
Abstract
Rapid progress has recently been made in the elucidation of the genetic basis of childhood-onset inherited generalized dystonia (IGD) due to the implementation of genomic sequencing methodologies. We identified four patients with childhood-onset IGD harboring novel disease-causing mutations in lysine-specific histone methyltransferase 2B gene (KMT2B) by whole-exome sequencing. The main focus of this paper is to gain novel pathophysiological insights through understanding the molecular consequences of these mutations. The disease course is mostly progressive, evolving from lower limbs into generalized dystonia, which could be associated with dysarthria, dysphonia, intellectual disability, orofacial dyskinesia, and sometimes distinct dysmorphic facial features. In two patients, motor performances improved after bilateral implantation of deep brain stimulation in the globus pallidus internus (GPi-DBS). Pharmacotherapy with trihexyphenidyl reduced dystonia in two patients. We discovered three novel KMT2B mutations. Our analyses revealed that the mutation in patient 1 (c.7463A > G, p.Y2488C) is localized in the highly conserved FYRC domain of KMT2B. This mutation holds the potential to alter the inter-domain FYR interactions, which could lead to KMT2B instability. The mutations in patients 2 and 3 (c.3596_3697insC, p.M1202Dfs*22; c.4229delA, p.Q1410Rfs*12) lead to predicted unstable transcripts, likely to be subject to degradation by non-sense-mediated decay. Childhood-onset progressive dystonia with orofacial involvement is one of the main clinical manifestations of KMT2B mutations. In all, 26% (18/69) of the reported cases have T2 signal alterations of the globus pallidus internus, mostly at a younger age. Anticholinergic medication and GPi-DBS are promising treatment options and shall be considered early.
Similar content being viewed by others

Introduction
Dystonia is characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive patterned movements and/or postures affecting limbs, trunk, neck, and the face [1]. The etiology of childhood-onset dystonia can be acquired or inherited. Owing to the power of next-generation sequencing, we face the challenge of an expanding spectrum of dystonia-associated genes. Furthermore, genetic diagnosis in dystonia may guide essential therapeutic decisions [2].
Despite comprehensive treatment options encompassing pharmacotherapy and supportive treatment, many children suffer from life-long severe impairment of function and participation in activities of daily living. Deep brain stimulation of the globus pallidus internus (GPi-DBS), a reversible neurostimulation inducing a change in electrophysiological activity in the GPi via implanted electrodes, has been shown to be an effective and safe treatment option in patients with pharmacorefractory inherited or idiopathic generalized or segmental dystonia [3].
Mutations in the lysine-specific histone methyltransferase 2B gene (KMT2B) have been discovered as a cause of childhood-onset dystonia (DYT28, OMIM #617284; [4, 5]). In our dystonia cohort, we identified four patients with KMT2B mutations and analyzed their clinical phenotype, disease course, and response to treatment with a detailed review of the literature. This data provides pathophysiological insights from neuroimaging and recommendations for therapeutic intervention.
Materials and methods
Patient collective
For this study, we included four patients with inherited generalized dystonia (IGD) due to KMT2B mutations. Three of them were recruited for a research project investigating the genetic basis of childhood-onset movement disorders through whole-exome sequencing (WES), one patient was already diagnosed in another center [6]. All parents and/or patients gave their informed consent. The study was approved by the ethics committee of the Medical Faculty, University Hospital Cologne, University of Cologne (17–096).
Genomic work-up
To uncover the underlying disease-causing mutations, WES was performed on an Illumina HighSeq 4000 Sequencer (Illumina, USA) with a paired-end 75 bp sequencing protocol according to the manufacturer’s best-practice protocol, after enrichment with the SureSelect Human All Exon V.6 (Agilent, USA) Kit [7] (see further details in Supplementary Material). On average, the mean coverage was 80-fold among the sequenced subjects (Supplementary Table 1 for detailed metrics). The genetic analysis of patient 4 had been previously described by Lange and colleagues [6]. Next generation sequence alignment and variant calling have been performed and described earlier ([8]; see also Supplementary Material). Variant filtering for rare pathogenic variants below an allele frequency of <0.1% following recessive and dominant inheritance models and their bioinformatic evaluation have been described earlier [9] and are described in Supplementary Material. Since a trio-WES was available for patient 1, we additionally performed filtering for de novo variants. Predicted phosphorylation sites were evaluated with NetPhos 3.1a (http://www.cbs.dtu.dk/services/NetPhos/). Ultimately, variants were scored based on the classification by the standards and guidelines of the American College of Medical Genetics and Genomics for the interpretation of variants [10].
Sanger sequencing was performed to confirm the mutations in KMT2B and to validate the de novo inheritance within the families (see Fig. 1). Parental blood samples were not available for patient 3.

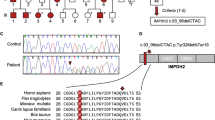
Pedigree and chromatogram of the variants in KMT2B in our three patients. Overview of KMT2B variants in all four patients. a Pedigree and chromatogram of the variant in KMT2B in patient 1. The parents are healthy. The patient showed a single base exchange (c.7463A>G, p.Y2488C). b Pedigree and chromatogram of the variant in KMT2B in patient 2. The parents are healthy. The patient showed an insertion and frameshift (c.3596_3697insC, p.M1202Dfs*22). We show the strand as it is mutated in the patient (above) and also without mutation (below) for comparison. c Pedigree and chromatogram of the variant in KMT2B in patient 3. The parents were not available for molecular genetic diagnoses. The patient showed a deletion (c.4229delA, p.Q1410Rfs*12)
Multiple sequence alignment
KMT2B sequence alignment between orthologs shown in Fig. 2 was performed by using the ClustalW algorithm and the FYRC conserved domain alignment for most diverse FYRC domain-containing proteins was obtained from the NCBI conserved domain database. For further technical alignment details with the accession number of proteins, see Supplementary Material.

Overview of KMT2B variants and FYRC domain conservation between KMT2B homologs. a KMT2B locus. Gene structure (NM_014727.1) with 37 exons is represented. Variants found in our four patients are labeled in red. b KMT2B protein structure (Q9UMN6) with domains. Positions of frameshift or missense mutations are located in important protein domains, which are labeled in red. AT-Hook: DNA-binding motif consisting of a conserved, palindromic, core sequence of proline-arginine-glycine-arginine-proline binding to AT (adenosine, tyrosine). Zf-CXXC: domain containing eight conserved cysteine residues binding to two zinc ions. PHD: plant homeodomain (PHD) finger is a C4HC3 zinc-finger-like motif found in nuclear proteins. Bromo ALL: approximately 110 amino acid protein domain recognizing acetylated lysine residues like N-terminal tails of histones. FYRN and FYRC: “FY-rich” domain N-terminal (FYRN) and “FY-rich” domain C-terminal (FYRC) sequence motifs are two poorly characterized phenylalanine/tyrosine-rich regions of around 50 and 100 amino acids found in a variety of chromatin-associated proteins. SET: 130–140 amino acid, evolutionary well-conserved sequence motif that was initially characterized in the Drosophila proteins Su(var)3–9, Enhancer-of-zeste, and Trithorax. Pro-SET: domain in the immediate vicinity to the SET domain. c Sequence alignment of top FYRC domains between KMT2B homologs, performed in CLC genomics workbench tool (protein accessions Human NP_055542.1; Mouse NP_001277502.1; Monkey XP_014979183.1; Cattle XP_015322911.1; Dog XP_003432729.3; Western Clawed Frog XP_002940678.2; Zebra Fish XP_009290151.1; Fruit Fly NP_599108.1; TBRG1 (model template NP_116200.2)
The structural modeling and interpretation of KMT2B mutations
Modeling the impact of p.Y2488C
As the structure of KMT2B’s FYRC domain was not available, we homology modeled it. First, the localization of mutated amino acids was visualized using Swiss-model for orientation (Supplementary Fig. 3) and indicated that modeling of inter-domain interactions would be required to gain an understanding of the structural impact. For homology modeling, HHpred was used to find the best template [11]. The FYR domain with 27% sequence identity to the human transforming growth factor beta regulator 1 (TBRG1, pdb id: 2WZO, [12]) was depicted to be the best template. Interestingly, in the case of TBRG1, the FYRN and FYRC elements are consecutive in a sequence, whereas in KMT2B they are separated by 628 amino acids.
By using 2WZO as a template, KMT2B-FYRC was modeled with Modeller [13]. HHpred and Modeller were utilized via the web form provided by the MPI Bioinformatics Toolkit (https://toolkit.tuebingen.mpg.de/; [14]).Structural comparison of the TBRG1-FYRC and KMT2B-FYRC domains revealed that p.Y2488C is located across KMT2B’s FYRN-FYRC inter-domain interface. Expanding on this, we decided to model the KMT2B-FYRN domain too. For this, we performed the same procedure as described above.
After acquiring KMT2B’s FYRN and FYRC models individually, they were aligned on TBRG1-FYR with the “super” command of Pymol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC). This resulted in the deduction of FYRN-FYRC’s dimeric orientation. In order to optimize the FYRN-FYRC inter-domain interface, the dimeric complex was refined with HADDOCK 2.2 web service (https://milou.science.uu.nl/services/HADDOCK2.2/haddockserver-refinement.html; [15]). The same procedure was followed for the p.Y2488C mutant of the FYRN-FYRC dimer where Y2488 was replaced with a cysteine. The interaction profile of each dimer is calculated with the PIC web-server (http://pic.mbu.iisc.ernet.in; [16]). For the step-by-step depiction of this workflow, please see Supplementary Fig. 4.
Modeling the impact of p.S1656del
For the mutation p.S1656del of patient 4, HHpred was used to find the best template to model PHD4 domain of human KMT2B. The Human Borjeson–Forssman–Lehmann Syndrome Associated Protein PHF6 (PDB id: 4NN2) with a sequence identity of 33% was depicted as the template to be fed into Modeller [17]. The top ranking four HHpred templates were chosen to investigate the PHD domain’s CXXXXC zinc-coordination motif (see Supplementary Fig. 5).
Results
Clinical presentation
All four patients manifested with a focal onset of dystonia—three out of four in the lower limbs between ages 2 and 6 years—and generalization of symptoms over time (see Table 1 and Supplementary Material for detailed patients’ histories). At school age, common features of all patients were generalized dystonia with distinct truncal, cervical, and oromandibular involvement resulting in opisthotonus, scoliosis, dystonic posturing of the limbs, torticollis, and dysarthria with spasmodic dysphonia, as well as impaired chewing and swallowing. Independent ambulation was lost between the ages 4 and 7 years for patients 1, 2, and 4. Potential triggers for an aggravation of symptoms were febrile infections (patients 1, 2, and 4), emotional distress (patient 2), and surgical interventions (patient 3). Patients 1, 2, and 4 are still under-aged, whereas patient 3 is now 47 years. Unlike the other patients, he shows a rather stable neurological status without a clear progression of disease (see video).
Cranial magnetic resonance imaging (cMRI) scans in patient 4 were unremarkable. In patient 1, a pallidal T2 hypointensity with a small central hyperintensity could be seen on the initial cMRI scan (see Supplementary Fig. 2A), which could not be detected on the follow-up cMRI scans 1 and 2 years later (see Supplementary Fig. 2B). In patient 3, a computed tomography scan at age 10 years and MRI scan at age 44 years (see Supplementary Fig. 2C) revealed decreased size of gyri, increased size of sulci, medial temporal atrophy, and broad cisterns, dilated third and fourth ventricles. Since there were no other previous MRI examinations in this patient and there are only mild clinical signs of mental decline, we refrain from speculative interpretations of this singular MRI finding.
Treatment trials with levodopa led to the deterioration of symptoms in patient 1, whereas patients 2 and 3 showed mild benefits. Patient 4 received pramipexole (dopamine-agonist) on top of a high dosage of trihexyphenidyl, which resulted in intolerable sedation and constipation. In patients 1 and 3, botulinum toxin was repeatedly injected into the lower limbs (pat. 1) and in the neck area (pat. 3) with only limited effect. Benzodiazepines brought alleviation during exacerbations in patients 1 and 2. With the introduction of high doses of trihexyphenidyl (30–40 mg/day), dystonia could be substantially reduced in patients 2 and 4 with distinct improvement of mobility. We assessed the severity of dystonia with the Burke–Fahn–Marsden Dystonia Rating Scale, BFMDRS [18] that includes a movement subscale covering nine body regions (eyes, mouth, speech and swallowing, neck, trunk, upper and lower extremity; BFMDRS-M scores 0–120, maximum score denotes severest dystonia) and disability subscale, consisting of daily activities reported by parents or patients (speech, handwriting, feeding, eating, swallowing, hygiene, dressing, and walking; BFMDRS-D scores 0–30, maximum score denotes complete dependency). In patient 2, a BFMDRS-M score of 75.5 in January 2017 dropped to 51 in February 2018 at 12 months follow-up.
Patient 1 was implanted with GPi-DBS at age 7 years leading to improvement of independent ambulation by significant reduction of dystonia. The BFMDRS-M score of 28.5 at the age of 7 years (pre-surgery) only dropped slightly to 26.5 at 14 days post-surgery operation. Further post-op BFMDRS-M scores were not available. At ages 10 and 12 years, she showed rapid deterioration of dystonia, which promptly resolved by replacing both electrodes and changing the implantable pulse generator. The current BFMDRS-M score, 11 years post-implantation, is now 58, which is mostly attributable to disease progression. When switching off the stimulation, a rapid deterioration of dystonia can be noticed. Patient 4 was also implanted with DBS-GPi at age 10 years leading to a reduction of dystonic posturing of the trunk, limbs, and neck (BFMDRS-M: pre-op 43 vs. 28 at 11 months post-op) as shown in Supplementary Fig. 6.
Genomic and structural analyses to dissect the impact of KMT2B mutations on protein structure
We performed WES to uncover the genetic etiology (see “Methods” and Supplementary Material for further details). In patient 1 (see Fig. 1a), we revealed a heterozygous single base exchange in KMT2B (c.7463A>G, p.Y2488C), which in turn leads to an amino acid change (tyrosine to cysteine) in the C-terminal phenylalanine/tyrosine-rich (FYRC) domain (see Fig. 2c). This novel mutation was not found in ClinVar, gnomAD, or ExAC browser. Our comparative sequence analysis indicated that KMT2B-FYRC including Y2488 position is very well conserved across different species (Fig. 2c) and conserved among other FYRC-containing proteins (Supplementary Fig. 1C).
As Y2488 was not reported or predicted to be a phosphorylation site (according to NetPhos 3.1a), we decided to investigate the structural impact of this mutation. The p.Y2488C mutation in patient 1 leads to a change in composition from an aromatic, polar, and large tyrosine to a non-aromatic, polar, and small cysteine. Modeling of the mutation p.Y2488C on FYRC indicated that mutation would be located across the hydrophobic FYRN–FYRC dimer interface (see “Methods”). Afterward, the modeling of the FYRN–FYRC dimer revealed that p.Y2488C is capable of disrupting the hydrophobic packing across the FYRN and FYRC interface (see Fig. 3). This is especially pronounced with the loss of direct hydrophobic contacts between amino acids L1753 (FYRN) and C2488 (FYRC) in the presence of the p.Y2488C mutation. So, as a result of p.Y2488C mutation, the hydrophobic FYRN–FYRC interface might get exposed to the hydrophilic solvent, leading to a destabilized inter-domain interaction. In line with our structural observation, the MutPred2 pathogenicity score for this particular variant (score = 0.844; reference 0, 1) estimates a rather significant pathogenicity likelihood for the amino acid variation p.Y2488C.

FYRN (light pink cartoon)–FYRC (lilac cartoon) wild-type and mutant dimer models. The dimer interface is kept intact through hydrophobic contacts (represented in spheres and sticks). a Y4288 of FYRC and L1753 of FYRN form direct hydrophobic interactions preventing the solvent to penetrate the inter-domain interface. b This hydrophobic shield is lost upon p.Y2488C mutation (depicted in gray spheres and stick)
In patient 2 (see Fig. 1b), we revealed a heterozygous insertion and frameshift in KMT2B (c.3596_3697insC, p.M1202Dfs*22). This novel mutation was also not found in ClinVar, the heterozygous allele frequencies were not available in gnomAD or ExAC browser. Sanger sequencing of parental DNAs confirmed the mutation to be very likely de novo. In patient 3 (see Fig. 1c), we revealed a deletion in KMT2B (c.4229delA, p.Q1410Rfs*12). This novel mutation was not found in ClinVar, gnomAD, or ExAC browser. Patients 2 and 3 showed truncated, probably unstable transcripts, which are predicted to be subject to degradation by nonsense-mediated decay (NMD) according to NMDEscPredictor, as these frameshifting indels are classified as being in NMD-competent regions (NMD+; [19]).
Patient 4 was added to this study because this patient received GPi-DBS implantation and the subsequent patient history after the GPi-DBS has previously not been described [6]. However, the genomic analysis of patient 4 had been reported earlier by Lange et al. [6] as a heterozygous, de novo, in-frame deletion in the Plant-Homeodomain type 4 of KMT2B (c.4966_4968TCCdel, p.S1656del; see Fig. 1b), which is predicted by MutationTaster and NMDEscPredictor to follow NMD with a splice site distance of 50 nucleotides. These NMD predictions were not confirmed experimentally by Lange et al. [6] because the KMT2B mRNA levels were not significantly altered. In order to clarify the effect of this mutation, we proceeded with the homology-based structural modeling of this PHD domain encompassing p.S1656del modification. This domain has four homologs (PDB IDs: 4NN2, 2LQ6, 5DAG, 5ERC), identified by HHpred. Structural alignment of these on our wild-type PHD structural model revealed that all of these structures have four amino acids between two cysteines (CXXXXC) responsible for coordinating a zinc ion (see Supplementary Fig. 5). CXXXXC was previously reported to be one of the prominent zinc-coordinating motifs [20]. Being at the tip of this zinc-coordinating loop (see Fig. 4a), deletion of S1656 results in the disorientation of this prominent motif (see Fig. 4b). So, the altered loop formation and impaired coordination of a zinc ion support a causative role of this deletion [6]. Our structural models are also in line with the predictions presented by Lange et al. [6].

Structural depiction of wild-type and mutant PHD domains. a Homology model of wild-type PHD domain. Possible zinc-coordinating residues are depicted in sticks (C1653, L1655, C1658, and C1682, respectively). Loop harboring CXXXXC motif (C1653-XXXX-C1658) is colored in pale red (CLSS). b Homology model of the mutant PHD domain. Deletion of S1656 results in the disorganization of the zinc-coordinating CXXXXC loop
Discussion
Clinical findings
In this study, we report four patients with KMT2B-associated childhood-onset dystonia expanding the genetic characterization and treatment responses to medication and GPi-DBS. So far, two cohorts and several case reports have been reported of patients with KMT2B mutations and symptom manifestation during the first decade of life [5, 21, 22], characterized by progressive dystonia and additional hyperkinetic features such as choreoathetosis or ballism of the limbs and trunk in most of the patients [23]. Emotional excitement, stress, infections, and surgical interventions seem to be potential triggers for the exacerbation of dystonia in the patients reported here. Additional symptoms such as microcephaly, developmental delay with mild cognitive impairment, and a characteristic facial appearance with an elongated face and bulbous nasal tip can be associated [22, 24]. The phenotypes of our patients share symptoms that have been previously reported in patients with KMT2B-associated dystonia, such as the onset in childhood with manifestation of dystonia predominantly in the lower extremities. In the following, we summarize the novel features observed in our patients:
-
1.
Disease course at higher age: The oldest patients with KMT2B-associated dystonia reported so far were 31 [25] and 39 years [26], with cervical dystonia. We report KMT2B-associated dystonia in an older patient aged 47 years with generalized dystonia and cervical involvement, too. While a previous report of a patient aged 31 years described a continuous disease progression [25], we only observed a mild progressive disease course in our patient 3, which appeared to be rather static since the patient developed cervical dystonia in the fifth decade of life.
-
2.
Response to anticholinergics: While patient 3 was started with anticholinergics at a rather late age, there was still some mild benefit. In patients 2 and 4, we started anticholinergics in the first decade of life and observed good responses concerning restoration of motor ability.
-
3.
Emergence of pallidal signal: In patient 1, we observed the emergence of a hypointensity in the globus pallidus internus with a central hyperintensity at the age of 6 years in T2-weighted cMRI sequences and its subsequent disappearance. While such a disappearance has previously been reported in a patient aged 17 years [26], we report the first instance of an emergence of a pallidal signal at a very young age.
Meyer et al. had reported 10 patients with dysmorphic features and deletions located at 19q13.11–19q13.12 (chr19:31,725,360–38,765,822) with a maximum size of 4.5 Mb [26]. The large mutations (deletions/duplications) could be associated with dysmorphic features including reduced growth, gastrointestinal and dermatological problems, whereas small mutations were only constrained to facial dysmorphia (elongated face, abnormal nasal tip) [22].
Pathophysiological insight from neuroimaging
On cMRI, subtle hypointensities on T2*-, diffusion-, and susceptibility-weighted sequences, particularly affecting the lateral part of GPi, have been reported in 17 out of 27 patients as an age-dependent MR-morphological sign that is especially accentuated at an early age [22]. To our knowledge, we report the 18th case out of 69 cases (26%) with T2-signal alterations of the GPi in KMT2B-associated dystonia [5, 23, 24, 27]. In one previously reported case, the pallidal signal was observed at 13 years and vanished at 17 years of age [22]. In line with these observations, a pallidal hypointensity with a small central hyperintensity could also be observed in patient 1 at 6 years and 10 months; however, neither in the previous nor in the subsequent MRI scans performed at 6, 8, and 10 years (see Supplementary Fig. 2A and 2B for cMRI examinations at the age of 6 years and 10 months and 10 years). It was interpreted as a neurodegenerative sign resembling the eye-of-the-tiger sign in Neurodegeneration with Brain Iron Accumulation (NBIA; [28]). Thus, our case does not only re-iterate the previously reported phenomenon of a vanishing pallidal signal but also shows the antecedent appearing of the pallidal signal [5, 23, 24, 27].
A review of the literature suggests that a hypointensity with central hyperintensity in T2 sequences has mainly been attributed to neuronal tissue injury in areas with high metabolic turnover [29]. Fluordeoxyglucose–positron emission tomographic studies have shown that temporal metabolic connectivity of GPi is particularly higher when compared to other brain areas [30]. T2-signal alterations are usually attributed to stroke, tumors, or mineral deposition, leading to restricted blood flow and neural tissue defects. The lenticulostriate arteries have previously been reported as supplying particularly high vascular blood flow through microvascular territories to the striatum and pallidum [31]. KMT2B mutations have been reported to change the epigenomic fate of neural transdifferentiation, including the expression of SLC40A1. Of note, SLC40A1 variants have been associated with hemochromatosis type 4 and severe iron (ferritin) deposition with associated changes in T2-basal ganglia signals [32]. However, deposition of other metal ions (such as zinc, manganese, or copper [33,34,35]) could also be involved. Nevertheless, further experimental investigations in animal models are needed to explore how KMT2B mutations lead to locally restricted neuronal damage in the basal ganglia.
We propose the following model for the age dependence of pallidal signals. The basal ganglia exhibit a particularly high metabolic rate [36] and a peak in developmental trajectories for global volumes in childhood and adolescence (pallidum 7.7 years for males and 9.5 years for females, striatum at 14.7 years for males and 12.1 for females; [37]). Meyer et al. also reported that mean age was significantly lower for patients with MRI abnormalities (11.7 years) than for those with normal brain scans (19.0 years) [22]. In other reports of neuro-imaging in KMT2B-associated dystonia, the patients were mostly in their late adolescence and showed no MRI abnormalities [5, 23, 24, 27]. We hypothesize that the pallidal signal might initially appear around the patients’ pallidal developmental peaks. However, subcortical structure growth is heavily dependent on hormonal changes during pubertal development, making it difficult to estimate individual growth trajectories [38].
This could also influence the vanishing of the pallidal signals on subsequent MRI scans of patient 1 and other reported cases. Meyer et al. had also reported a single case with decreasing pallidal hypointensity after 4 years [22]. Baumeister et al. have previously reported a vanishing pallidal signal in an NBIA patient and presumed resorption of deficient neurons as a leading cause of progressive pallidal atrophy [39]. Autophagy has been implicated in the process of clearing away dystrophic neurites [40] and iron deposition or other metal ions (such as zinc, manganese, or copper; [33,34,35]) have been reported to be cleared by phagocytosis.
Molecular impact of KMT2B mutations
KMT2B is a highly conserved gene that induces epigenetic posttranslational histone modifications (for mammalian conservation, see Supplementary Fig. 1A). MLL2/KMT2B proteins are responsible for the generation of H3K4me1/2/3 marks, thus inducing gene activation and regulation [41, 42]. Three patients of our collective showed previously unreported mutations in KMT2B. Patient 1 has a missense mutation (de novo c.7463A>G, p.Y2488C, see Fig. 1a) leading to an amino acid change (tyrosine to cysteine) in the FYRC domain of KMT2B (see Fig. 2b). In a broad taxonomic diversity, the FYRC domain is particularly found in chromatin remodelers, including MLL histone-methyltransferases and transforming growth factor beta regulator 1 (see Supplementary Fig. 1B). The highly conserved full-length MLL (mixed lineage leukemia) is a paralog and has been shown to be cleaved into N-terminal p320 (N320) and C-terminal p180 (C180) fragments [43] and the FYRN domain of N320 has been shown to directly interact with the FYRC and SET domains of C180. Thus MLL-N and MLL-C have been shown to heterodimerize through their FYRN/FYRC interaction domains and form a high-molecular-weight protein complex that is resistant to SIAH1/2-mediated degradation [44]. Regarding evolutionary conservation, human tyrosine 2488 is highly conserved in mammals and lower vertebrates (frog), as well as invertebrates (fly). Conservation of Y2488 in the fish was not observed (see Fig. 1c). Heterodimerization plays an important role in avoiding degradation and confer stability. Our in silico modeling supports the hypothesis that p.Y2488C may destabilize due to loss of FYRN/FYRC intra-domain hydrophobic interactions (see Fig. 3). Two patients with a similar clinical presentation have been reported to harbor a p.R2517W mutation in the FYRC domain, and the R2517 side chain is predicted to contribute to the binding to chromatin-remodeler WDR5 via a salt bridge [22].
The mutations in patients 2 and 3 probably lead to unstable transcripts (de novo c.3596_3697insC, p.M1202Dfs*22, see Fig. 1b; heterozygous c.4229delA, p.Q1410Rfs*12, see Fig. 1c), because they are predicted to follow NMD rule with a distance of 55 and 74 nucleotides, respectively, from splice sites. Although the 50-bp rule for NMD applies here, recent studies have shown that NMD follows a more complex set of rules [19]. According to NMDEscPredictor, both variants are predicted to be subject to degradation by NMD, as these frameshifting indels are classified as being in NMD-competent regions (NMD+; [19]). KMT2B haploinsufficiency has previously been described in patients with frameshift mutations [5]. Furthermore, it was shown experimentally in cultured T cells from another two patients with KMT2B mutations, leading to significantly decreased total KMT2B mRNA levels via NMD [45].
The mutation in patient 4, an in-frame deletion (de novo c.4966_4968TCCdel, p.Ser1656del, see Fig. 1c), is predicted by MutationTaster and NMDEscPredictor to follow NMD with a splice site distance of 50 nucleotides. However, KMT2B mRNA levels were not altered in sequence and quantitative expression analyses [6]. Based on comparative homology modeling, Lange and colleagues suspected that deletion of the serine residue alters loop formation and may lead to altered coordination of a zinc ion in the PHD finger, resulting in impaired DNA binding. Our own modeling of the in-frame deletion supports this hypothesis (see Figs. 2b and 4 and Supplementary Figs. 3 and 5).
The molecular pathophysiology of how KMT2B mutations lead to a movement disorder are currently emerging. Meyer et al. showed a statistically significant reduction in THAP1 expression in patient fibroblasts by immunoblotting and significantly reduced levels of dopamine 2 receptor (D2R) and an increase in tyrosine hydroxylase levels in cerebrospinal fluid immunoblotting [22]. THAP1 mutations are also associated with dystonia, also called DYT6 [46]. Barbagiovanni et al. could demonstrate that loss of KMT2B leads to massive transcriptional dysregulation of the gene expression program underlying the conversion of murine embryonic fibroblasts into induced neuronal cells and that KMT2B regulates the expression of transcripts initiating the maturation of neurons [32]. While Barbagiovanni et al. found a number of primary KMT2B targets dysregulated in Kmt2b−/− neurons had been associated with dystonia such as Gnao1 [2], they did not report to our knowledge a dysregulation Thap1 or Tor1a. Noteworthy is the fact that Barbagiovanni et al. found possible pathogenic mutations in 3 novel candidate genes (NOL4, SLC35F1, and SLC40A1) in a previously genetically unsolved cohort of dystonia patients [32] based on the differentially regulated transcripts from the Kmt2b−/− neurons. This supports the above-elaborated hypothesis that the pallidal hypointensity sign and damage is due to a local altered SLC40A1 expression and subsequent ferritin deposition.
In Kmt2b-knockdown mice with deficient memory learning, H3K4me3 levels were specifically decreased in the hippocampal Cornu Ammonis region [47]. KMT2B is ubiquitously expressed in the brain with higher levels not only in the cerebellum of humans but was also shown to be in the hippocampus and striatum of humans [22] and rats [45]. Striatal necrosis has been shown to disinhibit GABAergic projections to the GPi, which excited thalamocortical neurons and led to excessive motor symptoms by disinhibiting the thalamocortical pathway [48].
Therapeutic management
In patients 2 and 4, high doses of the oral anticholinergic agent trihexyphenidyl had beneficial effects at early stages of disease progression. Looking at the published data, trihexyphenidyl has been effective in 9 out of 14 patients, whereas other dystonia-specific medications were without any relevant effects [5, 6, 22, 49]. In the Tor1a mouse model of DYT1, trihexyphenidyl injections induced corticostriatal long-term depression (LTD) by activation of D2 receptors, which eventually led to the restoration of motor control [50]. Similarly, trihexyphenidyl may serve as a potent therapeutic option at an early stage of manifestation of KMT2B-associated dystonia in a similar manner to DYT1 by modulating corticostriatal LTD.
GPi-DBS showed significant beneficial effects in patients 1 and 4 by improving independent ambulation and slowing down disease progression. According to the previous literature, 12 out of 13 patients with KMT2B-associated dystonia and GPi-DBS showed a beneficial clinical response, while 1 patient only showed transient improvement [5, 22]. Recent studies indicate that a younger age at the time of surgery is associated with better DBS outcomes at latest follow-up in patients with IGD. Therefore, bilateral GPi-DBS should be considered early in patients with KMT2B-associated dystonia [5, 24].
Conclusion
Childhood-onset progressive dystonia with orofacial involvement seem to be key features of KMT2B mutations and should trigger genetic work-up. Identifying the underlying genetic etiology substantially improves the counseling of patients and families and may enable specific therapeutic strategies. Anticholinergic medication and GPi-DBS seem to be effective treatment options and should be considered early in these patients. To ease the unmet need for personalized treatment strategies, further research into pathophysiological insights are essential and required to decipher the molecular mechanisms of KMT2B-associated dystonia.
Change history
06 August 2019
Rapid progress has recently been made in the elucidation of the genetic basis of childhood-onset inherited generalized dystonia (IGD) due to the implementation of genomic sequencing methodologies. We identified four patients with childhood-onset IGD harboring novel disease-causing mutations in lysine-specific histone methyltransferase 2B gene (KMT2B) by whole-exome sequencing. The main focus of this paper is to gain novel pathophysiological insights through understanding the molecular consequences of these mutations.
The disease course is mostly progressive, evolving from lower limbs into generalized dystonia, which could be associated with dysarthria, dysphonia, intellectual disability, orofacial dyskinesia, and sometimes distinct dysmorphic facial features. In two patients, motor performances improved after bilateral implantation of deep brain stimulation in the globus pallidus internus (GPi-DBS). Pharmacotherapy with trihexyphenidyl reduced dystonia in two patients.
We discovered three novel KMT2B mutations. Our analyses revealed that the mutation in patient 1 (c.7463 A > G, p.Y2488C) is localized in the highly conserved FYRC domain of KMT2B. This mutation holds the potential to alter the inter-domain FYR interactions, which could lead to KMT2B instability. The mutations in patients 2 and 3 (c.3602dupC, p.M1202Dfs*22; c.4229delA, p.Q1410Rfs*12) lead to predicted unstable transcripts, likely to be subject to degradation by non-sense mediated decay.
Childhood-onset progressive dystonia with orofacial involvement is one of the main clinical manifestations of KMT2B mutations. In all, 26% (18/69) of the reported cases have T2 signal alterations of the globus pallidus internus, mostly at a younger age. Anticholinergic medication and GPi-DBS are promising treatment options and shall be considered early.
An amendment to this paper has been published and can be accessed via a link at the top of the paper
References
Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28:863–73.
Koy A, Cirak S, Gonzalez V, Becker K, Roujeau T, Milesi C, et al. Deep brain stimulation is effective in pediatric patients with GNAO1 associated severe hyperkinesia. J Neurol Sci. 2018;391:31–39.
Kupsch A, Benecke R, Müller J, Trottenberg T, Schneider G-H, Poewe W, et al. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. New Engl J Med. 2006;355:1978–90.
Lohmann K, Klein C Update on the Genetics of Dystonia. Current Neurology and Neuroscience Reports. 2017;17.
Zech M, Boesch S, Maier Esther M, Borggraefe I, Vill K, Laccone F, et al. Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia. Am J Hum Genet. 2016;99:1377–87.
Lange LM, Tunc S, Tennstedt S, Münchau A, Klein C, Assmann B, et al. A novel, in-frame KMT2B deletion in a patient with apparently isolated, generalized dystonia. Mov Disord. 2017;32:1495–97.
Wang H, Schänzer A, Kampschulte B, Daimagüler H-S, Logeswaran T, Schlierbach H, et al. A novel SPEG mutation causes non-compaction cardiomyopathy and neuropathy in a floppy infant with centronuclear myopathy. Acta Neuropathol Commun. 2018;6:83.
Wang H, Salter CG, Refai O, Hardy H, Barwick KES, Akpulat U, et al. Choline transporter mutations in severe congenital myasthenic syndrome disrupt transporter localization. Brain. 2017;140:2838–50.
Ghosh SG, Becker K, Huang H, Dixon-Salazar T, Chai G, Salpietro V, et al. Biallelic mutations in ADPRHL2, encoding ADP-ribosylhydrolase 3, lead to a degenerative pediatric stress-induced epileptic ataxia syndrome. Am J Hum Genet. 2018;103:431–39.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic acids Res. 2005;33:W244–8.
Garcia-Alai MM, Allen MD, Joerger AC, Bycroft M. The structure of the FYR domain of transforming growth factor beta regulator 1. Protein Sci. 2010;19:1432–8.
Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2016;86:2.9.1–2.9.37.
Alva V, Nam SZ, Soding J, Lupas AN. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic acids Res. 2016;44:W410–5.
van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, et al. The HADDOCK2.2 Web Server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720–25.
Tina KG, Bhadra R, Srinivasan N. PIC: protein interactions calculator. Nucleic acids Res. 2007;35:W473–6.
Liu Z, Li F, Ruan K, Zhang J, Mei Y, Wu J, et al. Structural and functional insights into the human Borjeson-Forssman-Lehmann syndrome-associated protein PHF6. J Biol Chem. 2014;289:10069–83.
Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scale for the primary torsion dystonias. Neurology. 1985;35:73–7.
Coban-Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. 2018;103:171–87.
Kluska K, Adamczyk J, Krezel A. Metal binding properties of zinc fingers with a naturally altered metal binding site. Metallomics. 2018;10:248–63.
Zech M, Jech R, Havránková P, Fečíková A, Berutti R, Urgošík D, et al. KMT2B rare missense variants in generalized dystonia: KMT2B Missense variants in dystonia. Mov Disord. 2017;32:1087–91.
Meyer E, Carss KJ, Rankin J, Nichols JME, Grozeva D, Joseph AP, et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat Genet. 2017;49:223–37.
Hackenberg A, Wagner M, Pahnke J, Zeitler P, Boltshauser E. Low voice, spasmodic dysphonia, and hand dystonia as clinical clues for KMT2B-associated early-onset dystonia. Neuropediatrics. 2018;49:356–56.
Gorman KM, Meyer E, Kurian MA. Review of the phenotype of early-onset generalised progressive dystonia due to mutations in KMT2B. Eur J Paediatr Neurol. 2018;22:245–56.
Klein C, Baumann H, Olschewski L, Hanssen H, Munchau A, Ferbert A, et al. De-novo KMT2B mutation in a consanguineous family: 15-Year follow-up of an Afghan dystonia patient. Parkinsonism Relat Disord. 2019. https://doi.org/10.1016/j.parkreldis.2019.03.018.
Meyer E, Carss KJ, Rankin J, Nichols JM, Grozeva D, Joseph AP, et al. Mutat histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat Genet. 2017;49:223–37.
Dai L, Ding C, Fang F. An inherited KMT2B duplication variant in a Chinese family with dystonia and/or development delay. Parkinsonism Related Disord. 2018. https://doi.org/10.1016/j.parkreldis.2018.08.021.
Lee JH, Gregory A, Hogarth P, Rogers C, Hayflick SJ. Looking deep into the eye-of-the-tiger in pantothenate kinase–associated neurodegeneration. Am J Neuroradiol. 2018;39:583–88. https://doi.org/10.3174/ajnr.A5514.
Harder SL, Hopp KM, Ward H, Neglio H, Gitlin J, Kido D. Mineralization of the deep gray matter with age: a retrospective review with susceptibility-weighted MR imaging. Am J Neuroradiol. 2008;29:176–83.
Tomasi DG, Shokri-Kojori E, Wiers CE, Kim SW, Demiral SB, Cabrera EA, et al. Dynamic brain glucose metabolism identifies anti-correlated cortical-cerebellar networks at rest. J Cereb Blood Flow Metab. 2017;37:3659–70.
Feekes JA, Cassell MD. The vascular supply of the functional compartments of the human striatum. Brain. 2006;129:2189–201.
Barbagiovanni G, Germain PL, Zech M, Atashpaz S, Lo Riso P, D’Antonio-Chronowska A, et al. KMT2B is selectively required for neuronal transdifferentiation, and its loss exposes dystonia candidate genes. Cell Rep. 2018;25:988–1001.
Harischandra DS, Ghaisas S, Rokad D, Zamanian M, Jin H, Anantharam V, et al. Environmental neurotoxicant manganese regulates exosome-mediated extracellular miRNAs in cell culture model of Parkinson’s disease: relevance to α-synuclein misfolding in metal neurotoxicity. Neurotoxicology. 2018;64:267–77.
Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci. 2017;18:598.
Wang P, Wang Z-Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res Rev. 2017;35:265–90.
Casanova MF, Araque JM. Mineralization of the basal ganglia: implications for neuropsychiatry, pathology and neuroimaging. Psychiatry Res. 2003;121:59–87.
Raznahan A, Shaw PW, Lerch JP, Clasen LS, Greenstein D, Berman R, et al. Longitudinal four-dimensional mapping of subcortical anatomy in human development. Proc Natl Acad Sci. 2014;111:1592–97.
Goddings A-L, Mills KL, Clasen LS, Giedd JN, Viner RM, Blakemore S-J. The influence of puberty on subcortical brain development. Neuroimage. 2014;88:242–51.
Baumeister FA, Auer DP, Hortnagel K, Freisinger P, Meitinger T. The eye-of-the-tiger sign is not a reliable disease marker for Hallervorden-Spatz syndrome. Neuropediatrics. 2005;36:221–2.
Jungbluth H. Autophagy - a fundamental cellular mechanism on the verge of clinical translation: editorial. Neuropathol Appl Neurobiol. 2015;41:598–600.
Ladopoulos V, Hofemeister H, Hoogenkamp M, Riggs AD, Stewart AF, Bonifer C. The histone methyltransferase KMT2B is required for RNA polymerase II association and protection from DNA methylation at the MagohB CpG island promoter. Mol Cell Biol. 2013;33:1383–93.
Shao G-B, Chen J-C, Zhang L-P, Huang P, Lu H-Y, Jin J, et al. Dynamic patterns of histone H3 lysine 4 methyltransferases and demethylases during mouse preimplantation development. In Vitro Cell Dev Biol Anim. 2014;50:603–13.
Hsieh JJD, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23:186–94.
Pless B, Oehm C, Knauer S, Stauber RH, Dingermann T, Marschalek R. The heterodimerization domains of MLL—FYRN and FYRC—are potential target structures in t(4;11) leukemia. Leukemia. 2011;25:663–70.
Kawarai T, Miyamoto R, Nakagawa E, Koichihara R, Sakamoto T, Mure H, et al. Phenotype variability and allelic heterogeneity in KMT2B-associated disease. Parkinsonism Relat Disord. 2018;52:55–61.
Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41:286–8.
Kerimoglu C, Sakib MS, Jain G, Benito E, Burkhardt S, Capece V, et al. KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell Rep. 2017;20:538–48.
Plotkin JL, Goldberg JA. Thinking outside the box (and arrow): current themes in striatal dysfunction in movement disorders. Neuroscientist. 2018. https://doi.org/10.1177/1073858418807887.
Fehlings D, Brown L, Harvey A, Himmelmann K, Lin J-P, Macintosh A, et al. Pharmacological and neurosurgical interventions for managing dystonia in cerebral palsy: a systematic review. Dev Med Child Neurol. 2018;60:356–66.
Dang MT, Yokoi F, Cheetham CC, Lu J, Vo V, Lovinger DM, et al. An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 ΔGAG knock-in mice. Behav Brain Res. 2012;226:465–72.
Acknowledgements
We would like to thank the patients and their families of this clinical series. This work was supported by the Deutsche Forschungsgemeinschaft Emmy Noether Grant to SC (CI 218/1–1). HSD and AK were supported by the Gerok program of the Faculty of Medicine, University of Cologne. AK received a research grant from the Dr. Hans Günther und Dr. Rita Herfort Stiftung. We furthermore thank the Regional Computing Center of the University of Cologne (RRZK) for providing computing time for the bioinformatics analyses on the DFG-funded High-Performance Computing (HPC) system CHEOPS as well as support. EK acknowledges the Alexander von Humboldt Foundation Return Fellowship.
Author information
Authors and Affiliations
Contributions
HSD analyzed clinical patient data, neuroimaging and molecular genetic data, and wrote the manuscript. SC, RS, AC, H-SD, KB, PN, JA, and HT analyzed molecular genetic data and critically reviewed the manuscript. GW, MS-R, KK, MK, AH, AP, AK, and BA analyzed clinical patient data and critically reviewed the manuscript. RS, AC, TK, EK, and SC performed the 3D protein modeling. AK and SC designed and supervised the study. SC obtained funding and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics statement
Informed consent was obtained from the patients for genetic investigations, recording, and publishing the disease-related information. The study was approved by the institutional review board of the Ethics Committee of the University Hospital of Cologne.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Dafsari, H.S., Sprute, R., Wunderlich, G. et al. Novel mutations in KMT2B offer pathophysiological insights into childhood-onset progressive dystonia. J Hum Genet 64, 803–813 (2019). https://doi.org/10.1038/s10038-019-0625-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0625-1
