
Abstract
Most of the 19 mitochondrial aminoacyl-tRNA synthetases (mt-aaRSs) involved in mitochondrial protein synthesis are already linked to specific entities, one of the exceptions being PARS2 mutations for which pathogenic significance is not finally validated. The aim of the study was to characterize the PARS2- related phenotype.
Three siblings with biallelic PARS2 mutations presented from birth with infantile spasms, secondary microcephaly, and similar facial dysmorphy. Mental development was deeply impaired with speech absence and no eye contact. A dilated cardiomyopathy and multiorgan failure developed in childhood at the terminal stage, together with mitochondrial dysfunction triggered by valproate administration.
Brain MRI showed progressive volume loss of the frontal lobes, both cortical and subcortical, with widening of the cortical sulci and frontal horns of the lateral ventricles. Hypoplasia of the corpus callosum and progressive demyelination were additional findings. Similar brain features were seen in three already reported PARS2 patients and seemed specific for this defect when compared with other mt-aaRSs defects (DARS2, EARS2, IARS2, and RARS2).
Striking resemblance of the phenotype and Alpers-like brain MRI changes with predominance of frontal cerebral volume loss (FCVL-AS) in six patients from three families of different ethnicity with PARS2 mutations, justifies to distinguish the condition as a new disease entity.
Similar content being viewed by others

Introduction
Mitochondrial aminoacyl-tRNA synthetases (mt-aaRSs) encoded by nuclear genes are essential enzymes involved in the translation of genetic information from DNA to the oxidative phosphorylation system complexes [1]. Already 17 out of 19 of these enzymes have been assigned to specific mitochondrial entities [2, 3]. For two remaining genes, WARS2 and PARS2 pathogenic significance was not finally validated [2] although, a causal relationship between these two genes and a human disease has already been proposed [4,5,6,7].
The aim of the study was to characterize the phenotype of PARS2 defect on the basis of own observation of the disease in three PARS2 mutated siblings and the available data in two literature reports. The PARS2 phenotypes were compared with other mt-aaRSs defects.
Material and methods
Three Polish siblings with PARS2 mutations were included into the study to describe clinical course of the disorder, reported previously in three other infants [5, 7]. All available clinical data of the six PARS2-mutated patients [5, 7] were analyzed to characterize the phenotype of the disorder more detailed.
Case reports
Individual 1
A boy (proband) was born at term, as the second child of nonconsanguineous parents (Fig. 1a), after an uneventful pregnancy and delivery, with the body mass of 3150 g, head circumference 34 cm, Apgar score 9. Prolonged jaundice was observed in the neonatal period. The patient's psychomotor development was markedly delayed from the very beginning. At the age of 4 months, severe seizures began. Myoclonic jerks and acoustic hypersensitivity were frequently observed. Neurological examination showed axial and limb hypotonia with brisk tendon reflexes and Babinski signs. Ophthalmologic examination showed pale optic disk and retinal rarefaction. Audiogram was normal. Hypsarrhythmia and burst-suppression pattern were described in EEG and West syndrome was diagnosed. At the age of 2 years, he was microsomic with severe secondary microcephaly, and facial dysmorphic features including slopping forehead, hypertelorism, prominent eyes with shallow set eyeballs, short nose with broad nasal bridge, anteverted nostrils, short philtrum, opened mouths, and small chin (Fig. 1b). MRI performed this time showed volume loss of the frontal lobes (not shown). Organic acidurias, aminoacidopathies, carnitine deficiency, congenital glycosylation disorders, lysosomal storage diseases were excluded. Plasma lactate concentration was at the normal range. During further observation, epilepsy was refractory to medication up to the age of 4 years when seizures receded. The patient did not display any significant development with speech absence and no eye contact. The only acquired ability was unsupported sitting at the age of 5–6 years. At 7 years, only slight progression of frontal brain atrophy was seen on brain MRI (Fig. 2a–d).

Pedigree and dysmorphic features in the family with pathogenic variants in PARS2. a The allelic status of each family members; symbols are as follows: filled affected, empty unaffected, dotted, heterozygous carrier, arrow proband, NA not analyzed. b Facial photos of affected individual 1, 2, and 3 at the age of 4, 6, and 2 years, respectively. A healthy brother (II-3) did not present of dysmorphic features and molecular study were not possible (no parental consent). (Color figure online)

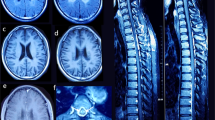
MR brain examinations in patients with PARS2 mutations: 7 years old boy (individual 1)—upper row, 4 years old girl (individual 2)—middle row, 3 months old boy (individual 3)—bottom row. Reduced volume of the frontal lobes (arrows on b,f) with widening of the cortical sulci in all patients and open Sylvian fissures in patient 3 (arrow on j). Thin corpus callosum in patient 1 and 2 (arrow on a), extremely thin in patient 3. Bilateral subdural hematomas in patient 3 (arrow on k). Pericerebral fluid spaces are widened over the frontal lobes. Frontal horns and bodies of the lateral ventricles are also widened
At the age of 7.5 years, seizures reappeared and valproic acid was administrated, but within few months it was withdrawn due to transient hypertransaminasemia up to 700 units (ref. < 50 units). 6 months later the boy suddenly deteriorated, and right-sided spastic paralysis and circulatory failure developed. Chest X-ray showed enlarged heart silhouette and ECHO revealed severe dilated cardiomyopathy. EEG disclosed general and multifocal abnormalities. Stroke-like lesions and basal ganglia involvement developed on brain MRI (Fig. 3). During this time lactic acid concentration ranged from markedly elevated (70 mg%) to normal levels (16.6, 11.5 mg%). Due to suspicion of mitochondrial disorder muscle biopsy was performed. It showed normal histological and histochemical picture and activities of respiratory chain complexes I–IV at the control range. Only citric synthase activity was increased (281 nmol/min/mg protein, ref. < 150). Molecular screening for common mutations in mtDNA and sequencing of POLG gene were negative. The patient improved partially on cardiological treatment and was discharged home. Due to the multiorgan insufficiency at the age of 8.5 years he died in the local hospital.

Follow-up MR brain examination in individual 1 at terminal stage, at the age of 8.5 years. Stroke-like lesions in the left caudate, lentiform nuclei and left frontal cortex (arrows)
Individual 2
The elder sister of the proband (Fig. 1a), born on time, after an uneventful pregnancy and delivery, with the body mass of 2640 g, head circumference 34 cm, and Apgar score 10. Prolonged neonatal jaundice was observed. The patient’s psychomotor development was markedly delayed from the very beginning. Epileptic seizures appeared for the first time at the age of 5 months. West syndrome was diagnosed with hypsarrythmic pattern in EEG. Neurological examination at the age of 1 year showed axial and limb hypotonia, brisk tendon reflexes, bilateral Babinski signs and secondary microcephaly. In addition, acoustic hypersensitivity, as well as myoclonic jerks were observed. Routine blood and CSF investigations were within normal values. At the age of 3 years, general tonic–clonic fits accompanied by general and multifocal abnormalities in EEG were observed. Pale, small optic disks were noted. When she was consulted by us at the age of 4 years, facial dysmorphic features similar to her brother and tendency to divergent squint were observed (Fig. 1b). MRI examination revealed the similar reduced volume of frontal lobes (Fig. 2e–h).
During follow-up time, no amelioration was observed, the patient’s psychomotor development was still markedly delayed, like in her older and younger brothers. Epilepsy was refractory to medication up to the age of 5 years. Occipitofrontal circumference at the age of 7 years was 45 cm (4.5 cm < 3 pct); she was also cachectic. No organomegaly was noted. At the age of 8 years her condition gradually deteriorated, and she died with signs of multiorgan insufficiency with dominant renal failure.
Individual 3
The youngest brother (Fig. 1a), born at term after an uneventful delivery, with head circumference 35 cm and Apgar score 10. In prenatal ultrasound examination his brain was assessed as ‘in preterm fetus’. He presented similar phenotype to his older siblings with dysmorphic features and secondary microcephaly (Fig. 1b). Myoclonic jerks were also observed. Reduced frontal lobes volume was found already in 3 months of age (Fig. 2i–l). West syndrome was diagnosed at the age of 6 months and antiepileptic drugs were administrated. Neurological examination performed repeatedly from the age of 2 years showed axial and limbs hypotonia, brisk deep reflexes and Babinski bilateral signs. Fundoscopic examination performed repeatedly showed in the beginning normal, but later slightly pale optic disks. The patient did not show any significant development. No organomegaly or cardiac structural and functional anomalies were found during 5 years of observation, but at the age of 6 mild thickening of left ventricle was detected. His weight and length gain were markedly disturbed.
Magnetic resonance imaging
Magnetic resonance (MR) examinations were performed in all the children with the use of 1.5 T scanners according to the routine brain protocol: axial SE/T1WI, axial, sagittal and coronal FSE/T2WI, axial FLAIR sequence, axial GRE/T2*WI or SWI, and DWI.
The results were assessed in relation to the brain MRI findings in 13 Polish cases with five other mt-aaRS defects (EARS2, DARS2, IARS2, RARS2, VARS2) [6, 8].
MRI parameters were analyzed with the use of gross definition of Leigh (LS) or Alpers (AHS) syndromes applied for mitochondrial patients by Sofou et al. [9]. Predominance of cerebral atrophy was qualified as AHS, and basal ganglia changes as LS. A definition of ‘AHS-like’ and ‘LS-like’ indicated the combination of both localizations.
Molecular study
Whole-exome sequencing
Genomic DNA was extracted by automated method (MagnaPure, Roche) from peripheral blood samples of the patients and their both parents. Whole-exome sequencing (WES) of the proband’s DNA was performed at the Department of Medical Genetics, Medical University of Warsaw, according to the established laboratory protocols. Prior to the library preparation, DNA samples were quantified using Qubit dsDNA HS Assay Kit (Life Technologies), and DNA degradation status was estimated by 1% agarose gel electrophoresis. About 50 ng of high quality genomic DNA was used for library construction with SureSelectQXT library prep kit (Agilent) and SureSelect Human All Exon V6 (Agilent) for whole exome enrichment. Each library was qualified using Bioanalyzer (Agilent), and quantified using Qubit (Life Technologies). Prior to the sequencing run, cluster amplification was carried out on cBot (Illumina) with TruSeq PE Cluster Kit v4 (Illumina). HiSeq1500 (Illumina) paired-end sequencing (2 × 100 bp) was performed using TruSeq SBS Kit v4 (Illumina) and high-output mode with the required mean sequence coverage ×100 (minimum ×20, GE20 ≥ 95%), four samples per lane.
WES data were analyzed using an in-house procedure, described in detail previously [6]. Briefly, generated reads were first merged and low-quality reads removed. Then the reads were aligned to the hg19 (GRCh37) reference human genome. Potential PCR duplicates and reads mapping to multiple genomic locations were removed. In variant calling, any call with the ratio smaller than 0.2 was assumed to be homozygous, and the rest - heterozygous. Alignments were viewed with Integrative Genomics Viewer v. 2.3.82. The detected variants were annotated using Annovar and converted to MS Access format for final manual analysis. All the non-coding and common variants (minor allele frequency above 0.01 in the general population) were discarded. The rare variants affecting the coding regions were filtered basing on the mode of inheritance and predicted consequences at the transcript level. The variants were prioritized according to the population frequency and the predicted effect on the protein. The potential consequences were defined according to the conservation of the affected amino acids and in silico predictions by using different algorithms.
The nomenclature of molecular variants follows the Human Genome Variation Society guidelines (HGVS, http://varnomen.hgvs.org/) using human PARS2 (MIM 612036; prolyl-tRNA synthetase 2, mitochondrial (putative) cDNA sequence: NM_152268.3 followed the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk).
The candidate pathogenic variants were verified in the proband, his affected siblings and parents by Sanger sequencing using BigDye Chemistry (Applied Biosystems).
Molecular analysis was performed after obtaining informed consent from the parents.
Functional assessment of molecular variant
Homologs of the human mitochondrial prolyl-tRNA synthetase (mt-PRS) were identified with a PSI-Blast [10] search (E-value threshold of 0.005) performed against the NCBI non-redundant protein sequence database. The collected sequences were filtered to include only sequences which belong to the phylum Chordata and which have a length of 400–600 amino acids. The sequences were then clustered with the cd-hit program [11] using 90% sequence identity threshold. The multiple sequence alignment of the mt-PRS family was derived using the pcma program [12]. Based on the result of the GeneSilico Metaserver [13] the prolyl-tRNA synthetase from Rhodopseudomonas palustris (Protein Data Bank id - 2i4l) [14] was selected as a template for homology modeling. The sequence-to-structure alignment between the target protein and the template (Supplementary Figure S1) was proposed using the consensus alignment approach and 3D assessment based on the results of FFAS, HHSearch and pdbblast [15]. Multiple sequence alignment of the family was also taken into consideration. The 3D model of the protein was built with MODELLER [16]. A model quality assessment was carried out using ProSA-web [17] and MolProbity [18]. The model refinement was carried out using molecular dynamics (MD) simulation using following procedure. The MD simulation was performed with the GROMACS [19] program using the GROMOS54A7 force field and SPC water model. The thickness of the water box was 1 nm. The simulation comprises 4 stages: an initial energy minimization was followed by a 100 ps NVT simulation in 300 K and 100 ps NPT simulation under pressure of 1 atm. The lowest energy conformation from 1 ns unconstrained production run was used for further analysis. Structure visualization was carried out with PyMOL (http://www.pymol.org).
Results
Phenotypic data of the three siblings with PARS2 mutations and three remaining individuals from two families reported in the literature are summarized in Table 1.
Perinatal period was generally uneventful in all individuals, but clear retardation of early development was obvious from the birth and suggested in fetus in one case. Prolonged jaundice occurred in two out of six patients. Feeding difficulties were observed in all cases, with impaired weight and height gain. Excessive length (+3, +5 SD) of an individual reported by Sofou et al. [5] is probably an accidental observation.
Central nervous system (CNS) involvement dominated the clinical course. It was demonstrated by infantile spasms of West type, with hypsarrhythmia, which usually did not responded to ACTH analog (Synacthen) administration. Myoclonic jerks were observed in some cases. Seizures started at the age of 2–5 months and were drug-resistant.
Secondary microcephaly of a marked degree developed in all individuals. Dysmorphic features of the face, similar in three siblings from family 1, included slopping forehead, hypertelorism, slanting up palpebral fissures, prominent eyes with shallow set eyeballs, short nose with anteverted nostrils, short philtrum, opened mouth and small chin (Fig. 1b). Such features were not mentioned in descriptions of individuals from families 2 or 3 (Table 1).
Mental development was deeply impaired, with speech absence and no eye contact. Optic atrophy developed in four individuals. Hearing loss was not reported, except for hyperacusis in two siblings from family 1. Motor development was stopped, with maximum milestones of sitting at the age of 6 (Individual 1). Muscle involvement was expressed by marked hypotonia (always associated with brisk tendon reflexes), which occurred in all six individuals.
Cardiovascular symptoms were absent during long-time observation. However, a dilated cardiomyopathy developed at the terminal stage in individuals 1 and 2 and 3, together with other parameters of mitochondrial dysfunction (hyperlactatemia, valproate sensitivity). Dilated left ventricular hypertrophy, enlarged heart and interstitial fibrosis were also found at autopsy of individual 4, reported by Sofou et al. [5]. Also in individual 3, mild hypertrophic cardiomyopathy started to develop at the age of 6. No liver and kidney involvement were present during infancy and early childhood, and the disease course was stable without fluctuation. However, in last months of life, a subacute deterioration developed in two siblings from family 1; at the critical stage, liver and renal failure developed, probably due to a cause-and-effect relationship with valproate administration.
Brain MRI of three affected siblings from family 1 (Fig. 2) showed atrophy (or hypoplasia?) of the frontal lobes, both cortical and subcortical, with widening of the cortical sulci and of the frontal horns of the lateral ventricles. Hypoplasia of the corpus callosum and progressive demyelination were additional findings. Similar MRI features were reported in remaining three PARS2 mutated individuals and showed in individuals 5 and 6 in Erratum to Mizuguchi et al. [5, 7, 20]. At the critical state of individual 1, numerous stroke-like lesions appeared in subsequent MRI (Fig. 3), most without true diffusion restriction as assessed on ADC maps as well as Leigh-like lesions in basal ganglia on T2.
Muscle biopsy performed in two individuals revealed a combined oxidative phosphorylation defect in one case [5] and increased activity of citric synthase in the second one.
Inheritance pattern in all three families was consistent with autosomal recessive mode.
Molecular findings
The sequencing run for the proband’s sample achieved 93,932,415 reads and the 20-fold coverage was 73.1%. Two missense variants, c.239 T > C, p.(Ile80Thr) and c.1091 C > G, p.(Pro364Arg), located in exon 2 of the PARS2 gene were identified in the proband (by WES) and his siblings (by Sanger sequencing). Parental studies confirmed trans allelic pattern of inheritance and compound heterozygosity for these changes (the first was inherited from the father and the second from the mother). The changes were previously reported by Pronicka et al. [6]. Isoleucine at position 80 was moderately conserved, but proline at position 364 was highly conserved amino acid (considering 11 species) located in aminoacyl-tRNA synthetase class II protein domain. Besides moderate physicochemical differences between Ile and Thr (Grantham dist.: 89), as well as Pro and Arg (Grantham dist.: 103), both changes were predicted to be deleterious by silico predictive algorithms: Mutation Taster, Poly Phen-2, SIFT and LRT. In addition, these were very rare molecular variants; only c.239 T > C was annotated in the ExAC and ESP databases with a minor allele frequency 0.001041 and 0.0014, respectively.
Assessment of the function of the molecular changes
The human mitochondrial prolyl-tRNA synthetase (mt-PRS) is a dimer of two identical protein monomers. Each monomer consists of the catalytic domain, responsible for activation of the proline amino acid and its transfer to the 3′-end of the cognate tRNA, and the anticodon-binding domain, responsible for recognition of the anticodon loop of the cognate tRNA (Fig. 4a, Supplementary Video S1) [14]. In the structure of the mitochondrial threonine-tRNA synthase (mt-TRS), which is also a member of class IIa aminoacyl-tRNA synthetases, each protein monomer binds a single tRNA moiety. However, the contacts between a given tRNA are not restricted solely to the residues of each individual monomer, but the catalytic domain of the neighboring monomer helps to stabilize the tRNA–protein complex [21].

Protein model and structural mapping of the disease-associated mutations in PARS2. Structure of mt-PRS homodimer. The catalytic and anticodon-binding domains of a single monomer are colored green and cyan, respectively. The location of the active site is marked with AMP molecule. The localization of mutations described in this work are colored red, while other mutations reported in Swedish and Japanese patients [5, 7, 9] (reason for showing these mutations) are marked blue (Fig. 4a). Multiple sequence alignment of the selected mt-PRS. Only regions next to herein reported mutation are shown. The number above the alignment corresponds to the amino acid position in the human protein sequence. The residues that mutations are the subject of this work are highlighted in cyan, Q76, which involvement in a stabilization of a protein–tRNA complex is discussed in the text, is highlighted in gray (Fig. 4b). The location of Ile80 and surrounding amino acids. Ile80 is a part of hydrophobic pocket which stabilize the conformation of a beta hairpin and surrounding helices, which are involved in formation of mt-PRS homodimer (Fig. 4c). The location of Pro364 and surrounding amino acids. Pro364 Arg mutation might result in changed conformation of the loop that join the catalytic and anticodon-binding domains (Fig. 4d). (Color figure online)
The multiple sequence alignment (MSA) demonstrates that both Ile80 and Pro364 are conserved residues among Chordata (MSA for selected organisms is shown in Fig. 4b). The Ile80, together with Met74, Leu75, Pro82, Trp88, and Leu198, is part of a network of conserved, hydrophobic residues (Fig. 4c). These amino acids most likely help to stabilize the conformation of the beta hairpin and nearby helices, which are in turn located at the interface between protein monomers. Furthermore, one of the helices harbors a Gln76 residue, which corresponds to Gln44 from the mt-TRS. In the mt-TRS–tRNA complex, Gln44 is in the close proximity to the tRNA, although it does not form a hydrogen bond with the tRNA. Considering that Gln76 is also a conserved residue, although in case of the mt-PRS form some organisms it can be substituted for a positively charged lysine, it might be plausible that this amino acid plays some role in stabilization of the mt-PRS-tRNA complex. The introduction of the Ile80Thr mutation most likely destabilizes the abovementioned interactions. As a result, the homodimer formation might be hampered and the stability of the protein–tRNA complex weakened. Interestingly, a mutations reported by Mizuguchi et al. [7] in infantile-onset neurodegenerative disorder, Val95Ile and Glu203Lys, are also located at the homodimer interface.
The Pro364 is located on the loop that joins the catalytic and anticodon-binding domains, and is in spatial proximity to Ile80, with the distance between their Cα atoms of nearly 19 Å (Fig. 4d). The amino acid is in the vicinity of hydrophobic Leu237, Gly238, Lys239, Ala351, Leu355, Trp363, and Leu367, which all seem to be involved in the stabilization of the loop that joins both domains of the mt-PRS. Thus,Fthe anticodon-binding domain might be correctly positioned relative to the catalytic domain. Introduction of a much larger and positively charged arginine, instead of the small proline, might force the loop to change its conformation and shift the position of the anticodon-binding domain relative to the catalytic domain. Thus the effective recognition of the cognate tRNA and the synthesis of Pro-tRNA might be disturbed. Further investigation is, however, required to fully understand the influence of both mutations on the protein stability and its function.
Discussion
Striking resemblance of the range of symptoms, clinical course, and outcome of the disorder associated with PARS2 mutations in six patients from three unrelated families of different ethnicity, justifies to separate the condition as a new disease entity. The phenotype included early developmental delay, infantile spasms, myoclonal jerks, hypsarrhythmia, severe postnatal microcephaly, drug-resistant epilepsy and neurological regression. Plasma lactate concentrations were not markedly elevated or not changed. Severely retarded patients may survive in the stabilized condition until childhood, when a deterioration develops in most of them, with symptoms of mitochondrial disease: cardiomyopathy, multiorgan failure and sensitivity to valproates. Surprisingly, brain MRI changes were nearly identical in all included individuals with PARS2 mutations, showing predominant frontal lobes hypoplasia/atrophy with addition of progressive demyelination of a various degree and corpus callosum hypotrophy [7, 20].
Dysmorphy of face is additional symptom observed in the pair of siblings with PARS2 defect, which deserve mentioning as the possible phenotypic feature. It includes slopping forehead, prominent eyes, broad nasal bridge. The dysmorphy is one of dubious symptoms in some mt-aaRSs defects, in this in MARS2 related phenotype (OMIM616430) and sometimes in RARS2 (OMIM611523), EARS2 (OMIM614924), IARS2 and VARS2 (OMIM616672).
In our country-wide reference center, the mitochondrial diseases related to the mt-aaRSs represented, as a total, about 7% of all probands diagnosed by WES [6]. All cases showed CNS impairment. The onset was at infancy, and the course progressive and severe, as shown above in the PARS2 defect. Dominating MRI changes may be arbitrarily assigned to the AHS-like or LS-like pattern (Table 2), with the presence of specific features for EARS2, RARS2 and DARS2 (Supplementary Material, Supplementary Table 1). The clear predominance of frontal lobe hypotrophy/atrophy was shown only in the patients with PARS2 defect (Table 2) which corresponds to dysmorphic facial features of slopping forehead visible in our PARS2-mutated siblings (Fig. 1b).
Although brain atrophy and microcephaly seem to be general features of CNS disorders caused by mt-aaRSs deficiencies, there is clear topographic difference on the brain MRI indicating that specific brain regions are involved in each separate aaRS disorder, in sufficiently differentiating way [22]. It goes far beyond the simple assignment to an AHS or LS pattern. Unique MRI features were already established for DARS2 [8], EARS2 [23], RARS2 [24], as seen in our material (Supplementary Material, Supplementary Table 1), and recently for AARS2 [25]. We suggest that the predominance of frontal lobes atrophy is the specific feature of PARS2. This may appear applicable in the future study of some human neurodegenerative disorders, e.g., ‘frontotemporal dementia’ of an unknown cause [26], searching next (milder) phenotype related to PARS2 variants. The process of discovering new phenotypes of mt-aaRSs is still ongoing [27].
The fascinating question is the occurrence of the phenotypic differences and similarities between various mt-aaRSs, as well as within a single mt-aaRS. Looking superficially, more similarities should be expected, due to canonical activity of mt-aaRSs, i.e., their participation in protein synthesis inside mitochondria. We assumed that the phenotype linked to mitochondrial prolyl-tRNA synthetase (PARS2) resembles this linked to mitochondrial prolyl-tRNA (MTTP). However contrary, the classical (mt-tRNA-related) phenotypes were observed in three MTTP mutated patients: a mitochondrial myopathy in two children [28, 29] and myoclonic epilepsy and ragged red fibers in an adult person [30].
Emerging evidences indicate that the physiological role of mt-aaRSs (and cytoplasmic aaRSs) goes far beyond the function of tRNA aminoacylation at the first step of protein synthesis [31]. Alternative functions of mt-aaRSs include a wide range of other poorly known metabolic pathways and cell signaling processes [31], and may explain, i.e., the differences of brain MRI topography linked to single mt-aaRSs.
Datt and Sharma, analyzing consequences of 63 known pathogenic variants of aaRSs (structural and functional), found that only 20% of all these changes impaired tRNA aminoacylation activity (via binding to ATP/amino-acid, tRNA or involvement in dimerization). Other mutations are localized in structural cores, at potential biomolecular interfaces or other annotated regions [32] leading to structural instability, or other not well recognized dysfunctions. Those unknown functions may be specific for particular mt-aaRSs, and responsible for topographic differences in CNS involvement assigned to them.
In our patients with PARS2 defect, functional analysis only partially foresees the mechanism of protein impairment. In the applied 3D model we showed that Ile80Thr may affect the formation of the mt-PRS homodimer, crucial for the enzyme’s function. A consequence of Pro364Arg seems to be shifting the relative position of the catalytic and anticodon-binding domains, which might impair the ability to correctly recognize the tRNAPro molecule. How these mutations affect the putative role of the mt-PRS in other metabolic pathways and cell signaling processes is still an open question.
One of the indisputable functions of mt-aaRSs is the participation in RNA expression and proceeding [22]. It may be speculated that the studies of RNA pathways in relevant parts of the brain may reveal the specificity of the role of particular mt-aaRSs. The RNA has been recently subjected to intensive investigations. Some experiments suggest that the presence of mt-aaRS fragments can abolish harmful effect of mt-tRNA mutations [33, 34]. This offers the chance to find methods of casual therapy of mt-aaRSs defects in the future.
Conclusion
Depending on available data of six patients from three families, we describe a phenotype linked to PARS2 pathological variants. This phenotype includes: early epileptic encephalopathy, infantile spasms, and Alpers-like brain MRI changes with predominance of frontal cerebral volume loss (FCVL-AS). Combined OXPHOS deficiency was moderately expressed or absent. Significance of facial dysmorphy and hearing impairment require careful investigation of consecutive patients with PARS2 mutations.
References
Bonnefond L, Fender A, Rudinger-Thirion J, Giege R, Florentz C, Sissler M. Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry. 2005;44:4805–16.
Mayr JA, Haack TB, Freisinger P, Karall D, Makowski C, Koch J, et al. Spectrum of combined respiratory chain defects. J Inherit Metab Dis. 2015;38:629–40.
Konovalova S, Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol Genet Metab. 2013;108:206–11.
Theisen, BE, Rumyantseva, A, Cohen, JS, Alcaraz, WA, Shinde, DN, Tang, S et al. Deficiency of WARS2, encoding mitochondrial tryptophanyl tRNA synthetase, causes severe infantile onset leukoencephalopathy. Am J Med Genet A. 2017;173:2505-10.
Sofou K, Kollberg G, Holmstrom M, Davila M, Darin N, Gustafsson CM, et al. Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol Genet Genom Med. 2015;3:59–68.
Pronicka E, Piekutowska-Abramczuk D, Ciara E, Trubicka J, Rokicki D, Karkucinska-Wieckowska A, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med. 2016;14:174.
Mizuguchi T, Nakashima M, Kato M, Yamada K, Okanishi T, Ekhilevitch N, et al. PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J Hum Genet. 2017;62:525–9.
Tylki-Szymanska A, Jurkiewicz E, Zakharova EY, Bobek-Billewicz B. Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation: high outcome variation between two siblings. Neuropediatrics. 2014;45:188–91.
Sofou K, Moslemi AR, Kollberg G, Bjarnadottir I, Oldfors A, Nennesmo I, et al. Phenotypic and genotypic variability in Alpers syndrome. Eur J Paediatr Neurol. 2012;16:379–89.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9.
Pei J, Sadreyev R, Grishin NV. PCMA: fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics. 2003;19:427–8.
Kurowski MA, Bujnicki JM. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 2003;31:3305–7.
Crepin T, Yaremchuk A, Tukalo M, Cusack S. Structures of two bacterial prolyl-tRNA synthetases with and without a cis-editing domain. Structure. 2006;14:1511–25.
Ginalski K, Rychlewski L. Protein structure prediction of CASP5 comparative modeling and fold recognition targets using consensus alignment approach and 3D assessment. Proteins. 2003;53 Suppl 6:410–7.
Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–91.
Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–410.
Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21.
Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–54.
Mizuguchi T, Nakashima M, Kato M, Yamada K, Okanishi T, Ekhilevitch N, et al. Erratum: PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J Hum Genet. 2017;62:587
Holman KM, Wu J, Ling J, Simonovic M. The crystal structure of yeast mitochondrial ThrRS in complex with the canonical threonine tRNA. Nucleic Acids Res. 2016;44:1428–39.
Abbott JA, Francklyn CS, Robey-Bond SM. Transfer RNA and human disease. Front Genet. 2014;5:158.
Steenweg ME, Ghezzi D, Haack T, Abbink TE, Martinelli D, van Berkel CG, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–94.
Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–62.
Lakshmanan R, Adams ME, Lynch DS, Kinsella JA, Phadke R, Schott JM, et al. Redefining the phenotype of ALSP and AARS2 mutation-related leukodystrophy. Neurol Genet. 2017;3:e135.
Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. 2015;7:171.
Linnankivi T, Neupane N, Richter U, Isohanni P, Tyynismaa H. Splicing defect in mitochondrial seryl-tRNA synthetase gene causes progressive spastic paresis instead of HUPRA syndrome. Hum Mutat. 2016;37:884–8.
Moraes CT, Ciacci F, Bonilla E, Ionasescu V, Schon EA, DiMauro S. A mitochondrial tRNA anticodon swap associated with a muscle disease. Nat Genet. 1993;4:284–8.
Morel G, Bannwarth S, Chaussenot A, Cano A, Fragaki K, Ait-El-Mkadem S, et al. A new mutation in the mitochondrial tRNAPro gene associated with early-onset neuromuscular phenotype and ragged-red fibers. Neuromuscul Disord. 2016;26:885–9.
Blakely EL, Trip SA, Swalwell H, He L, Wren DR, Rich P, et al. A new mitochondrial transfer RNAPro gene mutation associated with myoclonic epilepsy with ragged-red fibers and other neurological features. Arch Neurol. 2009;66:399–402.
Pang YL, Poruri K, Martinis SA. tRNA synthetase: tRNA aminoacylation and beyond. Wiley Interdiscip Rev RNA. 2014;5:461–80.
Datt M, Sharma A. Evolutionary and structural annotation of disease-associated mutations in human aminoacyl-tRNA synthetases. BMC Genom. 2014;15:1063.
Perli E, Giordano C, Tuppen HA, Montopoli M, Montanari A, Orlandi M, et al. Isoleucyl-tRNA synthetase levels modulate the penetrance of a homoplasmic m.4277T > C mitochondrial tRNA(Ile) mutation causing hypertrophic cardiomyopathy. Hum Mol Genet. 2012;21:85–100.
Perli E, Fiorillo A, Giordano C, Pisano A, Montanari A, Grazioli P, et al. Short peptides from leucyl-tRNA synthetase rescue disease-causing mitochondrial tRNA point mutations. Hum Mol Genet. 2016;25:903–15.
Cassandrini D, Cilio MR, Bianchi M, et al. Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J Inherit Metab Dis. 2013;36:43–53.
Acknowledgements
This work was supported by the CMHI grants 134/13, 136/13, 216/12 and EU Structural Funds, project POIG.02.01.00-14-059/09. ML and DP are supported by the Polish National Science Centre (Grant Number 2014/15/B/ST6/05082) and Foundation for Polish Science (TEAM to DP).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The author declare that they have no conflict of interest.
Additional information
Elżbieta Ciara, Dariusz Rokicki, and Michal Lazniewski contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Ciara, E., Rokicki, D., Lazniewski, M. et al. Clinical and molecular characteristics of newly reported mitochondrial disease entity caused by biallelic PARS2 mutations. J Hum Genet 63, 473–485 (2018). https://doi.org/10.1038/s10038-017-0401-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0401-z
This article is cited by
-
Four pedigrees with aminoacyl-tRNA synthetase abnormalities
Neurological Sciences (2022)
-
Mild phenotype of glutaric aciduria type 1 in polish patients – novel data from a group of 13 cases
Metabolic Brain Disease (2019)
-
Valproic acid
Reactions Weekly (2018)
