
Abstract
Hereditary leukonychia (also known as porcelain nails or white nails) is a genetic disorder. It may exist as an isolated feature or associated with other cutaneous or systemic disorders. Although a number of genes have been described to cause leukonychia, still the underlying genetic etiologies of many cases remain unknown. Here, we report a Pakistani family presenting leukonychia and koilonychia nails in mother and five of her kids. All the affected individuals had white to pale nails in appearance exhibiting complete and partial leukonychia, respectively. Similarly, nails of finger and toe appeared brittle and concave, showing the characteristics features of koilonychia. Whole exome sequencing and subsequent Sanger sequencing identified a pathogenic novel missense mutation (c.1390G>A, p.Glu464Lys) in PLCD1, co-segregating with the disorder in an autosomal dominant pattern. In silico prediction tools supported the pathogenicity of the identified mutation. Literature review determined that mutations in PLCD1 only cause leukonychia. Therefore, our findings add another pathogenic variant to the PLCD1 mutation pool causing leukonychia that would help to understand the underlying molecular mechanism.
Similar content being viewed by others


Introduction
Leukonychia, or white opacity of nails, is a common disease in humans for decades with both acquired and hereditary conditions [1,2,3,4,5]. The acquired leukonychia is mainly related to the number of factors such as drugs, injury, infections, and the deficiency of calcium [2,3,4,5]. Hereditary leukonychia may present as an isolated feature or associated with other integumentary or systematic abnormalities [5,6,7,8,9,10,11]. After the first description of leukonychia by Gutman, several other studies have been conducted to investigate the underlying molecular mechanism [12, 13]. Mutations in Gap junction alpha-1 (GJA1) and the Phospholipase C, delta-1 (PLCD1) genes were described to cause isolated leukonychia [5,6,7,8]. Farooq et al. and Mir et al. found two different autosomal recessive mutations in PLCD1 with isolated leukonychia in Jordanian and Pakistani families, respectively [5, 8]. Kiuru et al. also reported PLCD1 mutations, segregating in autosomal recessive and autosomal dominant manners with isolated leukonychia in four Pakistani families. In addition, they determined the localization of PLCD1 in fetal nails and also detected its mRNA in foreskin, keratinocytes, and fibroblast, suggesting a vital role for PLCD1 [7]. Noteworthy, PLCD1 is the enzyme required for calcium (Ca2+) signal transduction in a number of tissues and its importance in the manifestation of hereditary leukonychia has recently been investigated [14,15,16,17]. Nomikos et al. determined significant divergent enzymatic activities for leukonychia-linked mutant PLCD1, validating that mutations in PLCD1 cause isolated leukonychia [17].
Leukonychia in syndromic condition has also been reported in a number of studies [11, 18, 19]. A familial Bart–Pumphrey Syndrome-associated leukonychia was described, co-segregating with autosomal recessive missense mutation in Gap junction beta-2 (GJB2) gene [18]. Similarly, an autosomal dominant missense mutation in Desmoplakin (DSP) gene instigated leukonychia with Carvajal/Naxos syndrome [11]. Leukonychia was also accompanied with familial peeling skin syndrome, caused by autosomal recessive loss of function mutations in Calpastatin (CAST) gene [19]. To date, few cases of sporadic leukonychia with koilonychia, characterized by white, thin, and spoon-shaped nails have been reported [20,21,22]. However, the genetic mechanisms that lead to leukonychia with koilonychia remain unrevealed.
The current study was aimed to determine the genetic etiology of leukonychia and koilonychia in a Pakistani family. Whole exome sequencing (WES) data analysis narrowed-down the five potential candidates. Subsequent Sanger sequencing revealed an autosomal dominant variant PLCD1 (c.1390G>A, p.Glu464Lys ) co-segregated with leukonychia. Thus, our findings further validated that pathogenic mutation in PLCD1 causes leukonychia.
Materials and methods
Subjects
A Pakistani family comprised of parents (II-1 and II-2), their five sons (III-1, III-2, III-3, III-4, and III-5), and three daughters (III-6, III-7, and III-8) was recruited for this study. Written and informed consent form, approved by the Institutional Ethical Committee of University of Science and Technology of China (USTC), was obtained from all the affected and control family members.
DNA extraction
Peripheral blood samples were collected in ethylene diamine tetra acetic acid (EDTA) tubes from all the participants through 5 ml sterile syringes manufactured by Becton Dickinson (BD) suppliers, Pakistan, using standard protocol approved by World Health Organization (WHO) [23]. Genomic DNA was extracted from the collected blood samples of all the participants using FlexiGene DNA kit, according to the manufacturer’s manual (QIAGEN, 51206). The DNA concentration was tested through OD260 (Thermo Fisher NanoDrop 1000 spectrophotometer) followed by agarose gel electrophoresis and ethidium bromide staining to evaluate the quality.
Whole exome sequencing and variants filtration
For exome capturing, Agilent SureSelect Human All Exon v5 Kit (Agilent, Santa Clara, CA, USA) was used. Captured libraries were constructed for family members, II-1, II-2, III-3, III-4, and III-5, as instructed by the manufacturer. Sequencing was carried out by Hiseq2000 platform (Illumina, San Diego, CA, USA). Clean reads were mapped to the human reference genome (hg19) by Burrows–Wheeler Alignment tool [24]. The variants were discovered and annotated by Genome Analysis Toolkit and ANNOVAR, respectively. A schematic representation of the filtering process is described in Supplementary Fig. 1. First, variants that co-segregated with the disease in autosomal recessive, autosomal dominant, or X-linked dominant were kept for further analysis. Second, variants with minor allele frequencies (MAF) greater than 5% in 1000 Genomes Project (ftp://ftp.1000 genomes.ebi.ac.uk/vol1/ftp) were eliminated. Next, variants having mutations in untranslated regions or introns were also excluded. For remaining variants, we did literature survey. Subsequently, Sanger sequencing was performed to verify the selected variants in all the available family members (Table S1, S2). Sequences of primers are listed in Table S3.
In silico analysis
The conservation of affected amino acids in the present study and from previous reports was predicted using phylogenetic software MEGA6 version 6.0 (Fig. 2e, Table S5) [25]. For predicting the effect of the mutation identified in the study, on protein structure, the protein sequences of both wild-type (WT) and mutant PLCD1 (P51178) were input separately into SWISS-MODEL tool (https://swissmodel.expasy.org/). The blast resulted in seven models with different sequence identity for both WT and mutant proteins. The models with 90.6% sequence identity were considered for final results (Fig. 2f). The functional pathogenicity of the mutation was assessed via 12 deleterious prediction tools in ANNOVAR (Table S4).
Results
Clinical features
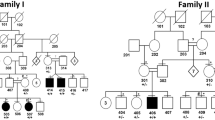
In this part of the study, we recruited a Pakistani family, displaying leukonychia and koilonychia nails from a remote region of Pakistan (Fig. 1a). The family comprised of parents, five sons, and three daughters. The affected individuals (II-2, III-1, III-2, III-3, III-5, and III-7) of the two-generation family presented leukonychia with koilonychia since birth (Fig. 1b). The appearance of the finger nail plates was chalky and white, indicating leukonychia. However, toe nails had partial leukonychia with yellowish coloration in the distal parts of the nails. In addition, both finger and toe nails had concave or spoon-shaped appearance, a condition known as koilonychia. The surfaces of finger and toe nails appeared smooth and rough, respectively. No other abnormalities of the skin or its appendages were observed in the affected individuals.

A family presents leukonychia with koilonychia. a. Pedigree of the family presenting leukonychia and koilonychia. Squares and circles represent males and females, respectively. Filled symbols represent individuals affected from leukonychia and koilonychia, clear symbols represent normal individuals, and diamonds indicate multiple individuals without sex information. b–g. Representative images of finger and toe nails. b, c. Normal finger and toe nails from an individual (III-4). d. Typical severe koilonychia nails from mother (II-2) fingers. Flattened plate with everted upward and concave edges are prominent, giving rise to the characteristic “spoon” shape. The nails are thin and entire whitening including lunula is observed. e. Mother (II-2) toe nails displayed incomplete leukonychia with yellowish coloration in the distal part of the plates of all nails. Chalky white coloration can also be observed in the few nails. f. Finger nails from the individual (III-5) exhibited thin and spoon-shaped. Nail plates are flattened while edges are everted upwards with complete leukonychia. g. Toe nails from individual III-5: thin, flattened, and yellowish discoloration of all the nails is observed. The field size of third and fourth toe nails are also reduced
Identification of autosomal dominant missense mutation (c.1390G>A, p.Glu464Lys) in PLCD1
To find the causative variant(s) underlying leukonychia in the subject family, we performed WES for father (II-1), mother (II-2), and three brothers (III-3, III-4, and III-5) (Fig. 1a). Given that the phenotypes are present in two generations in the family, three possible inheritance patterns: autosomal recessive, autosomal dominant, and X-linked dominant were assumed. The causative mutations were identified by filtering the data according to (i) the frequency of the mutations (MAF < 0.05), (ii) mutations that are potentially protein damaging, and (iii) an extensive literature survey. The validation of filtered candidate variants (Table S1) was performed by Sanger sequencing. Consequently, the result revealed that only PLCD1 variant co-segregated with the disorder in an autosomal dominant pattern in the family (Fig. 2a, S2, and Table S2).

Identification and predicted effect of the PLCD1 missense mutation. a. Representative chromatograms showing normal and affected individuals. NM, Normal; A, adenine; C, cytosine; G, guanine; T, thymine. b. The mutation in PLCD1 occurs in exon 11, which causes G to A substitution corresponding to genomic DNA (gDNA) position at nucleotide (nt) 20054 (NM_006225). c. In transcript (mRNA), the mutated site is present at nt 1390. d. Diagram elaborates the PLCD1 protein domains. The protein comprises an N-terminal pleckstrin homology (PH) domain, two EF hand motifs, catalytic domain X, catalytic domain Y, and a C-terminal domain C2 (pdb code: 1DJG). In mutant protein, lysine replaces the glutamic acid at position 464 (P51178), which lies in XY linker between X and Y domains of the protein. e. Sequence alignment shows that glutamic acid (the affected amino acid) indicated with an arrow, is the member of highly conserved residues group present in XY linker of the protein (highlighted in the box). f. SWISS-MODEL tool predicts the structural changes caused by the mutation, indicated in the red box. Red arrows in panels (a–d) indicate mutation site
The c.1390G>A caused the glutamic acid replacement with lysine at position 464 in PLCD1 protein, p.Glu464Lys (Fig. 2d). The sequence alignment revealed that the affected amino acid is the component of highly conserved group of residues across the species, localizing in the XY linker of protein (Fig. 2e). The XY linker of the protein (Fig. 2d) contains Ca2+-binding site, thus implying that this mutation could impair Ca2+-dependent enzymatic activity [26]. The consequence of mutation on protein structure was checked with SWISS-MODEL tool (https://swissmodel.expasy.org/). Resultantly, an altered structure of the mutant protein was predicted (Fig. 2f), displaying that the identified mutation caused structural pathogenicity. Further, 12 deleterious prediction software were checked, seven of which predicted lethality for the mutation (Table S4). Altogether, these results elaborated that the PLCD1 mutation (c.1390G>A, p.Glu464Lys), segregating in an autosomal dominant manner, was pathogenic and strongly associated with nail disorder in the family. Interestingly, the previous studies determined only leukonychia with PLCD1 mutations [5, 7, 8].
Discussion
In the current study, we identified a novel missense mutation in PLCD1 resulting in leukonychia (Fig. 1). Comprehensively, the six family members (II-2, III-1, III-2, III-3, III-5, and III-7) displayed leukonychia (white and pale appearance) with koilonychia (concave and everted edges) nails [20]. Our WES data analysis and Sanger sequencing identified a novel PLCD1 mutation (c.1390G>A, p.Glu464Lys) causing leukonychia. Remarkably, the PLCD1-associated leukonychia phenotype is consistent with the previous reports (Fig. S3, Table S5) [5, 7, 8].
PLCD1 maps to the short arm of chromosome 3 (3p22.2), consists of 15 exons, and spans a genomic region of 22.265 kb on the minus strand [9]. The gene encodes two isoforms of 777 and 756 amino acids. To date, six pathogenic mutations have been described in PLCD1 gene causing leukonychia (Fig. S3, Table S5). These could broadly be categorized into pre-mature termination codon (PTC) and missense mutations. Our screening of the subject family added another novel variant (c.1390G>A, p.Glu464Lys) to the PLCD1 mutation pool (Fig. 2, S2). Noteworthy, all the missense mutations including the one identified in this study, inherited in an autosomal dominant pattern. By contrast, all the previously reported PTC followed the autosomal recessive mode of inheritance. This happens occasionally that recessive and dominant mutations in the same gene produce similar phenotype [7]. It could possibly be explained by two phenomena: the first, for normal function, some genes and/or pathways require only one WT copy whereas, others require two copies as proposed by Smirnov and Cheung [27]. Kiuru et al. suggested the other possibility; the missense mutations may imply a dominant negative effect on the WT allele with complete loss of function [7]. Indeed, it would be interesting to investigate which one of these phenomena is related to the PLCD1 autosomal dominant mutations in leukonychia.
The PLCD1 belongs to the ubiquitous family of phospholipase C (PLC) enzymes. The protein contains an N-terminal pleckstrin homology (PH) domain, a pair of EF hand motifs, the X and Y catalytic domains, and a C2 domain at C-terminal. Both X and Y catalytic domains are linked by a Ca2+-binding site containing auto-inhibitory, flexible, and unstructured XY linker (Fig. 2) [26, 28, 29]. Recently, the leukonychia-linked PLCD1 mutations (p.Cys209Arg, p.Ala574Thr, and p.Ser740ArgfsX19) lying on different domains were assessed for the effect on the protein’s biophysical and biochemical properties (Fig. S3, Table S5). The p.Cys209Arg mutation, residing in EF hand1 motif resulted in slightly increased thermal stability of the enzyme. Contrastingly, the p.Ala574Thr mutant protein (mutation present in Y domain) demonstrated obvious upsurge of catalytic activity. Whereas, the p.Ser740ArgfsX19 mutation, which localized on C2 domain, caused deleterious effects on protein stability and activity [17]. Comparing with them, our identified mutation is located in XY linker (highly conserved region; Fig. 2e), which gave rise to an acidic glutamate replacement with basic lysine residue (Fig. 2). As predicted, this mutation caused structural changes (Fig. 2); thus, it would be fascinating to determine whether it indeed affects the Ca2+-dependent enzymatic activity of PLCD1.
Although thePLCD1 expression has been shown in nail matrix, the mechanism that how the PLCD1 disruption causes leukonychia, is yet unknown. Notably, the familial autosomal dominant leukonychia was previously mapped on chromosome region: 12q13, by linkage analysis that contains genes for type II (basic) cytokeratins and hard keratins [30]. This suggests the requirement of PLCD1 for keratins expression; the disruption of which leads to leukonychia. The proposed hypothesis is further supported by Nakamura et al. who demonstrated the FOXN1 upstream regulation of PLCD1 [31]. Importantly, FOXN1 acts as a regulator for the hard keratin genes transcription and proved to be essential for nail differentiation [32, 33]. Kiuru et al. determined the expression of PLCD1 in follicle matrix and hair bed. Contrary to them, we, as well as previous PLCD1 studies did not find any hair abnormality in the affected individuals [7]. This may possibly be explained by its compensation with other members of phosphoinositide-specific phospholipase C family [7].
Although several genes have been described to cause leukonychia, PLCD1 is the most extensively studied gene. All the previously reported PLCD1 mutations did not exhibit any other associated phenotype [5, 7, 8]. Interestingly, the family reported here, also presented koilonychia both in finger and toe nails along with leukonychia. A number of cases of leukonychia with koilonychia are described but the genetic cause has not been determined so far. Studies on multiple familial cases are required to understand the genetic etiology for the co-occurring of the aforementioned phenotypes. Moreover, the genes/loci for isolated koilonychia and the underlying molecular mechanism of leukonychia caused by PLCD1 mutations are yet unrevealed. Filling these gaps certainly would help to understand the association of leukonychia with koilonychia.
In summary, it is concluded that the novel mutation c.1390G>A, p.Glu464Lys in PLCD1 causes leukonychia,thus, further fortified the pathogenicity of PLCD1 mutations.
References
Assadi F. Leukonychia associated with increased blood strontium level. Clin Pediatr (Phila). 2005;44:531–3.
Pathipati AS, Ko JM, Yost JM. A case and review of congenital leukonychia. Dermatol Online J. 2016;22(10):6.
Tosti A, Iorizzo M, Piraccini BM, Starace M. The nail in systemic diseases. Dermatol Clin. 2006;24:341–7.
Yoruk A, Yukselgungor H. Chemotherapy induced transverse leukonychia in children. Int J Dermatol. 2003;42:468–9.
Mir H, Khan S, Arif MS, Ali G, Wali A, Ansar M, et al. Mutations in the gene phospholipase C, delta-1 (PLCD1) underlying hereditary leukonychia. Eur J Dermatol. 2012;22:736–9.
Wang H, Cao X, Lin Z, Lee M, Jia X, Ren Y, et al. Exome sequencing reveals mutation in GJA1 as a cause of keratoderma–hypotrichosis–leukonychia totalis syndrome. Hum Mol Genet. 2015;24:243.
Kiuru M, Kurban M, Itoh M, Petukhova L, Shimomura Y, Wajid M, et al. Hereditary leukonychia, or porcelain nails, resulting from mutations in PLCD1. Am J Hum Genet. 2011;88:839–44.
Farooq M, Kurban M, Abbas O, Obeidat O, Fujikawa H, Kibbi AG, et al. A novel mutation in the PLCD1 gene, which leads to an aberrant splicing event, underlies autosomal recessive leuconychia. Br J Dermatol. 2012;167:946–9.
Ishikawa S, Takahashi T, Ogawa M, Nakamura Y. Genomic structure of the human PLCD1 (phospholipase C delta 1) locus on 3p22->p21.3. Cytogenet Cell Genet. 1997;78:58–60.
Richard G, Brown N, Ishida-Yamamoto A, Krol A. Expanding the phenotypic spectrum of Cx26 disorders: Bart–Pumphrey syndrome is caused by a novel missense mutation in GJB2. J Invest Dermatol. 2004;123:856–63.
Boule S, Fressart V, Laux D, Mallet A, Simon F, de Groote P, et al. Expanding the phenotype associated with a desmoplakin dominant mutation: Carvajal/Naxos syndrome associated with leukonychia and oligodontia. Int J Cardiol. 2012;161:50–2.
Kruse WT, Cawley EP, Cotterman CW. Hereditary leuconychia totalis. J Invest Dermatol. 1951;17:135–40.
Le CY, Steff M, Croue A, Filmon R, Verret JL, Le CC. Hereditary leukonychia totalis, acanthosis-nigricans-like lesions and hair dysplasia: a new syndrome? Eur J Med Genet. 2009;52:229–33.
Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988;334:661.
Suh P, Park J, Manzoli L, Cocco L, Peak JC, Katan M, et al. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 2008;41:415–34.
Kadamur G, Ross EM. Mammalian phospholipase C. Annu Rev Physiol. 2013;75:127–54.
Nomikos M, Thanassoulas A, Beck K, Theodoridou M, Kew J, Kashir J, et al. Mutations in PLCδ1 associated with hereditary leukonychia display divergent PIP2 hydrolytic function. FEBS J. 2016;283:4502–14.
Krol A. Expanding the phenotypic spectrum of Cx26 disorders: Bart–Pumphrey syndrome is caused by a novel missense mutation in GJB2. J Invest Dermatol. 2004;123:856.
Lin Z, Zhao J, Nitoiu D, Scott CA, Plagnol V, Smith FJ, et al. Loss-of-function mutations in CAST cause peeling skin, leukonychia, acral punctate keratoses, cheilitis, and knuckle pads. Am J Hum Genet. 2015;96:440–7.
Kwon NH, Kim JE, Cho BK, Jeong EG, Park HJ. Sporadic congenital leukonychia with koilonychia. Int J Dermatol. 2012;51:1400–2.
Brown PJ, Padgett JK, English J 3rd. Sporadic congenital leukonychia with partial phenotype expression. Cutis. 2000;66:117–9.
Baran R, Martinon J. Association of total leukonychia and koilonychia. Bull Soc Fr Dermatol Syphiligr. 1967;74:571.
World Health Organization. WHO guidelines on drawing blood: best practices in phlebotomy. 2010.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Essen L-O, Perisic O, Cheung R, Katan M, Williams RL. Crystal structure of a mammalian phosphoinositide-specific phospholipase Cδ. Nature. 1996;380:595–602.
Smirnov DA, Cheung VG. ATM gene mutations result in both recessive and dominant expression phenotypes of genes and microRNAs. Am J Human Genet. 2008;83:243–53.
Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell . 1995;83:1037–46.
Essen L-O, Perisic O, Lynch DE, Katan M, Williams RL. A ternary metal binding site in the C2 domain of phosphoinositide-specific phospholipase C-δ1. Biochemistry. 1997;36:2753–62.
Norgett E, Wolf F, Balme B, Leigh I, Perrot H, Kelsell D, et al. Hereditary ‘white nails’: a genetic and structural study. Br J Dermatol. 2004;151:65–72.
Nakamura Y, Ichinohe M, Hirata M, Matsuura H, Fujiwara T, Igarashi T, et al. Phospholipase C-δ1 is an essential molecule downstream of Foxn1, the gene responsible for the nude mutation, in normal hair development. FASEB J. 2008;22:841–9.
Schlake T, Schorpp M, Maul‐Pavicic A, Malashenko A, Boehm T. Forkhead/winged‐helix transcription factor Whn regulates hair keratin gene expression: molecular analysis of the nude skin phenotype. Dev Dyn. 2000;217:368–76.
Mecklenburg L, Paus R, Halata Z, Bechtold LS, Fleckman P, Sundberg JP. FOXN1 is critical for onycholemmal terminal differentiation in nude (Foxn1nu) mice. J Invest Dermatol. 2004;123:1001–11.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31501202, 31771668, and 31501199), the Fundamental Research Funds for the Central Universities (WK2340000069), and CAS-TWAS President’s PhD Fellowship Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, T., Khan, M., Yousaf, A. et al. Whole exome sequencing identifies a novel dominant missense mutation underlying leukonychia in a Pakistani family. J Hum Genet 63, 1071–1076 (2018). https://doi.org/10.1038/s10038-018-0491-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0491-2
