to increased risk of certain cancers in Japanese. However, there are limited efforts to identify such variants [71, 72].
Despite the various obstacles, the use of variants as biomarkers in personalized medicine has more great potential. Although most research focuses on specific genes and cancers, including BRCA1/2 for breast and ovarian cancer and MLH1, MSH2, and MSH6 for colorectal cancer, more recently, other genes and diseases have been investigated due to the expansion of multi-gene panels. For instance, in as late as 2018, 5.5% of pancreatic cancer patients were found to have germline pathogenic variants in BRCA1/2, as well as ATM, CDKN2A, TP53, and MLH1 (odds ratio = 2.6–12.3) [73]. These proportions and odds ratio are comparable to those of breast cancer. Among metastatic pancreatic cancer patients with germline pathogenic variants in BRCA1/2, progression-free survival was reported longer after treatment with a PARP inhibitor in 2019 [74]. Therefore, since other cancers and genes would have such possibilities for personalize medicine with rare variants, further investigations are needed.
Population screening for large-scale precision prevention is another potential approach [75]. Although the ethical, social, and legal implications should be carefully considered, several population screening studies have already been conducted mainly in specific high-risk populations, including Ashkenazi Jews. Even a clinical trial of genomic screening of newborn babies is being running [76]. How these new techniques are introduced into society will require careful consideration. Data accumulation is indispensable for this consideration.
Future perspectives
As described above, rare variants play unique roles in the genetics of complex diseases. It is worth noting that the four roles described above are not mutually exclusive. Rare variants in BRCA1/2 played all roles, although the third role was different. A deeper understanding of the function of BRCA1/2 has led to the development of a new concept, denoted as synthetic lethality [77]. Since NGS will continue to be used to identify new rare variants, these unique roles are likely to become more important. However, several barriers remain to be overcome.
Sample size
Rare variants require much larger sample sizes than common variants to obtain a sufficiently high statistical power. For instance, when the effect size of a variant is 0.1 (corresponding to an odds ratio of ~1.2) phenotyping standard deviation units, a common variant with MAF = 10% needs ~10,000 individuals to obtain genome-wide significance at P = 5 × 10−8 with 80% statistical power. MAF of variant = 1% and 0.1% requires ~100,000 and 1 million individuals, respectively. Despite improvement ideas in sample collection [78], genome-sequencing methods [79], and data analysis [80], achieving such large volumes remains a challenge. To overcome this, various methods are used, including statistical analysis [28], imputation [7], target sequencing, and the use of other populations.
As described above, several methods to determine the effects of rare variants in different scenarios have been developed to increase statistical power. However, more care is needed in rare variants than common variants. Association analysis with rare variants is influenced by various factors, including the geographical area of the samples, the timing of sampling, sequencing coverage, and the selection of qualifying variants [28]. Among these, the timing of DNA sampling requires further explanation. One reason is due to age-associated somatic variants in myeloid cancer-associated genes, such as DNMT3A, TET2, ASXL1, and TP53 in DNA extracted from blood [81]. The prevalence of somatic variants increases with age, from 0% in individuals in their 20 s to 29.4% in individuals over 100 years old. Therefore, somatic TP53 variants frequently confound genetic testing results, although they are intended to analyze germline variants [82]. Another example is treatment-induced somatic variants. Chemotherapy-induced somatic variants in PPM1D, which caused pseudo-associations between breast and ovarian cancers, and variants in PPM1D because patients received chemotherapy to induce somatic variants in PPM1D [83]. Therefore, the statistical analysis of rare variants requires more care from sampling.
Imputation has evolved by increasing the number of samples and variants, as well as including various populations. Recent public reference panels include UK10K projects (3781 samples, 42.0 million variants, European), Haplotype Reference Consortium (32,470 samples, 40.4 million variants, predominantly European), and Trans-Omics for Precision Medicine (60,039 samples, 239.7 million variants, multiethnic) [84]. An important resource is a website used to perform imputation [8]. Imputation is a computer-intensive task, and not all researchers have access to the servers needed for imputation. Thanks to this website, the number of variants identified from SNP arrays can be increased to enable the analysis of rare variants by imputation.
Target sequencing is used to sequence specific regions of interest based on prior knowledge. The target sequencing of functionally candidate genes was frequently conducted in the 1990s and 2000s before GWAS, although it has been criticized for its low replication rate [85, 86]. However, when focusing on positional candidate genes located in GWAS loci, the rate improves, most likely due to the selection of genes by GWAS increasing the possibility that a target gene is causal for disease. Indeed, the target sequencing of candidate genes identified by GWAS has led to the identification of rare variants associated with diseases with reasonable p values (Table 1). Target sequencing could be used to analyze rare variants in a much greater number of samples than WGS and whole-exome sequencing, and thereby reveal the contribution of rare variants with better statistical power.
Another possibility is to use other populations. Even if a certain variant in one gene needs a huge number of individuals to obtain significant associations between variants in one gene and diseases in one population, another population might have a higher frequency of such a variant or more frequent variants with similar functional impact in the same gene to have better statistical power. A typical example is the association between a LoF variant (p.Arg684Ter) in TBC1D4 and type 2 diabetes in Greenland [87]. The allele frequency of this variant was 17% in this population, but 0.003% in other European populations. Therefore, this variant was not identified to be associated with type 2 diabetes and related phenotypes, most probably because p.Arg684Ter was neither genotyped nor imputed in previous GWAS using European populations. For the same disease, GWAS in Japanese populations identified GLP1R, which was previously missed due to lower frequency of variants in European populations [88]. Using the same concept, GWAS with multi-ethnic and admixed populations was conducted to show substantial benefits for fine-mapping and insight on clinical implications [89]. In addition, population specific custom SNP arrays have been used, including Infinium Asian Screening Array, Axiom Japonica Array, and Infinium Multi-Ethnic AMR/AFR. They include population-specific rare variants. These efforts will also compensate for inequitable access to precision medicine in minority populations with a disproportionately higher burden of chronic conditions.
Estimation of the functional impact of a rare variant
In all four roles of rare variants, it is always important to select functional causal variants. If nonfunctional variants are included, statistical power decreases and functional analysis may be in the wrong direction. Even LoF requires filtering and manual curation for removing artefacts not to dilute association signals [90, 91]. For nonsynonymous variants, several in silico programs, such as SIFT, PolyPhen, Condel, and CADD, provide an estimation of the impact of each variant. However, the resulting estimation of each variant may not be sufficiently accurate [92]. Functional assays have been developed to estimate the functional impact of rare variants. Nonsynonymous variants in BRCA1/2 have been frequently assayed, focusing on the homology-directed DNA repair function, embryonic stem cell viability, transcriptional activation, drug-sensitivity, protein–protein interaction, and splicing [93], as their functional impact is directly linked to changes in clinical management. However, these efforts have not been successful in decreasing variants with unknown functional impacts due to the fact that functional assays focus on specific functions in BRCA1/2, which do not mimic full function in vivo [93]. However, the accurate classification of variants in BRCA1 using saturation genome editing has been recently reported [93]. In this study, the functional impact of ~4000 variants was assessed, and was almost perfectly concordant with the established clinical interpretation of pathogenicity in ClinVar. A similar strategy could be applied to other genes, although the experimental condition needs optimization according to the genes being studied. In addition, while the clinical interpretation of a large number of variants in BRCA1/2 has been already submitted to ClinVar [60] and could be used as “ground truth positive” to optimize experiments, a limited number of variants in other genes have been deposited. Therefore, the sharing of clinical interpretation data on variants in other genes is indispensable for the development of high-throughput functional assays in other genes.
Beyond coding regions
The unique roles of rare variants have been mainly played in coding regions. However, there is evidence that rare variants in non-coding regions have a large impact on gene expression and disease. A large deletion at the 3′-end of EPCAM is known to cause allele-specific epigenetic silencing of the neighboring DNA mismatch repair gene MSH2, leading to Lynch syndrome [94]. Hernandez et al. reported that singletons contribute to ~25% of cis eQTL heritability across genes [95]. A variant in the 5′ untranslated region that is known to result in the methylation-associated silencing of BRCA1 is dominantly inherited in some families affected by breast and ovarian cancer [96]. The association between genetic variants and multi-omics data, including transcriptome, post-transcriptional regulation, epigenome, protein post-translation modification, metabolome, and microbiome data, has helped to improve our understanding of rare variants in non-coding regions [97]. However, estimating the impact of rare variants on target genes at the single-base resolution remains a challenge. Nevertheless, a new model with a novel experimental approach, CRISPRi-FlowFISH, has been proposed for interpreting the functions of variants in non-coding regions [98]. In addition, various in silico prediction tools for non-coding regions are being developed, including regBase [99], RegSNPs-intron [100], and GRAM [101]. Overall, a better understanding of variants in coding and non-coding regions and single variant annotation across whole genome would take advantage of population-based sequencing data to provide great benefits to human health.
Integration of genetic and non-genetic information
One of the goals in the characterization of variants is to provide diagnosis and forecast of future disease risk. In this context, researchers will have to consider all genetic variants across the entire genome, including structural variants such as copy number variations, insertion, inversions, and translocations [102]. The functional impact of each variant discussed above should also be included in the calculation. In addition, electronic health record including digital image, data from health monitoring device, and other environmental exposure might be considered alongside. Artificial intelligence is expected to deal with all the information [103]. However, there are various challenges and limitations, including regulatory issues, interpretability, and data and machine bias. Therefore, large-scale training and validation datasets about genomics, electronic health record and other information are needed for artificial intelligence to integrate genetic and non-genetic information to provide diagnosis and forecast of future disease risk.
Conclusion
Rare variants play unique roles in the genetics of complex diseases in humans, including as hypothesis-free evidence of gene causality, a precise target of functional analysis for understanding disease mechanisms, a new target for drug development, and a genetic marker with high disease risk for personalized medicine (Fig. 1). Advances in WGS will continue to allow for the identification of rare variants, where a better estimation of the functional impact of each rare variant across the whole genome will provide paramount benefits to human health.
References
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20:467–84.
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi S, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–24.
Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther. 2011;13:101.
Voight BF, Kang H, Ding J, Palmer CD, Sidore C, Chines PS, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793.
Bahcall O. COGS project and design of the iCOGS array. Nat Genet. 2013. https://doi.org/10.1038/ngicogs.4.
Das S, Abecasis GR, Browning BL. Genotype imputation from large reference panels. Annu Rev Genom Hum Genet. 2018;19:73–96.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, et al. A brief history of human disease genetics. Nature. 2020;577:179–89.
Kowalski MH, Qian H, Hou Z, Rosen JD, Tapia AL, Shan Y, et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15:e1008500.
Venkataraman GR, Rivas MA. Rare and common variant discovery in complex disease: the IBD case study. Hum Mol Genet. 2019;28:R162–R69.
Hunter DJ, Altshuler D, Rader DJ. From Darwin’s finches to canaries in the coal mine—mining the genome for new biology. N Engl J Med. 2008;358:2760–63.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6.
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604.
Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58.
Consortium TWTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78.
Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62.
Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2008;41:56–65.
Study C, Houlston RS, Webb E, Broderick P, Pittman AM, Bernardo M, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008;40:1426–35.
Spain SL, Barrett JC. Strategies for fine-mapping complex traits. Hum Mol Genet. 2015;24:R111–9.
Huang H, Fang M, Jostins L, Umicevic Mirkov M, Boucher G, Anderson CA, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature. 2017;547:173–78.
Momozawa Y, Mni M, Nakamura K, Coppieters W, Almer S, Amininejad L, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet. 2011;43:43–7.
Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–73.
Luo Y, de Lange KM, Jostins L, Moutsianas L, Randall J, Kennedy NA, et al. Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nat Genet. 2017;49:186–92.
Bomba L, Walter K, Soranzo N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017;18:77.
Povysil G, Petrovski S, Hostyk J, Aggarwal V, Allen AS, Goldstein DB. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet. 2019;20:1–13.
Singh T, Walters JTR, Johnstone M, Curtis D, Suvisaari J, Torniainen M, et al. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat Genet. 2017;49:1167–73.
Allen AS, Bellows ST, Berkovic SF, Bridgers J, Burgess R, Cavalleri G, et al. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 2017;16:135–43.
Momozawa Y, Dmitrieva J, Theatre E, Deffontaine V, Rahmouni S, Charloteaux B, et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat Commun. 2018;9:2427.
Motegi T, Kochi Y, Matsuda K, Kubo M, Yamamoto K, Momozawa Y. Identification of rare coding variants in TYK2 protective for rheumatoid arthritis in the Japanese population and their effects on cytokine signalling. Ann Rheum Dis. 2019;78:1062–69.
Toma C, Torrico B, Hervás A, Valdés-Mas R, Tristán-Noguero A, Padillo V, et al. Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol Psychiatry. 2013;19:784–90.
Gelfman S, Dugger S, de Moreno C, Ren Z, Wolock CJ, Shneider NA, et al. A new approach for rare variation collapsing on functional protein domains implicates specific genic regions in ALS. Genome Res. 2019;29:809–18.
McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–12.
Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–45.
Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Brüning JC, et al. Inactivation of the Fto gene protects from obesity. Nature 2009;458:894–98.
Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42:1086–92.
Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014;507:371–75.
Cao Z, Conway KL, Heath RJ, Rush JS, Leshchiner ES, Ramirez-Ortiz ZG, et al. Ubiquitin Ligase TRIM62 regulates CARD9-mediated anti-fungal immunity and intestinal inflammation. Immunity. 2015;43:715–26.
Corbin LJ, Tan VY, Hughes DA, Wade KH, Paul DS, Tansey KE, et al. Formalising recall by genotype as an efficient approach to detailed phenotyping and causal inference. Nat Commun. 2018;9:711.
Dendrou CA, Cortes A, Shipman L, Evans HG, Attfield KE, Jostins L, et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci Transl Med. 2016;8:363ra149.
King EA, Davis WJ, Degner JF. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019;15:e1008489.
Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–60.
Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12:581–94.
Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–22.
DeBoever C, Tanigawa Y, Lindholm ME, McInnes G, Lavertu A, Ingelsson E, et al. Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study. Nat Commun. 2018;9:1612.
Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–39.
Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352:474–77.
Feagan BG, Sandborn WJ, D’Haens G, Panés J, Kaser A, Ferrante M, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2017;389:1699–709.
Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372:2243–57.
Cohen SA, Pritchard CC, Jarvik GP. Lynch syndrome: from screening to diagnosis to treatment in the era of modern molecular oncology. Annu Rev Genom Hum Genet. 2019;20:1–15.
Trinder M, Francis GA, Brunham LR. Association of monogenic vs polygenic hypercholesterolemia with risk of atherosclerotic cardiovascular disease. JAMA Cardiol. 2020;5:390–99.
Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom M-J, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317:2402–16.
Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–33.
Daly MB, Pilarski R, Yurgelun MB, Berry MP, Buys SS, Dickson P, et al. NCCN guidelines insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 1.2020. J Natl Compr Canccer Netw. 2020;18:380–91.
Momozawa Y, Iwasaki Y, Parsons MT, Kamatani Y, Takahashi A, Tamura C, et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat Commun. 2018;9:4083.
Fujita M, Liu X, Iwasaki Y, Terao C, Takata S, Inai C, et al. Population-wide screening for germline variants of hereditary cancer genes in 12K unselected Japanese colorectal cancers and 27K controls. https://www.biorxiv.org/content/10.1101/2020.03.15.989947v1.full.pdf. 2020.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8.
Wappenschmidt B, Becker AA, Hauke J, Weber U, Engert S, Kohler J, et al. Analysis of 30 putative BRCA1 splicing mutations in hereditary breast and ovarian cancer families identifies exonic splice site mutations that escape in silico prediction. PLoS ONE. 2012;7:e50800.
Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98:1067–76.
Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517–24.
Rebbeck TR, Friebel TM, Friedman E, Hamann U, Huo D, Kwong A, et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum Mutat. 2018;39:593–620.
Roa BB, Boyd AA, Volcik K, Richards SC. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet. 1996;14:185–87.
Maxwell KN, Domchek SM, Nathanson KL, Robson ME. Population frequency of germline BRCA1/2 mutations. J Clin Oncol. 2016;34:4183–85.
Consortium C. CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet. 2004;74:1175–82.
Zhang G, Zeng Y, Liu Z, Wei W. Significant association between Nijmegen breakage syndrome 1 657del5 polymorphism and breast cancer risk. Tumour Biol. 2013;34:2753–57.
Cybulski C, Wokołorczyk D, Kluźniak W, Jakubowska A, Górski B, Gronwald J, et al. An inherited NBN mutation is associated with poor prognosis prostate cancer. Br J Cancer. 2012;108:461–8.
Momozawa Y, Iwasaki Y, Hirata M, Liu X, Kamatani Y, Takahashi A, et al. Germline pathogenic variants in 7,636 Japanese patients with prostate cancer and 12,366 controls. J Natl Cancer Inst. 2020;112:369–76.
Liu X, Takata S, Ashikawa K, Aoi T, Kosugi S, Terao C, et al. Prevalence and spectrum of pathogenic germline variants in Japanese Patients with early-onset colorectal, breast, and prostate cancer. JCO Precis Oncol. 2020;4:183–91.
Hayano T, Matsui H, Nakaoka H, Ohtake N, Hosomichi K, Suzuki K, et al. Germline variants of prostate cancer in Japanese families. PLOS ONE. 2016;11:e0164233.
Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018;319:2401–09.
Golan T, Hammel P, Reni M, Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–27.
Manchanda R, Lieberman S, Gaba F, Lahad A, Levy-Lahad E. Population screening for inherited predisposition to breast and ovarian cancer. Annu Rev Genom Hum Genet. 2020;21:373–412.
Ceyhan-Birsoy O, Murry JB, Machini K, Lebo MS, Yu TW, Fayer S, et al. Interpretation of genomic sequencing results in healthy and Ill newborns: results from the BabySeq Project. Am J Hum Genet. 2019;104:76–93.
Lord CJ, Tutt A, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–70.
DeBoever C, Tanigawa Y, Aguirre M, McInnes G, Lavertu A, Rivas MA. Assessing digital phenotyping to enhance genetic studies of human diseases. Am J Hum Genet. 2020;106:611–22.
Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, Schloss JA, et al. DNA sequencing at 40: past, present and future. Nature. 2017;550:345–53.
Zhang D, Zhao L, Li B, He Z, Wang GT, Liu DJ, et al. SEQSpark: a complete analysis tool for large-scale rare variant association studies using whole-genome and exome sequence data. Am J Hum Genet. 2017;101:115–22.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Weitzel JN, Chao EC, Nehoray B, Tongeren LR, LaDuca H, Blazer KR, et al. Somatic TP53 variants frequently confound germ-line testing results. Genet Med. 2018;20:809–16.
Pharoah PDP, Song H, Dicks E, Intermaggio MP, Harrington P, Baynes C, et al. PPM1D Mosaic truncating variants in ovarian cancer cases may be treatment-related somatic mutations. J Natl Cancer Inst. 2016;108:djv347.
Sazonovs A, Barrett JC. Rare-variant studies to complement genome-wide association studies. Annu Rev Genom Hum Genet. 2018;19:97–112.
Tabor HK, Risch NJ, Myers RM. Opinion: candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet. 2002;3:391–7.
Ioannidis JPA, Tarone R, McLaughlin JK. The false-positive to false-negative ratio in epidemiologic studies. Epidemiology. 2011;22:450–56.
Moltke I, Grarup N, Jørgensen ME, Bjerregaard P, Treebak JT, Fumagalli M, et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature. 2014;512:190–93.
Suzuki K, Akiyama M, Ishigaki K, Kanai M, Hosoe J, Shojima N, et al. Identification of 28 new susceptibility loci for type 2 diabetes in the Japanese population. Nat Genet. 2019;51:379–86.
Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–18.
Minikel E, Karczewski KJ, Martin HC, Cummings BB, Whiffin N, Rhodes D, et al. Evaluating drug targets through human loss-of-function genetic variation. Nature. 2020;581:459–64.
Cummings BB, Karczewski KJ, Kosmicki JA, Seaby EG, Watts NA, Singer-Berk M, et al. Transcript expression-aware annotation improves rare variant interpretation. Nature. 2020;581:452–58.
Mahmood K, Jung C-H, Philip G, Georgeson P, Chung J, Pope BJ, et al. Variant effect prediction tools assessed using independent, functional assay-based datasets: implications for discovery and diagnostics. Hum Genom. 2017;11:10.
Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–22.
Kuiper RP, Vissers L, Venkatachalam R, Bodmer D, Hoenselaar E, Goossens M, et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat. 2011;32:407–14.
Hernandez RD, Uricchio LH, Hartman K, Ye C, Dahl A, Zaitlen N. Ultrarare variants drive substantial cis heritability of human gene expression. Nat Genet. 2019;51:1349–55.
Evans DGR, van Veen EM, Byers HJ, Wallace AJ, Ellingford JM, Beaman G, et al. A dominantly inherited 5′ UTR variant causing methylation-associated silencing of BRCA1 as a cause of breast and ovarian cancer. Am J Hum Genet. 2018;103:213–20.
Ye Y, Zhang Z, Liu Y, Diao L, Han L. A multi-omics perspective of quantitative trait loci in precision medicine. Trends Genet. 2020;36:318–36.
Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat Genet. 2019;51:1664–69.
Zhang S, He Y, Liu H, Zhai H, Huang D, Yi X, et al. regBase: whole genome base-wise aggregation and functional prediction for human non-coding regulatory variants. Nucleic Acids Res. 2019;47:e134.
Lin H, Hargreaves KA, Li R, Reiter JL, Wang Y, Mort M, et al. RegSNPs-intron: a computational framework for predicting pathogenic impact of intronic single nucleotide variants. Genome Biol. 2019;20:254.
Lou S, Cotter KA, Li T, Liang J, Mohsen H, Liu J, et al. GRAM: A GeneRAlized Model to predict the molecular effect of a non-coding variant in a cell-type specific manner. PLoS Genet. 2019;15:e1007860.
Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli L, et al. A structural variation reference for medical and population genetics. Nature. 2020;581:444–51.
Dias R, Torkamani A. Artificial intelligence in clinical and genomic diagnostics. Genome Med. 2019;11:70.
Diogo D, Kurreeman F, Stahl EA, Liao KP, Gupta N, Greenberg JD, et al. Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am J Hum Genet. 2013;92:15–27.
Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. 2013;45:1366–70.
Heart N, Institute B. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl J Med. 2014;371:22–31.
Flannick J, Thorleifsson G, Beer NL, Jacobs SBR, Grarup N, Burtt NP, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014;46:357–63.
Cuyvers E, Roeck A, den Bossche T, Cauwenberghe C, Bettens K, Vermeulen S, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol. 2015;14:814–22.
Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19:571–77.
Momozawa Y, Akiyama M, Kamatani Y, Arakawa S, Yasuda M, Yoshida S, et al. Low-frequency coding variants in CETP and CFB are associated with susceptibility of exudative age-related macular degeneration in the Japanese population. Hum Mol Genet. 2016;25:5027–34.
Khera AV, Won H-H, Peloso GM, O’Dushlaine C, Liu D, Stitziel NO, et al. Association of Rare and common variation in the lipoprotein lipase gene with coronary artery disease. JAMA. 2017;317:937–46.
Choi S, Weng L-C, Roselli C, Lin H, Haggerty CM, Shoemaker BM, et al. Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA. 2018;320:2354–64.
Dressen A, Abbas AR, Cabanski C, Reeder J, Ramalingam TR, Neighbors M, et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: a candidate gene sequencing study. Lancet Respir Med. 2018;6:603–14.
Grarup N, Moltke I, Andersen MK, Dalby M, Vitting-Seerup K, Kern T, et al. Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes. Nat Genet. 2018;50:1–3.
Cochran NJ, Geier EG, Bonham LW, Newberry SJ, Amaral MD, Thompson ML, et al. Non-coding and loss-of-function coding variants in TET2 are associated with multiple neurodegenerative diseases. Am J Hum Genet. 2020;106:632–45.
Hamilton PJ, Campbell NG, Sharma S, Erreger K, Hansen HF, Saunders C, et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry. 2013;18:1315–23.
Steri M, Orrù V, Idda LM, Pitzalis M, Pala M, Zara I, et al. Overexpression of the cytokine BAFF and autoimmunity risk. N Engl J Med. 2017;376:1615–26.
Chang J, Zhong R, Tian J, Li J, Zhai K, Ke J, et al. Exome-wide analyses identify low-frequency variant in CYP26B1 and additional coding variants associated with esophageal squamous cell carcinoma. Nat Genet. 2018;50:338–43.
Nissim S, Leshchiner I, Mancias JD, Greenblatt MB, Maertens O, Cassa CA, et al. Mutations in RABL3 alter KRAS prenylation and are associated with hereditary pancreatic cancer. Nat Genet. 2019;51:1308–14.
Jiang SH, Athanasopoulos V, Ellyard JI, Chuah A, Cappello J, Cook A, et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat Commun. 2019;10:2201.
Dwivedi O, Lehtovirta M, Hastoy B, Chandra V, Krentz NAJ, Kleiner S, et al. Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat Genet. 2019;51:1596–606.
Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. 2020;16:97–107.
Whiffin N, Armean IM, Kleinman A, Marshall JL, Minikel EV, Goodrich JK, et al. The effect of LRRK2 loss-of-function variants in humans. Nat Med. 2020;26:869–77.
Wang X-J, Xu X-Q, Sun K, Liu K-Q, Li S-Q, Jiang X, et al. Association of rare PTGIS variants with susceptibility and pulmonary vascular response in patients with idiopathic pulmonary arterial hypertension. JAMA Cardiol. 2020;5:677–84.
Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97.
Singh D, Cadden P, Hunter M, Collins L, Perkins M, Pettipher R, et al. Inhibition of the asthmatic allergen challenge response by the CRTH2 antagonist OC000459. Eur Respir J. 2012;41:46–52.
Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh WM, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–50.
Goldberg YP, Pimstone SN, Namdari R, Price N, Cohen C, Sherrington RP, et al. Human Mendelian pain disorders: a key to discovery and validation of novel analgesics. Clin Genet. 2012;82:367–73.
Sabatine MS. PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol. 2019;16:155–65.
Hall J, Dennler P, Haller S, Pratsinis A, Säuberli K, Towbin H, et al. Genomics drugs in clinical trials. Nat Rev Drug Discov. 2010;9:988.
Acknowledgements
This work was supported by AMED under Grant Number JP19kk0305010, JP20ck0106402, JP19cm0106605, and 20ck0106553. We thank Ms. Misaki Mizukoshi in the Laboratory for Genotyping Development, RIKEN Center for Integrative Medical Sciences for the illustrations of Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Momozawa, Y., Mizukami, K. Unique roles of rare variants in the genetics of complex diseases in humans. J Hum Genet 66, 11–23 (2021). https://doi.org/10.1038/s10038-020-00845-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-020-00845-2
This article is cited by
-
FiTMuSiC: leveraging structural and (co)evolutionary data for protein fitness prediction
Human Genomics (2024)
-
PASTRY: achieving balanced power for detecting risk and protective minor alleles in meta-analysis of association studies with overlapping subjects
BMC Bioinformatics (2024)
-
Genetic polymorphisms associated with adverse pregnancy outcomes in nulliparas
Scientific Reports (2024)
-
The genetic landscape of substance use disorders
Molecular Psychiatry (2024)
-
Genetic determinants of plasma protein levels in the Estonian population
Scientific Reports (2024)
