
Abstract
We report a family with progressive myoclonic epilepsy who underwent whole-exome sequencing but was negative for pathogenic variants. Similar clinical courses of a devastating neurodegenerative phenotype of two affected siblings were highly suggestive of a genetic etiology, which indicates that the survey of genetic variation by whole-exome sequencing was not comprehensive. To investigate the presence of a variant that remained unrecognized by standard genetic testing, PacBio long-read sequencing was performed. Structural variant (SV) detection using low-coverage (6×) whole-genome sequencing called 17,165 SVs (7,216 deletions and 9,949 insertions). Our SV selection narrowed down potential candidates to only five SVs (two deletions and three insertions) on the genes tagged with autosomal recessive phenotypes. Among them, a 12.4-kb deletion involving the CLN6 gene was the top candidate because its homozygous abnormalities cause neuronal ceroid lipofuscinosis. This deletion included the initiation codon and was found in a GC-rich region containing multiple repetitive elements. These results indicate the presence of a causal variant in a difficult-to-sequence region and suggest that such variants that remain enigmatic after the application of current whole-exome sequencing technology could be uncovered by unbiased application of long-read whole-genome sequencing.
Similar content being viewed by others
Introduction
Progressive myoclonic epilepsy (PME) is a clinically heterogeneous group of rare neurodegenerative disorders characterized by myoclonus, seizures, ataxia, and progressive cognitive impairment. The genetic etiology of PME varies. Numerous autosomal recessive diseases are associated with PME, such as neuronal ceroid lipofuscinoses (NCLs), Lafora disease, Unverricht–Lundborg disease, sialidosis, and Gaucher disease [1]. Genetic analysis, histological examination, and enzyme testing need to be considered to establish the diagnosis because of genetic heterogeneity and clinical overlap among these neurodegenerative disorders. Regardless of the strategic approach for diagnosis, a substantial proportion of cases remain unresolved. Whole-exome sequencing (WES) can effectively screen 99% of the entire set of RefSeq genes covering about 58 Mb of the human genome, without specific prior knowledge. In fact, the diagnostic yield upon applying WES reached 31% (26/84 cases) of unresolved PME cases that had previously undergone genetic investigation. However, the remaining 69% of cases present a genetic challenge [2]. These findings suggest that certain types of pathogenic variation evade detection by the currently available genetic analysis.
Recent advanced studies using multi-platform approaches with de novo assembly revealed unprecedented structural variants (SVs) in the human genome, although their pathogenic roles remain unknown [3,4,5,6]. Improving the capacity of long-read sequencing technology can provide an opportunity to study the pathogenic role of such newly recognized SVs, especially in cases in which the genetic cause of disease has not been resolved. In fact, Merker et al. recently reported a 2.1-kb pathogenic deletion for autosomal dominant Carney complex using long-read Whole-genome sequencing (WGS). This variant had not been recognized by initial genetic screening [7]. However, the application of long-read sequencing for disease research remains at an early stage [8, 9]. Here, PacBio long-read-only analysis was used in a family with unresolved PME that was negative in WES. Long-read sequencing ensures unbiased coverage even in GC-rich repetitive sequences, which enables the identification of previously unidentified pathogenic SVs. This paper presents the utility of this approach for medical research.
Materials and methods
Ethics approval
Written informed consent for inclusion in the study was obtained from all participants. This study was approved by the Institutional Review Board of Yokohama City University School of Medicine.
Case reports
A 20-year-old female (II-2) is the second child born to nonconsanguineous Japanese parents. She has a healthy older brother (II-1). She was delivered at full term without any complications. Her development was normal until 4 years of age, when she developed myoclonus and ataxia. She also had generalized tonic clonic seizures. A diffuse spike pattern was observed on EEG. Her seizures were refractory to multiple anti-epileptic drugs. Her development was regressive and she became bedridden. At 8, 9, and 16 years, brain MRI showed progressive diffuse atrophy (cortical, white matter, caudate, thalamus, and cerebellum) and thin corpus callosum (Supplementary Fig. S1). She had visual impairment with optic atrophy, but no cherry red spot. Both electron microscopy of skin biopsy and lysosomal enzyme activity were normal.
III-2, a 13-year-old boy, is a younger brother of II-2. He was born at 36 weeks of gestation without any complications. His psychomotor development was mildly delayed. He had neurological features similar to those of his affected sister. At 4 years of age, he developed myoclonus, ataxia, and generalized tonic clonic seizures. EEG showed occipital-dominant spikes. His seizures were refractory to multiple anti-epileptic drugs. He gradually showed developmental regression and became bedridden. Brain MRI at 9 years showed diffuse atrophy (cortical, white matter, caudate, thalamus, and cerebellum). Corpus callosum was thin and the lateral ventricle was enlarged (Supplementary Fig. S1). Lysosomal enzyme activity was normal.
Clinical findings of the affected siblings (II-2 and II-3) are summarized in Table 1.
SMRTbell library preparation
Genomic DNA was extracted from peripheral blood leukocytes using QuickGene DNA whole-blood kit (Kurabo) for II-2 and three controls. The size and integrity of genomic DNA were assessed by pulse-field agarose gel electrophoresis and the DNA concentration was measured by a Qubit fluorometer (Life Technologies). Seven micrograms of genomic DNA in a 150-µl volume was fragmented using g-TUBE (Covaris) by centrifugation at 1500 × g for 2 min twice. Recovered DNA was purified and concentrated using AMpure PB magnetic beads (Pacific Biosciences).
SMRTbell Template Prep Kit 1.0 SPv3, Sequel Binding Kit 2.0, SMRTbell Clean Up Column v2 Kit, and MagBead Kit v2 (Pacific Biosciences) were used for SMRTbell library construction. SMRTbell template DNA/polymerase complex was used for sequencing on the PacBio Sequel system.
Five micrograms of fragmented DNA was subjected to SMRTbell library preparation in accordance with the manufacturer’s instructions (Procedure & Checklist >20 kb Template Preparation Using BluePippin Size-Selection System for Sequel Systems; Pacific Biosciences).
The resulting SMRTbell template was size-selected by BluePippin (Sage Science) and enriched for DNA fragments of >10 kb in size. Extraction conditions were set as follows: 0.75% DF Marker S1 high-pass 6–10 kb vs3 with a base-pair threshold start value (BP start) of 10,000. The size-selected library was purified by AMpure PB and then subjected to a DNA damage repair reaction. SMRTbell template DNA was annealed with Sequencing Primer v3 at 20 °C for 1 h. For polymerase binding, primer-annealed SMRTbell template DNA was incubated at 30 °C for 4 h with Sequel Polymerase 2.0. SMRTbell template DNA/polymerase complex was then purified using SMRTbell Clean Up Column. The purified complex was diluted to achieve an on-plate loading concentration of 20 pM, and then mixed and incubated with MagBead at 4 °C for 1 h to prepare MagBead-bound SMRTbell complex. This complex was loaded onto Sequel SMRT Cell 1 M v2 and sequenced using Sequel Sequencing Kit 2.0. Data were collected for 6 h for each SMRT cell.
Data analysis using SMRT analysis module provided by SMRT link
Four SMRT cells were used for II-2, control 2, and control 3, which generated mean genome-wide coverage of 6×, 8×, and 6×, respectively. For control 1, mean coverage of 13× was obtained by using 10 SMRT cells. Raw statistics on the sequencing performance is described in Supplementary Table S1.
Secondary analysis using base-called data was performed on SMRT analysis v5.1.0. Structural variants were called using PBSV with the default settings, an application provided by SMRT analysis. Minimum SV length, minimum reads that support SV, and minimum percentage of variant reads were set to 50 bp, two reads, and 20%, respectively. PBSV called two types of SV, insertion and deletion. When comparing the insertion calls among different individuals, regions up to 50 bp in length might be misaligned due to high sequence error rates of long-read sequencing; such inaccuracies were thus ignored and grouped into a single unit with the same/similar SVs. The resequencing application provided by SMRT analysis was used to summarize the mapping statistics in order to evaluate the data quality because PBSV does not generate such metrics (Supplementary Table S1).
Southern blot analysis
Ten micrograms of genomic DNA was digested with EcoRV to confirm CLN6 deletion (chr15: 68,518,038–68,530,471). Digested DNA was run on a 0.8% agarose gel (w/v) in 1.0×TBE and transferred to a positively charged nylon membrane using capillary transfer. The Digoxigenin-labeled DNA probe was generated using the primers listed in Supplementary Table S2, according to the manufacturer’s instructions (Merck).
Whole-exome sequencing (WES)
WES was performed in II-2 and her parents. DNA samples were captured by SureSelectXT Human All Exon V5 (Agilent Technologies) and sequenced on the Illumina HiSeq2500 with 101-bp paired-end reads (Illumina). Short-reads were aligned to the human reference genome (GRCh37/hg19) using Novoalign v3.02.13. PCR duplicates were removed using Picard. Local realignments around indels and base quality score recalibration were performed with the Genome Analysis Toolkit (GATK) 3.2-0. Variants were called by GATK UnifiedGenotyper. For examining copy number alterations, eXome-Hidden Markov Model (XHMM) and Nord method were used (See below in DNA copy number analysis). Variants were screened for based on an autosomal recessive model as described previously [10]. The mean read depth of protein-coding regions ranged from 82.5×, 52.9×, and 40.4× for II-2, I-1, and I-2, respectively. The variants that fulfilled the following criteria were considered for further analysis: (1) variants with minor allele frequency (MAF) <1% in the Exome Sequencing Project (ESP6500), Exome Aggregation Consortium (ExAC), and in-house 575 Japanese exome datasets; (2) possible pathogenicity based on variant type (nonsense, missense, frameshift, and splice site), with computational prediction of a deleterious effect on protein function by SIFT, Polyphen-2, and MutationTaster; (3) biallelic variants, at least one of which was deleterious under an autosomal recessive model; and (4) variants found in a gene whose pathogenic variant causes epilepsy as reported in the literature.
Haplotype analysis
Fluorescently labeled PCR primers from the Linkage Mapping Set v2.5 (Applied Biosystems) were used for microsatellite markers. The PCR amplicons were run on an ABI 3130xl Genetic Analyzer. The fragment analysis was performed using ABI Prism GeneScan analysis software. For SNP haplotype, genotype data were extracted from trio exomes. SNPs with fewer than 30 reads were discarded from the analysis. Two heterozygous SNPs (chr15: 68,486,358 and chr15: 68,925,781) flanking the 440-kb homozygous block in II-2 were confirmed by Sanger sequencing. Haplotype frequency from phase 3 of the 1000 Genomes Project (G_A_C_C_T_A_C_C_T_T for rs11855587_ rs4777035_ rs2271724_ rs2271722_ rs7167822_ rs2292745_ rs7168069_ rs6494733_ rs2306023_ rs4777049) was determined using NIH LDlink 3.2.0 web-based tools (https://ldlink.nci.nih.gov/).
DNA copy number analysis
Copy number variants (CNVs) were investigated using WES data. eXome-Hidden Markov Model (XHMM) was used for genome-wide screening [11]. A total of 31 known genes for PME were also tested by the Nord method (ASAH1, CACNB4, CASR, CLN3, CLN5, CLN6, CSTB, EFHC1, EPM2A, FOLR1, GABRA1, GABRD, GLDC, GOSR2, HEXA, HEXB, KCNC1, KCTD7, MYBPC1, NEU1, NHLRC1, NOL3, NPC1, NPC2, POLG, PPT1, PRICKLE1, PRICKLE2, SCARB2, SGCE, and TPP1) [12].
Real-time quantitative PCR (qPCR)
Rotor-Gene SYBR Green kit was used for real-time quantification of genomic DNA, with amplification monitored on the Rotor-Gene cycler system (Qiagen). DNA copy number was measured by the standard curve method with specific primers (Supplementary Table S2). DNA copy number of targets (exons 1 and 2 of CLN6) was normalized using an internal control region (chr9: 130,425,699–130,425,759) and then additionally relative to an unrelated control individual.
Evaluation of the CLN6 deletion in a Japanese cohort (n = 3,936)
Japanese individuals in Tohoku Medical Megabank Genome Reference Panel (n = 3,552)
The heterozygous 12.4-kb deletion (chr15: 68,518,090–68,530,472) was investigated using short-read WGS data of 3,552 Japanese individuals. FASTQ files of each sample were aligned to the human reference genome (GRCh37), which contains the revised Cambridge Reference Sequence (rCRS), unlocalized/unplaced contigs, human gamma herpesvirus 4 sequence (NC_007605), and a decoy sequence (hs37d5) by using BWA-MEM version 0.7.12 with the “-K 10000000” option in addition to the default options to reduce any differences when we performed calculations with multiple threads. We checked this by calculating ratios of the normalized read count in the region to the median of the normalized read count, and observed that the ratios were virtually in a normal distribution with mean = 1.0 and standard deviation = 0.059, and that no individuals had ratios <0.7, where individuals with heterozygosity are expected to have the ratio = 0.5.
Healthy individuals from Tokyo Healthy Control (THC; n = 384)
A total of 418 healthy individuals from Tokyo Healthy Control (THC) with the HiSeq X sequencer (150 paired-end sequencing protocol) were used in this analysis. THC included healthy Japanese individuals residing in and around Tokyo, Japan. Alignment was performed using BWA-MEM (ver. 0.7.5a-r405) with the default option to the human reference genome (GRCh37/hg19) with decoy sequences (hs37d5) and NC_007605 (Human Gamma Herpesvirus 4). For mitochondria, a major haplotype in Japanese, JN253391 (http://www.ncbi.nlm.nih.gov/nuccore/JN253391), was used. The complete fasta file (hg19_tommo_v2.fa) is available from the iJGVD website (https://ijgvd.megabank.tohoku.ac.jp/download/tommo_hg19_v2/) [13]. As a sample QC for CNV analysis, we calculated the coefficient of variation (CV) of the read depth to the reference genome except for inaccessible regions, that is, N in fasta format. As a result, thirty-four samples with CV>0.9 were excluded for the downstream analysis. To estimate the copy number status for the qualified 384 samples, mapped reads within chr15 from 68,518,090 to 68,530,472 were normalized with the total mapped reads in autosomal regions (refer to the normalized region score). To estimate the ploidy of each sample, the normalized region scores were classified into 0, 1, and 2 copies by selecting the nearest one.
Short-read Whole-genome sequencing (WGS)
WGS was performed for II-2 and control 2. The WGS libraries were generated using TruSeq DNA PCR Free Sample Prep Kit (Illumina) and sequenced on the Illumina HiSeq2500 with 150-bp paired-end reads (Illumina) by Macrogen Japan. Alignment was applied using BWA-MEM (ver. 0.7.12) to the human reference genome (GRCh37/hg19). SV analysis using 37.94 × and 29.69 × coverage WGS data for II-2 and control, respectively, was performed by BreakDancerMax-1.1r112 [14].
Web resources
OMIM, http://www.omim.org/
LDlink, https://ldlink.nci.nih.gov/
SIFT, http://sift.jcvi.org/
Polyphen-2, http://genetics.bwh.harvard.edu/pph2/
MutationTaster, http://www.mutationtaster.org/
ESP6500, http://evs.gs.washington.edu/EVS/
ExAC, http://exac.broadinstitute.org/
HGMD, http://www.biobase-international.com/product/hgmd
Tohoku Medical Megabank Genome Reference Panel, https://jmorp.megabank.tohoku.ac.jp/
Results
The similarity of the severe neurodegenerative phenotypes of two affected siblings was highly suggestive of a genetic etiology in this family (Fig. 1a, Table 1, and Case Reports). We initially performed trio-based WES and searched for pathogenic single-nucleotide variants (SNVs). No probable causative variants remained after variant filtering including known PME genes, such as PRICKLE1, EPM2A, NHLRC, KCTD, SCARB2, GOSR2, KCNC1, CERS1, LMNB2, PRDM8, NEU1, and 13 genes for NCLs. No causal CNVs were also detected by WES-based CNV analysis (Supplementary Fig. S2).

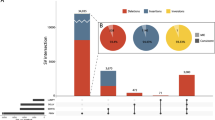
Familial pedigree and pathogenic SV detection by PacBio long-read sequencing. a Pedigree and segregation of a 12.4-kb deletion involving CLN6. + , wild type; del, 12.4-kb deletion involving CLN6. Southern blot analysis to confirm the presence of a 12.4-kb deletion involving CLN6. + , 23.9-kb EcoRV fragment as expected from the reference sequence (GRCh37/hg19); del, ~11.5-kb EcoRV fragment due to the 12.4-kb CLN6 deletion. b SV prioritization to narrow down pathogenic candidates. c Case-only (II-2) deletions overlapping with RefSeq genes were classified by their relative position and size. CDS protein-coding sequence, UTR untranslated sequence. d Comparison of short-read WES data and PacBio long-read WGS data around CLN6. Data for one unrelated control individual are also shown as a control for WES and PacBio. Arrow indicates CLN6 exon 1, which was rarely covered by short-reads. EcoRV EcoRV restriction sites. The EcoRV fragment with the expected size of 23,964 bp in Southern blotting is shown by a bidirectional arrow. PacBio subreads are shown at the bottom. PacBio long-read data were aligned to the human genome reference sequence (GRCh37/hg19) by the long-read alignment program NGMLR powered by the PBSV application in SMRT Link v5.1.0. Forward and reverse complement strands are shown by pink and gray rectangles, respectively. The sites of deletion and insertion are shown by black connecting lines and longitudinal blue boxes, respectively
Further investigation using PacBio single-molecule, real-time (SMRT) sequencing [15] was performed to complement the short-read sequencing technology, in particular to investigate intermediate-size SVs (50 bp–50 kb) in repetitive and GC-rich regions. Genomic DNA from II-2 and three unrelated control individuals (controls 1, 2, and 3) was sequenced by PacBio Sequel. Average read depth was 6×, 13×, 8×, and 6× for II-2, and controls 1, 2, and 3, respectively (Supplementary Table S1). The mean subread length and subread N50 were 9,744 bp (range: 8,102–10,442 bp) and 15,520 bp (range: 12,439–16,725 bp), respectively (Supplementary Table S1). PacBio long-read data were analyzed using PBSV, which is a structural variant caller for PacBio reads. A total of 7,216 deletions and 9,949 insertions (≥50 bp) were called in II-2 (Fig. 1b). We initially excluded the probable nonpathogenic SV calls that were shared with any of three control individuals. A total of 1,451 deletions and 3,002 insertions remained as case-only SVs in II-2 (Supplementary Fig. S3). Considering the pathogenic impact of genetic change, we further selected the SVs based on their positions in the genes (Fig. 1c, Supplementary Fig. S3 and Supplementary Table S3). The total numbers of SV calls overlapping with RefSeq genes, exons, and protein-coding sequences were 1,981 (658 deletions and 1,323 insertions), 106 (37 deletions and 69 insertions), and 50 (18 deletions and 32 insertions), respectively (Fig. 1b). Among the 50 SV calls affecting protein-coding sequences, five (two deletions and three insertions) were linked to an OMIM entry involving an autosomal recessive phenotype (Table 2). Surprisingly, a 12.4-kb deletion call spanning the first coding exon of CLN6 was found, which is causal of ceroid lipofuscinosis, neuronal, 6 (CLN6, OMIM #601780) [16, 17]. This deletion leads to loss of the initiation codon of CLN6, which is supposed to be a null variant. The clinical features of the affected siblings were compatible with CLN6 (Case Reports and Table 1). The pathogenicity of the other four SV calls, C4A (C4a deficiency, OMIM #614380), MYO7A (deafness, autosomal recessive 2, OMIM #600060, and Usher syndrome, type 1B, OMIM #276900), PEX5 (peroxisome biogenesis disorder 2 A, OMIM #214110), and MEGF8 (Carpenter syndrome 2, OMIM #614976) was less likely when considering the patients’ phenotype and the heterozygous genotype call of these variants, except for C4A (Table 2). In C4A deficiency, comprehensive defects do not occur because C4A’s function is complemented by its homolog, C4B. C4A and C4B are mapped within a segmental duplication (chr6: 31,948,309–31,967,262), so a large 32-kb deletion involving C4A and C4B could not be confirmed. Nonetheless, deficiencies in complement components would be unlikely to explain the neuronal degeneration.
CLN6 deletion was supported by three PacBio reads without any read supporting the reference allele (Fig. 1d). The findings showed that the deletion spanned 12,433 bp, including complete deletion of exon 1 of CLN6 (chr15: 68,518,038–68,530,471). We initially evaluated the relative DNA copy number using real-time quantitative PCR. By comparing the amplification signal of exon 1 (deleted) with that of exon 2 (not deleted), we confirmed homozygous and heterozygous deletion in the affected individuals (II-2 and II-3) and their parents (I-1 and I-2), respectively (Supplementary Fig. S4). To further validate the size and position of the deletion, we performed Southern blot analysis. A 23.9-kb EcoRV fragment of the wild type and an 11.5-kb deletion variant (23.9–12.4 kb) were detected, indicating the reliable size estimate obtained by PacBio reads (Fig. 1a). This deletion variant cosegregated with disease based on autosomal recessive inheritance. Alternative ATG codon usage is possible, but RT-PCR analysis revealed a lack of CLN6 expression, confirming that the variant was pathogenic (Supplementary Fig. S5).
Using positional information from PacBio PBSV calls, we designed unique long-range PCR primers amplifying the breakpoint junction (Fig. 2a). The junction fragment was amplified when using DNA from the other affected sibling (homozygous deletion) and the carrier parents (heterozygous deletion), but not from control DNA (Fig. 2b). Sanger sequencing of the junction fragment identified the Alu elements, AluSq2 and AluSc5, on both sides of the deletion, which is consistent with the PBSV calls (Fig. 2c).

Characterization of a 12.4-kb deletion at the CLN6 locus. a Deletion call (hatched box, chr15: 68,518,038–68,530,471) by PBSV application in SMRT Link v5.1.0 with three supportive PacBio subreads is shown. Forward and reverse complement strands are shown by pink and gray rectangles, respectively. Breakpoint PCR primers were designed to amplify 17,090- and ~4,697-bp products in wild-type and CLN6 deletion alleles, respectively. Repetitive sequences (SINE, LINE, LTR, DNA repeat elements, and simple repeats) in the RepeatMasker track of the UCSC genome browser are shown at the bottom. Arrows indicate the locations of recombination at Alu elements (AluSq2 and AluSc5). b An approximately 4.7-kb breakpoint junction fragment was amplified in PME family members (I-1, I-2, II-2, and II-3), but not in a control. c The location of the breakpoint junction was determined at nucleotide resolution. Sanger sequencing confirmed DNA recombination between AluSq2 (chr15: 68,518,029–68,518,332) and AluSc5 (chr15: 68,530,473–68,530,764). The breakpoint junction was identical in II-2 and their parents. The recombined sequence is highlighted in red. The gray reference Alu sequences denote mismatching bases compared with the sample sequence
All junction fragments from both patients and their parents were identical at the nucleotide level (Fig. 2c), raising the possibility that the paternal and maternal alleles were derived from an ancestral founder. Haplotype analysis on chromosome 15 using microsatellite markers and single-nucleotide polymorphisms (SNPs) showed that the CLN6 deletion was located at a ~440-kb homozygous block, suggesting a very old founder, if any (Supplementary Table S4). This 440-kb SNP haplotype (G_A_C_C_T_A_C_C_T_T) is common in East Asian populations, based on data from the 1000 Genomes Project (frequency of 0.1359 in East Asians, compared with 0.0008, 0.0187, 0.0276, and 0 in Africans, admixed Americans, South Asians, and Europeans, respectively) [18]. As such, heterozygous healthy carriers such as I-1 and I-2 may exist in the Japanese population and contribute to the risk of NCL. However, no heterozygote was found in 3,552 Japanese individuals in the Tohoku Medical Megabank Genome Reference Panel (https://jmorp.megabank.tohoku.ac.jp/201808/variants) [13, 19, 20]. We observed the read count by using deep WGS data after normalizing with total read counts for each individual and confirmed that no individuals had significantly low read counts among them. We also confirmed that no individuals had clipped reads around breakpoints in the top 10 low-read-count individuals. In another cohort, the ploidy of 384 Japanese subjects residing in and around Tokyo, Japan (Tokyo Healthy Control) were also estimated and classified into 0, 1, and 2 copies for CLN6 region (chr15: 68,518,038–68,530,471). All 384 samples were categorized into two copies, so we concluded that negative for the heterozygous deletion variant in the 384 Japanese samples. In summary, the deletion was not found in 3,936 (3,552 + 384) control individuals, indicating that it is indeed extremely rare.
Discussion
Technological innovations in genomic analysis, such as the development of microarrays and short-read next-generation sequencing (NGS), have improved our understanding of human genomic variants. PacBio SMRT sequencing using the Sequel system is now capable of reading >10-kb DNA and is an appropriate method to investigate the genetic cause in unresolved cases. Indeed, we and others have reported that long-read WGS is potentially useful for evaluating the known and novel repeat expansions in a genome [21, 22].
Using 6 × PacBio SMRT sequencing data, a total of 17,165 SVs (7,216 deletion and 9,949 insertion calls) were initially called in II-2. As is the case with short-read NGS analysis, variant filtering is beneficial for prioritizing pathogenic SVs. Notably, 79.9% (5765 of 7216 deletion calls) and 69.8% (6947 of 9949 insertion calls) of calls could be excluded from the candidates when comparing the SV calls from the three unrelated control individuals (Fig. 1b). Comparison to multiple control datasets was undoubtedly useful, as it was found that comparisons to one (control 1), two (controls 1 and 2), and three (controls 1, 2, and 3) control individuals resulted in 2,658, 1,830, and 1,451 unique deletion calls being left (Supplementary Fig. S3). For the purpose of reducing the number of candidate of diseases-causing mutations, it would be extremely beneficial if a public database for SVs were available, like The Exome Aggregation Consortium (ExAC) for SNVs [23].
WES data can be used for detecting CNVs. For example, XHMM evaluates normalized read-depth of WES data and detect smaller CNVs (<100 kb in size) than microarray [11]. Moreover, Nord method using the combined sliding-window read-depth and split-read analysis could precisely detect even a single exon deletion [12]. However both methods completely missed the homozygous CLN6 deletion (Supplementary Fig. S2), probably due to the scanty read coverage against CLN6 exon 1 with high GC content (77.6%) even in controls (Fig. 1d and Supplementary Table S5) [24]. By contrast, PacBio long reads showed uniform coverage (Fig. 1d), which improved the variant detection in GC-rich regions containing multiple repetitive elements. Even low coverage (6× on a genome-wide basis) with only three reads in the CLN6 region increased the power to investigate SVs that remained unrecognized by WES. Moreover, PacBio long reads correctly mapped the recombination junction at specific Alu elements (AluSq2 and AluSc5). Although there were only three reads, the long sequences of the reads conferred excellent mappability and ensured the robust detection of SVs.
Sequence similarity and physical distance in the genome are critical determinants of recombination events [25]. A recombination event was observed in this study between two Alu repetitive elements with 82% similarity, AluSq2 (chr15: 68,518,029–68,518,332) and AluSc5 (chr15: 68,530,182–68,530,472), which are located within 12.4 kb of each other. These parameters are ideal for recombination events and indeed Alu/Alu-mediated recombination (AAMR) occurred at the A-box of the Alu element (Supplementary Fig. S6), where recombination events occurred with high frequency [26]. Hence, the 12.4-kb deletion in this study had characteristics typical of being the result of an AAMR event. There are more than one million copies of Alu elements per haploid constituent of the human genome and AAMR is thought to contribute to a certain proportion of human genetic diseases [27, 28]. In fact, at least 219 AAMR events at the nucleotide level have been documented in the literature [26]. However, their exact frequency is unclear because appropriate analytical technologies are lacking. Nonetheless, approximately 75% of these events are below 57 kb in size, which is generally too small for microarray analysis, but too large for short-read sequencing technology [26]. Moreover, these regions are difficult to sequence using current short-read sequencing because of biased read coverage. Thus, these types of variants have been poorly investigated in the genome. Long-read sequencing has the potential to fully cover such intermediate-size variants by spanning repetitive sequences and directly using split-read mapping for SV detection rather than indirect inference, such as depth of coverage and the paired read approach [29]. Similar SVs as a result of AAMR events may underlie the genetic causes of currently-unexplained diseases and should be solved by developing long-read WGS.
Recent studies have suggested the advantage of PCR-free short-read WGS over WES to improve the coverage of GC-rich regions [30]. Short-read NGS is now routinely used with high accuracy and throughput for SNV, so SV analysis using short-read WGS is valuable when implemented in the current analytical pipeline, in particular with respect to the cost. This prompted us to investigate II-2 using short-read WGS after confirming the 12.4-kb deletion by long-read WGS. As previously reported, short-read WGS increased the coverage at GC-rich regions and enabled the detection of a 12.4-kb CLN6 deletion by manual inspection (Supplementary Fig. S7a). This deletion was also called by the BreakDancer program, which provides genome-wide SV calls from paired-end sequencing reads (Supplementary Fig. 7b) [14]. Both long- and short-read WGS should become alternative methods to the currently well-established WES. However, further studies are needed to compare these developing methods in much larger samples. The cost (sequencing and storage of data), computational burden, and handling of the large number of variants remain issues to consider for future clinical implementation.
References
Ramachandran N, Girard JM, Turnbull J, Minassian BA. The autosomal recessively inherited progressive myoclonus epilepsies and their genes. Epilepsia. 2009;50:29–36.
Muona M, Berkovic SF, Dibbens LM, Oliver KL, Maljevic S, Bayly MA, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet. 2015;47:39–46.
Huddleston J, Chaisson MJP, Steinberg KM, Warren W, Hoekzema K, Gordon D, et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017;27:677–85.
Chaisson MJP. Multi-platform discovery of haplotype-resolved structural variation in human genomes. bioRxiv. 2017; https://doi.org/10.1101/193144
Seo JS, Rhie A, Kim J, Lee S, Sohn MH, Kim CU, et al. De novo assembly and phasing of a Korean human genome. Nature. 2016;538:243–7.
Pendleton M, Sebra R, Pang AWC, Ummat A, Franzen O, Rausch T, et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat Methods. 2015;12:780–6.
Merker JD, Wenger AM, Sneddon T, Grove M, Zappala Z, Fresard L, et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet Med. 2018;20:159–63.
Reiner J, Pisani L, Qiao W, Singh R, Yang Y, Shi L, et al. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet-Biedl Syndrome 9 (BBS9) deletion. NPJ Genom Med. 2018;3:3.
Hoijer I, Tsai YC, Clark TA, Kotturi P, Dahl N, Stattin EL, et al. Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum Mutat. 2018;39:1262–72.
Mizuguchi T, Nakashima M, Kato M, Yamada K, Okanishi T, Ekhilevitch N, et al. PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J Hum Genet. 2017;62:525–9.
Miyatake S, Koshimizu E, Fujita A, Fukai R, Imagawa E, Ohba C, et al. Detecting copy-number variations in whole-exome sequencing data using the eXome Hidden Markov Model: an ‘exome-first’ approach. J Hum Genet. 2015;60:175–82.
Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genom. 2011;12:184.
Yamaguchi-Kabata Y, Nariai N, Kawai Y, Sato Y, Kojima K, Tateno M, et al. iJGVD: an integrative Japanese genome variation database based on whole-genome sequencing. Hum Genome Var. 2015;2:15050.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81.
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–8.
Wheeler RB, Sharp JD, Schultz RA, Joslin JM, Williams RE, Mole SE. The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am J Hum Genet. 2002;70:537–42.
Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, et al. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet. 2002;70:324–35.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–7.
Tadaka S, Saigusa D, Motoike IN, Inoue J, Aoki Y, Shirota M, et al. jMorp: Japanese Multi Omics Reference Panel. Nucleic Acids Res. 2018;46:D551–D7.
Kuriyama S, Yaegashi N, Nagami F, Arai T, Kawaguchi Y, Osumi N, et al. The Tohoku Medical Megabank Project: design and mission. J Epidemiol. 2016;26:493–511.
Zeng S, Zhang MY, Wang XJ, Hu ZM, Li JC, Li N, et al. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. Journal of medical genetics. 2018; e-pub ahead of print 2018; https://doi.org/10.1136/jmedgenet-2018-105484
Mizuguchi T, Toyota T, Adachi H, Miyake N, Matsumoto N, Miyatake S. Detecting a long insertion variant in SAMD12 by SMRT sequencing: implications of long-read whole-genome sequencing for repeat expansion diseases. J Hum Genet 2018; e-pub ahead of print 2018; https://doi.org/10.1038/s10038-018-0551-7
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–9.
Wang RW, Lee CS, Haber JE. Position effects influencing intrachromosomal repair of a double-strand break in budding yeast. PLoS ONE. 2017;12:e0180994.
Song X, Beck CR, Du R, Campbell IM, Coban-Akdemir Z, Gu S, et al. Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 2018;28:1228–42.
Kim S, Cho CS, Han K, Lee J. Structural variation of Alu element and human disease. Genom Inform. 2016;14:70–7.
Price AL, Eskin E, Pevzner PA. Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res. 2004;14:2245–52.
Pirooznia M, Goes FS, Zandi PP. Whole-genome CNV analysis: advances in computational approaches. Front Genet. 2015;6:138
Carss KJ, Arno G, Erwood M, Stephens J, Sanchis-Juan A, Hull S, et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet. 2017;100:75–90.
Acknowledgements
We would like to thank all the subjects for participating in this study. We also thank N. Watanabe, T. Miyama, M. Sato, and K. Takabe for their technical assistance. We are also grateful to Edanz (www.edanzediting.co.jp) for editing the English text of a draft of this manuscript. This work was supported by AMED under grant numbers JP18ek0109280, JP18dm0107090, JP18ek0109301, JP18ek0109348, JP18km045205, and JP18kk020500; JSPS KAKENHI under grant numbers JP17K15630, JP17H01539, JP17K10080, and JP17K15630; the Ministry of Health, Labour, and Welfare; and Takeda Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Mizuguchi, T., Suzuki, T., Abe, C. et al. A 12-kb structural variation in progressive myoclonic epilepsy was newly identified by long-read whole-genome sequencing. J Hum Genet 64, 359–368 (2019). https://doi.org/10.1038/s10038-019-0569-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0569-5
This article is cited by
-
Targeted long-read sequencing identified a causal structural variant in X-linked nephrogenic diabetes insipidus
BMC Medical Genomics (2024)
-
Advances in sequencing technologies for amyotrophic lateral sclerosis research
Molecular Neurodegeneration (2023)
-
Long-read sequencing revealing intragenic deletions in exome-negative spastic paraplegias
Journal of Human Genetics (2023)
-
Deciphering the exact breakpoints of structural variations using long sequencing reads with DeBreak
Nature Communications (2023)
-
SvAnna: efficient and accurate pathogenicity prediction of coding and regulatory structural variants in long-read genome sequencing
Genome Medicine (2022)
