
Abstract
Autosomal recessive cerebellar ataxias (ARCAs) are clinically and genetically heterogeneous neurological disorders. Through whole-exome sequencing of Japanese ARCA patients, we identified three index patients from unrelated families who had biallelic mutations in ERCC4. ERCC4 mutations have been known to cause xeroderma pigmentosum complementation group F (XP-F), Cockayne syndrome, and Fanconi anemia phenotypes. All of the patients described here showed very slowly progressive cerebellar ataxia and cognitive decline with choreiform involuntary movement, with young adolescent or midlife onset. Brain MRI demonstrated atrophy that included the cerebellum and brainstem. Of note, cutaneous symptoms were very mild: there was normal to very mild pigmentation of exposed skin areas and/or an equivocal history of pathological sunburn. However, an unscheduled DNA synthesis assay of fibroblasts from the patient revealed impairment of nucleotide excision repair. A similar phenotype was very recently recognized through genetic analysis of Caucasian cerebellar ataxia patients. Our results confirm that biallelic ERCC4 mutations cause a cerebellar ataxia-dominant phenotype with mild cutaneous symptoms, possibly accounting for a high proportion of the genetic causes of ARCA in Japan, where XP-F is prevalent.
Similar content being viewed by others
Introduction
Cerebellar ataxia (CA) is a clinically and genetically heterogeneous neurological disorder. Recent advances in whole-exome sequencing (WES) have contributed significantly not only to the discovery of novel mutated genes [1], but also to the identification of genetic causes of CA, even in unrecognized phenotypes [2], and to the comprehensive genetic diagnosis of CA patients [3, 4].
In this study, we identified three index Japanese patients from unrelated families who exhibited young adolescent- or midlife-onset CA with biallelic single nucleotide variants (SNVs) of ERCC4; two of these patients were found through WES analysis of 55 unrelated Japanese patients with autosomal recessive or sporadic CA and spastic paraplegia (SPG) of unknown cause. All four patients described in this study showed similar phenotypic features of CA, with choreiform involuntary movements and brain atrophy that included the cerebellum and brainstem. Although recessive loss-of-function mutations in ERCC4 have been known to cause xeroderma pigmentosum complementation group F (XP-F) [5], Cockayne syndrome (XP-F/CS) [6], and Fanconi anemia (XP-F/CS/FA) phenotypes [6, 7], our patients lacked subjective skin symptoms as well as morphological, developmental, and hematological abnormalities. However, a careful history and examination revealed mild skin findings consistent with xeroderma pigmentosum (XP). Furthermore, we conducted an unscheduled DNA synthesis (UDS) assay of fibroblasts from the patient and revealed that removal of ultraviolet (UV)-induced DNA damage was impaired. While previous reports have described a low incidence of XP-F with neurological involvement [8], very recently a CA-dominant phenotype was described as a rare cause of autosomal recessive CA (ARCA) [9]. Our study also indicates that biallelic ERCC4 mutations cause a CA-dominant phenotype with mild symptoms of XP, and may account for a high proportion of the genetic causes of ARCA in Japan.
Materials and methods
This study included 55 unrelated Japanese patients with autosomal recessive or sporadic CA (n = 23) or SPG (n = 32), with no mutations in genes known to cause either condition. Clinical information, radiological images, and blood samples were obtained from the patients and their family members after written informed consent was provided. Experimental protocols were approved by the Institutional Review Board of Yokohama City University Faculty of Medicine. All experiments were performed in accordance with institutional guidelines.
To identify pathogenic mutations, we performed WES on all 55 patients. Genomic DNA was processed using the SureSelect Human All Exon Kit v3~v5 (Agilent Technologies, Santa Clara, CA), and sequenced on an Illumina HiSeq 2000/2500 with 101-bp paired-end reads. Alignment, variant calling, and annotation were performed using Novoalign (http://www.novocraft.com/), Picard (http://picard.sourceforge.net/), Genome Analysis Toolkit (https://www.broadinstitute.org/gatk/index.php), and ANNOVAR (http://www.openbioinformatics.org/annovar/), as described previously [10]. For each patient, SNVs and insertion/deletions (indels) were extracted based on information regarding rare variants with a minor allele frequency <1% in dbSNP137, located in exons or splice sites (within 10 bp of the boundaries), and with a frequency <1% in exome data from 575 “in house” Japanese controls. Among these, we included frameshift, nonsense, or missense SNVs that were either homozygous or potentially compound heterozygous and were predicted to be disruptive to protein function when analyzed with Polyphen2 [11] and SIFT [12]. We carefully checked variants in common genes shared in two or more patients.
UDS assays were performed on cultured skin fibroblasts obtained from a normal control subject, patient 1, and a patient with XP-A, as described previously [13]. Briefly, cells were plated in multiwell plates (96-well) and UV-irradiated (20 J/m2 of 254 nm UVC). Cells were incubated for 4 h in medium supplemented with 5 μM 5-ethynyl-2′-deoxyuridine (EdU) after UV irradiation. Incorporated EdU was conjugated with Alexa Fluor 488 azide and nuclei were stained with DAPI. Fluorescent (Alexa Fluor 488 azide and DAPI) images were obtained and data were analyzed using the ArrayScan VTI (Thermo Fisher Scientific, Waltham, MA).
The minimal erythema dose (MED) for UVB was measured using UV source Dermaray-100 (Toshiba Medical, Tokyo, Japan), with FL20S·E-30/DMR lump for patient 1, 2 and 3, or Dermaray-320 (Toshiba Medical) with FL32S·E-12/DMR lump for patient 4.
Results
ERCC4 mutations in CA patients
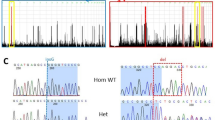
We found a homozygous missense ERCC4 variant in one patient (patient 3) and potentially compound heterozygous variants of ERCC4 in another patient (patient 1) with a similar CA phenotype, both from the Yokohama City University cohort (n = 55). Patient 2, who was the sister of patient 1 and whose primary symptom was secondary amenorrhea, had the same variants in ERCC4. We also found another homozygous missense ERCC4 variant in one patient (patient 4) who was included in the additional CA cohort (n = 11). Although biallelic loss-of-function mutations in ERCC4 have been known to cause XP-F, XP-F/CS, and XP-F/CS/FA phenotypes, the four patients described here did not demonstrate skin symptoms, developmental abnormalities, or anemia as critical clinical features. Sanger sequencing of the patients and the parents of patients 1 and 2 confirmed that the mutations were either homozygous or compound heterozygous, and were thus compatible with an autosomal recessive model (Fig. 1a). Patients 1, 3, and 4 were negative for the genetic alterations associated with spinocerebellar ataxia (SCA) 1, SCA2, SCA3, SCA6, SCA7, SCA12, SCA17, SCA 31, and dentatorubral-pallidoluysian atrophy. Among the detected variants, c.2395C>T [p.Arg799Trp] in ERCC4, which was found in patients 1, 2, and 4, has been reported as a pathogenic mutation in XP-F [14], while c.577del [p.Trp193Glyfs*6], found in patients 1 and 2, results in protein truncation, indicating that these variants were definitively pathological. In patient 3, c.715G>C [p.Glu239Gln] in ERCC4 occurred in a highly conserved amino acid (Fig. 1b) and has been consistently predicted to be disruptive to protein function when analyzed with multiple tools, including Polyphen2, SIFT, and Mutation Taster [11, 12, 15]. The minor allele frequency (MAF) of the c.2395C>T mutation was 0.0052 (3/575) in our “in-house” Japanese control exome, and 0.00051 (62/121398) in the Exome Aggregation Consortium (ExAc) database (http://exac.broadinstitute.org/), respectively. The c.577del and c.715G>C mutations were not found in our “in-house” Japanese control exome or in the ExAc database. Patients 1, 3, and 4 did not have any homozygous or potentially compound heterozygous pathological mutations in genes related to other ARCAs or to autosomal recessive hereditary spastic paraplegias.

Clinical manifestations of patients with ERCC4 mutations. a Pictures of patients’ exposed skin areas and their family pedigrees (Patients 1–4) are shown, as well as an electropherogram from Sanger sequencing. The arrow indicates the index patient in each family. Patients 3 and 4 were children born to first-cousin parents. The father of patients 1 and 2 has retinitis pigmentosa, their sister died from glioblastoma, and two sisters of patient 4 died from cancer. b Two missense mutations occurred at evolutionarily conserved amino acids
Clinical presentations
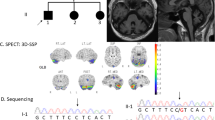
The patients’ clinical presentations are summarized in Table 1. Early developmental milestones were normal in all cases. Age of disease onset ranged from 25 to 43 years. Initial symptoms were gait unsteadiness, dysarthria, and/or involuntary movements. In addition, initially mild cognitive decline gradually worsened. Neurological symptoms were very slowly progressive. On neurological examination, all patients showed impairment of smooth eye pursuit, dysarthria, and limb and truncal ataxia. Three patients showed choreiform involuntary movements of the neck, tongue, and upper and lower extremities. Patient 1 exhibited very mild choreiform movements in her upper extremities. All patients’ tendon reflexes were slightly increased. Laboratory tests were essentially normal, including blood count. A nerve conduction study disclosed no abnormalities. Brain MRI in all patients showed brain atrophy, especially in the cerebellum and brainstem, along with dilation of the ventricles. The brain atrophy was progressive in an age-dependent manner (Fig. 2). The electroencephalogram (EEG) of patient 3, who had a seizure history, showed abnormal spikes on photic stimulation. None of the patients experienced problems due to their cutaneous symptoms. During re-evaluation, a history of severe sunburn in childhood was confirmed in three patients, and normal to mildly abnormal solar lentigines and freckles on exposed skin areas were confirmed in all patients (Fig. 1a). The MED for UVB showed slight hypersensitivity to UVB in patients 2, 3, and 4 (Table 1). A UDS assay of fibroblasts from patient 1 revealed decreased level of UDS (Fig. 3), indicating impaired function of the nucleotide excision repair (NER) system.

Brain MRI of patients. Axial sections of T2-weighted images and sagittal sections of T1-weighted images are shown. Left panels show MRI results of Patient 1 at 31 years of age (upper) and at 37 years of age (lower). Right panels show MRI results of Patient 3 at 62 years of age (upper) and of Patient 4 at 66 years of age (lower). Age-dependent atrophy of the brain, including the cerebellum and brainstem, is observed

UDS assay of fibroblasts. Fibroblasts from patient 1 show reduced UDS level compared to control cells (triplicate; filled bars, 20 J/m2 UVC; open bars, no UVC)
Discussion
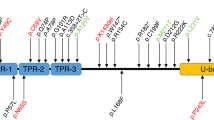
ERCC4 encodes XPF, a component of ERCC1/XPF endonuclease, which is mainly involved in incision of DNA damages in NER and interstrand crosslink (ICL) repair, and its mutations have been found in XP-F, XP-F/CS, and XP-F/CS/FA phenotypes [5,6,7]. A previous study demonstrated that impairment of global genome NER (GG-NER), transcription-coupled NER (TC-NER), and ICL repair were related to the XP, CS, and FA phenotypes, respectively [6]. Mutations of ERCC4 variably affect these repair functions and cause diverse phenotypes [6, 7]. Neurological symptoms have been identified in several XP-F patients with ERCC4 mutations [14, 16], but their incidence seems to be very low [8]. In accordance with the neurological presentation of XP-F patients described in previous reports [14, 16], patients in this study showed cognitive deterioration, CA, choreiform involuntary movements, and brain atrophy. Of note, none of the patients considered their skin manifestations to be symptoms of a disease, and indeed, the nature of these lesions remained unclear until genetic diagnosis. Such skin findings are likely to be missed unless the clinician has a low threshold for diagnostic evaluation. To confirm that the skin symptoms were due to a pathological condition, we conducted a UDS assay to evaluate NER function in patient 1, and revealed that this CA-dominant XP-F patient showed decreased level of UDS. Furthermore, three of the four patients were hypersensitive to UVB, indicating that the patients were at potential risk of skin disorders such as skin cancer. Very recently, two individuals in a Caucasian cohort of CA patients were found to have ERCC4 mutations with chorea, minimal skin symptoms, and cerebellar and cortical atrophy on MRI, and thus ERCC4 mutations were reported as a rare cause of ARCA [9, 17]. Our cases similarly showed age-dependent, progressive atrophy of the cerebellum and brainstem, thus further characterizing the clinical phenotype and MRI findings in CA-dominant XP-F. As of this writing, eight cases, including the four in this study, have been reported as having XP-F with neurological involvement; [9, 14, 16] six out of eight have the c.2395C>T [p.Arg799Trp] mutation in at least in one allele. Considering the allele frequency of the c.2395C>T [p.Arg799Trp] mutation in Japanese control exome data (MAF = 0.0052), it is likely that the disease is underdiagnosed in CA cohorts, especially in Japan, because of the mild skin phenotype. Loss-of-function mutations of genes related to DNA repair pathways, for instance TDP1, APTX, and ATM, have been known to cause ARCA [18]. When complexed with ERCC1, XPF nuclease is known to incise damaged DNA strands 5′ to the lesion in the process of NER, and is one of the major mechanisms for repairing DNA damage [18]. ERCC4 mutations should be recognized as a potential cause of ARCA.
In conclusion, we found biallelic ERCC4 mutations in three families with adult-onset CA. ERCC4 mutations should be considered to be an important cause of adult-onset cerebellar ataxia and cognitive decline with choreiform movements as well as cerebral and brainstem atrophy. Elevated cancer risk in these patients may also be important in their clinical management.
References
Hammer MB, Eleuch-Fayache G, Schottlaender LV, Nehdi H, Gibbs JR, Arepalli SK, et al. Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am J Hum Genet. 2013;92:245–51.
Pyle A, Griffin H, Yu-Wai-Man P, Duff J, Eglon G, Pickering-Brown S, et al. Prominent sensorimotor neuropathy due to SACS mutations revealed by whole-exome sequencing. Arch Neurol. 2012;69:1351–4.
Doi H, Ohba C, Tsurusaki Y, Miyatake S, Miyake N, Saitsu H, et al. Identification of a novel homozygous SPG7 mutation in a Japanese patient with spastic ataxia: making an efficient diagnosis using exome sequencing for autosomal recessive cerebellar ataxia and spastic paraplegia. Intern Med. 2013;52:1629–33.
Hammer MB, Eleuch-Fayache G, Gibbs JR, Arepalli SK, Chong SB, Sassi C, et al. Exome sequencing: an efficient diagnostic tool for complex neurodegenerative disorders. Eur J Neurol. 2013;20:486–92.
Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–22.
Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet. 2013;92:807–19.
Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92:800–6.
Tofuku Y, Nobeyama Y, Kamide R, Moriwaki S, Nakagawa H. Xeroderma pigmentosum complementation group F: report of a case and review of Japanese patients. J Dermatol. 2015;42:897–9.
Carre G, Marelli C, Anheim M, Geny C, Renaud M, Rezvani HR, et al. Xeroderma pigmentosum complementation group F: a rare cause of cerebellar ataxia with chorea. J Neurol Sci. 2017;376:198–201.
Miyatake S, Okamoto N, Stark Z, Nabetani M, Tsurusaki Y, Nakashima M, et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J Hum Genet. 2017;62:741–6.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Nakazawa Y, Sasaki K, Mitsutake N, Matsuse M, Shimada M, Nardo T, et al. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat Genet. 2012;44:586–92.
Sijbers AM, van Voorst Vader PC, Snoek JW, Raams A, Jaspers NG, Kleijer WJ. Homozygous R788W point mutation in the XPF gene of a patient with xeroderma pigmentosum and late-onset neurologic disease. J Invest Dermatol. 1998;110:832–6.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Moriwaki S, Nishigori C, Imamura S, Yagi T, Takahashi C, Fujimoto N, et al. A case of xeroderma pigmentosum complementation group F with neurological abnormalities. Br J Dermatol. 1993;128:91–94.
Marelli C, Guissart C, Hubsch C, Renaud M, Villemin JP, Larrieu L, et al. Mini-exome coupled to read-depth based copy number variation analysis in patients with inherited ataxias. Hum Mutat. 2016;37:1340–53.
Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Prog Neurobiol. 2011;94:166–200.
Acknowledgements
We thank the patients and their families for their participation in this study. This work was supported by grants from Research on Measures for Intractable Diseases (NM); Comprehensive Research on Disability Health and Welfare (NM); the Strategic Research Program for Brain Science (NM); the Initiative on Rare and Undiagnosed Diseases in Pediatrics (NM); the Initiative on Rare and Undiagnosed Diseases for Adults (NM) from the Japanese Agency for Medical Research and Development; a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Grants-in-Aid for Scientific Research (A (NM)], B (NMi, HS), and C (HD, SM, MN)); the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems (NM) from the Japanese Science and Technology Agency; and grants from the Ministry of Health, Labor and Welfare (NM), the Takeda Science Foundation (NMi, HS, NM), and The Ichiro Kanehara Foundation for the Promotion of Medical Science & Medical Care (SM).
Author contributions
HD wrote the main manuscript and prepared the figure and the table. HT, NM, and FT revised the manuscript and gave conceptual advice. HD, SK, SM, SN, MK, AK, RK, and KO collected the clinical data and samples. HD, SM, RF, SI, SK, KT, MT, KT, MN, YT, NMi, and HS conducted the analysis of the genetic data.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Doi, H., Koyano, S., Miyatake, S. et al. Cerebellar ataxia-dominant phenotype in patients with ERCC4 mutations. J Hum Genet 63, 417–423 (2018). https://doi.org/10.1038/s10038-017-0408-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0408-5
This article is cited by
-
Hornerin deposits in neuronal intranuclear inclusion disease: direct identification of proteins with compositionally biased regions in inclusions
Acta Neuropathologica Communications (2022)
-
Autosomal recessive adult onset ataxia
Journal of Neurology (2022)
-
Long-read sequencing identifies the pathogenic nucleotide repeat expansion in RFC1 in a Japanese case of CANVAS
Journal of Human Genetics (2020)
-
The Classification of Autosomal Recessive Cerebellar Ataxias: a Consensus Statement from the Society for Research on the Cerebellum and Ataxias Task Force
The Cerebellum (2019)
