
Abstract
Ionizing radiation released by the atomic bombs at Hiroshima and Nagasaki, Japan, in 1945 caused many long-term illnesses, including increased risks of malignancies such as leukemia and solid tumours. Radiation has demonstrated genetic effects in animal models, leading to concerns over the potential hereditary effects of atomic bomb-related radiation. However, no direct analyses of whole DNA have yet been reported. We therefore investigated de novo variants in offspring of atomic-bomb survivors by whole-genome sequencing (WGS). We collected peripheral blood from three trios, each comprising a father (atomic-bomb survivor with acute radiation symptoms), a non-exposed mother, and their child, none of whom had any past history of haematological disorders. One trio of non-exposed individuals was included as a control. DNA was extracted and the numbers of de novo single nucleotide variants in the children were counted by WGS with sequencing confirmation. Gross structural variants were also analysed. Written informed consent was obtained from all participants prior to the study. There were 62, 81, and 42 de novo single nucleotide variants in the children of atomic-bomb survivors, compared with 48 in the control trio. There were no gross structural variants in any trio. These findings are in accord with previously published results that also showed no significant genetic effects of atomic-bomb radiation on second-generation survivors.
Similar content being viewed by others


Introduction
Previous studies of atomic-bomb (A-bomb) survivors, notably the “Life Span Study” conducted by the Radiation Effects Research Foundation, have demonstrated many late health effects of the resulting radiation in humans [1], including increased risks of leukemia and other cancers [2, 3]. This suggests that radiation from the explosions generated gene variants in somatic cells in atomic-bomb survivors. However, ionizing radiation has also been shown to induce genetic/hereditary effects in experimental animals such as fruit flies and mice [4, 5], attributable to gene variants in germ cells (oogonia and spermatogonia), leading to concern over the potential hereditary effects of atomic bomb-related radiation. Numerous studies have addressed this question by examining heritable genetic effects among children of atomic-bomb survivors, including the incidence of hereditary anomalies [6], variants and enzymatic activity of proteins in erythrocytes [7], minisatellite [8] and microsatellite variants [9], and risk of death [10]. Although there is currently no clear evidence for any hereditary effects of atomic-bomb radiation, these approaches only analysed part of the inheritance phenomenon induced by genomic changes.
The technique of next-generation sequencing (NGS) has undergone rapid, recent developments, making it possible to detect single nucleotide changes in the genome, and vast numbers of genomic changes in an individual. Kong et al. used NGS to address the de novo variant rate in offspring, and pointed out the importance of the father’s age [11]. They demonstrated the use of detecting single nucleotide changes in the whole genome as an approach for investigating heritable gene variants. We therefore used NGS to assess the heritable genetic effects in the children of atomic-bomb survivors by examining de novo genomic changes.
Materials and methods
Contacting subjects and sampling
We mailed a self-administered questionnaire to 2229 atomic-bomb survivors who received regular health check-ups at the Nagasaki Atomic Bomb Casualty Council Health Management Center. The questionnaire sought information about the survivor’s family members, and asked for permission to conduct a detailed telephone survey as a second step. Returning the response (by mail) was optional. After receiving the responses, we conducted a telephone survey of the survivors who consented to additional investigations, as explained in the questionnaire. The telephone survey gathered patient information related to the atomic-bomb explosion, such as their distance from the hypocenter and acute radiation symptoms, and the medical histories of all family members. We then extracted families in which no member had a history of chemo/radiation therapy or hematologic disorders, and contacted these survivors to explain the details of the study in relation the candidate family members (father, mother, and child [‘trio’]). We collected samples from each trio after obtaining written informed consent. Permission for this procedure (contact, questionnaire, sample collection) was obtained from the Mayor of Nagasaki City. Three trios in which the father, but not the mother, was exposed to atomic-bomb radiation within 1.5 km from the hypocenter were enrolled in this study, together with a control trio unexposed to atomic-bomb radiation.
Peripheral blood samples (10 mL) were collected and genomic DNA was extracted from blood mononuclear cells using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). All study procedures were approved by the Committee for the Ethical Issues on Human Genome and Gene Analysis at Nagasaki University. Genome analyses were carried out after the obtaining written informed consent from all participants.
WGS and variant detection
Genomic DNA (3 µg) was sheared into fragments about 400 base pairs (bp) long using an S2 focused-ultrasonicator (Cavaris, Woburn, MA, USA) and cleaned using Agencourt AMPure® XP (Beckman Coulter, CA, USA). Genomic DNA fragment libraries for sequencing were constructed using a TruSeq DNA PCR-free Sample Prep Kit (Illumina, San Diego, CA, USA) and data were obtained using the HiSeq 2500 system (Illumina). Raw data files (101 + 101 base paired-end reads) were converted to FASTQ files using the Bcl2Fastq software (Illumina) version 1.8.4. The hg19 human genome reference sequence of canonical chromosomes (mitochondrial genome and chromosomes 1–22, X, and Y) downloaded from UCSC Genome browser FTP server [12] was prepared for read mapping.
To detect single nucleotide variants (SNVs) and short insertions/deletions (indels) by whole-genome sequencing (WGS), reads in FASTQ files were subjected to mapping and base quality-score recalibration, based on information on SNV sites not registered in dbSNP version 136 mapped using the NovoAlign software version 3 (Novocraft, Selangor, Malaysia). The generated BAM files were subjected to position-wise sorting and marking polymerase chain reaction (PCR) and optical deduplication (dedup) using the NovoSort software version 1.3 (Novocraft). The sorted and deduped BAM files were then processed with Genome Analysis Tool Kit (GATK) software package version 3.4–46 [13]. The workflow based on GATK best practice recommendations [14, 15] involved local realignment of reads around indels using RealignerTargetCreator and IndelRealigner with known indel sites (Mills_and_1000G_gold_standard) in the GATKbundle package version 2.8. Variant calling was performed by HaplotypeCaller with a default setting to generate genomic VCF (GVCF) files. GVCF files obtained from 12 individuals in the four trio families were converted into a genotyped VCF file using GATK-GenotypeGVCFs. Variant quality-score recalibration of biallelic SNV sites was performed using GATK-VariantRecalibrator, GATK-ApplyRecalibration, and GATK-SelectVariants. The following files in GATKbundle were used to specify which SNV call sets should be used to build the recalibration model: “hapmap_3.3.hg19.sites.vcf” for true site training with prior likelihood Q15, “1000G_omni2.5.hg19.sites.vcf” for true site training with prior likelihood Q12, and “1000G_phase1.snps.high_confidence.hg19.sites.vcf” for non-true site training with prior likelihood Q10. The following annotations were used to evaluate the likelihood of SNVs being true positives: coverage (DO), QUAL value by depth (QD), Fishier strand (FS), strand odds radio (SOR), mapping quality rank sum test (MQRankSum), read position rank sum test (ReadPosRankSum), and RMS mapping quality (MQ). The tranche filter level was 99.5%, which was expected to retrieve 99.5% of true variants from the true site training sets. This number was more stringent in terms of specificity than the threshold of 99.9% recommended in the GATK documents.
Finally, we referred GQ (genotype quality) score and QUAL (quality score for assertion of alternative alleles) that calculated by HaplotypeCaller bundled in GATK to pick up de novo SNV candidates. Most likely genotype likelihood score in individual at the interested site is set lik(X), and secondary likely score is set lik(Y), and GQ = −10 × log10(likelihood ratio lik(Y)/lik(X)). Maximum score is capped at 99 even if score is over 99. So, positive selection landmark score is essentially comparable to Kong’s method [11], criterion (3) the likelihood ratio lik(AR)/lik(RR) is above 1010. QUAL represents the score to show probability for existence of variant at the interested site calculated by GATK.
As shown in Fig. 1, de novo SNVs fulfilling the following criteria in each trio were selected for validation using an in-house Ruby script: (1) a child’s SNV site was successfully genotyped in all individuals and (2) the variant is unique in all individuals. “Blacklist genomic regions” potentially affected by misalignment of short reads and genes highly variable by somatic recombination/variant were built using regions in the following tracks of UCSC Genome Browser: (1) RepeatMasker [16] track restricted to repeating elements classified as Alu or L1; (2) segmental duplication [17] track; and (3) GENCODE [18] basic version19 track restricted to immunoglobulin-coding genes (IGHV*, IGLV*, and IGKV*), T-cell receptor-coding genes (TRAV*, TRBV*, TRGV*, and TRDV*), and olfactory receptor genes. The blacklist genomic regions covered about 30.11% (932,124,869 bp/3,095,693,983 bp) of non-N nucleotides in the hg19 human genome assembly and were applied to remove common somatic variant sites using BEDTOOLS software package version 2.22.1 [19]. Deduped sample statistics of “depth of coverage” at outside of “blacklist genomic regions” or putative unique region, were calculated using GATK-DepthOfCoverage and in-house scripts using R [20] and the ggplot2 library [21]. We adopted two criteria of highly reliable SNVs for validation: GQ ≥ 90, and a combination of GQ < 90 and QUAL ≥ 200.

Flowchart showing filtering step
Structural variants were detected by mapping FASTQ files using the BWA software package version 0.6.2 [22] followed by sorting and deduping with NovoSort for input files in the Meerkat software package version 0.185 [23]. Structural variants were detected and filtered using Meerkat with recommended parameters for 60–80x genomes in Meerkat’s manual. De novo structural variants were detected by considering the child as the tumour genome and parents as normal genomes for Meerkat settings. To avoid misalignment of short reads from HiSeq2500 on the human genome, results in the regions of Alu sequences [24] and segmental duplication regions [17] were omitted by repeat masking [25].
Confirmation of de novo SNV variants
Amplification primers were designed using DNA Hotspot designs on the Ion AmpliSeq designer web server (Thermo Fisher Scientific, Waltham, MA, USA) and used for multiplex amplification to confirm de novo SNV variants in trios. Sequence data were obtained using MiSeq system (Illumina). Multiplex PCR amplification was performed on 100 ng genomic DNA using KAPA2G FAST Multiplex Kits (KAPA Biosystems, MA) with AmpliSeq-designed primers. The PCR conditions were as follows: initial denaturation and enzyme activation for 3 min at 95 °C, followed by 25 thermal cycles of denaturation for 15 s at 95 °C, annealing for 60 s at 55 °C, and extension for 60 s at 72 °C. After amplification, uracil DNA glycosylase (final concentration 0.05 U/µL) and endonuclease IV (final concentration 0.1 U/µL) were added directly to the reaction mixture and incubated at 37 °C for 30 min to separate uracil-containing sites. The mixture was cleaned using Agencourt AMPure® XP (Beckman Coulter), and a DNA fragment library was constructed using a KAPA HTP library construction kit for Illumina with adaptor oligonucleotides compatible with the Illumina pair-end index sequencing protocol (KAPA).
MiSeq (300 + 200 base paired-end reads) raw data files were converted to FASTQ files using the MiSeq Reporter software (Illumina) version 2.3.32. The hg19 human genome reference sequence was used as a read-mapping template, as for WGS except for the deduplication process. Readable sites had >200 depth, but some sites could not be amplified by multiplex PCR.
Confirmation of de novo structural variants
We tried to detect de novo structural variants by variant-specific PCR. PCR primers for validation were designed by Meerkat’s primers.pl script invoking the Primer3 software package version 2.6 [26] and BLAT software version 36 [27]. The repeat-masking function of the script was applied by invoking with the “-rl” option. The designed amplicons were ≤500 bp in size. Primers were designed automatically to amplify the structural variation site within 500 base pair length. Primer pairs were used for amplification only when at least one of primer in a pair has no repetitive sequence. Structural variant-specific PCR amplification was performed using KOD FX Neo Taq DNA polymerase (Toyobo, Osaka, Japan), according to the manufacturer’s instructions. PCR products were separated by agarose gel electrophoresis and evaluated for amplification specificity.
Confirmation of de novo copy number alterations
De novo copy number alterations (CNA) were screened by using the cn.MOPS software package [28] on the R language [29]. Twelve BAM files, 4 trios, were analyzed simultaneously in the cn.Mops function. A window size for read depth calculation was set 10 kb width. CNA loci detected in single samples were selected by visual inspection referring “Normalized Read Counts” and “Read Count Ratio” data plots output from cn.MOPS.
Results
Trio characteristics
At the time of the atomic-bomb explosion, the exposed fathers in the FPMX01, FPMX03, and FPMX06 trios were 16, 9, and 6 years old, respectively. In the interview, all the survivors (fathers) stated that they had experienced acute symptoms such as hair loss (Table 1) starting around the beginning of September 1945, almost 3 weeks after the bombing. Hematological data at the time of sampling were normal in all cases, except for one father (FPMX03 trio) who showed mild thrombocytopenia (127 × 103/μL). No individuals had anemia or neutropenia.
Quality control of sample WGS data
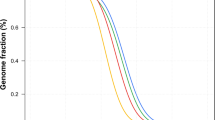
The quality of the WGS data, including library construction, sequencing, mapping, and fraction of duplication, were controlled by analysing (Table 2) the depth of coverage of the sample data. In the mean of all samples, about 7.7% of the whole human genome are not analysed because of no coverage of sequence, even with unique sequence. A frequency distribution line graph (Fig. 2) was plotted for depths of coverage ≥5, and depth of coverage curve indicates that each sample has enough coverage of sequence and did not show any deviated read depth.

Frequency distribution of depth of coverage in putative unique regions in entire genome. X axis indicates depth of coverage from 5–180; Y axis indicates base-wise count of reads
De novo SNVs in children of atomic-bomb survivors
Analyses focused on SNVs rather than indel variants in unique sequences in the human genome, because it was difficult to evaluate the existence of indels due to a lack of assessment criteria. De novo variant candidates were selected by GQ and QUAL (simple quality) scores calculated in GATK HaplotypeCaller. The results are shown in Table 3. The total numbers of de novo SNVs were 62 in FPMX01, 81 in FPMX03, 42 in FPMX06, and 48 in FCMC10. These results suggest that the incidence of de novo SNVs was not increased evidently in the children of atomic-bomb survivors.
Number of de novo gross structural variants and copy number alterations in children of atomic-bomb survivors
The number of amplification primer sets and amplification results using Meerkat with selected structural variant-specific primers are shown in Table 4. Some of the primer sets could amplify structural polymorphisms shared between parent(s) and child, but none of the primers could amplify de novo structural variant sites.
In cn.MOPS analyses, de novo CNA were not identified. Some loci showed de novo CNA in children, but those were included in the loci in which “normalized read count” and “Read Count Ratio” were variable and continuous values among samples. Because copy number alteration analyses using cn.MOPS include ~10 kb size alterations and chromosomal abnormality, we can conclude that relatively large genomic deletion/duplication is not frequent in children from A-bomb survivors.
Discussion
WGS analysis failed to provide evidence of an increase in de novo SNVs in children of atomic-bomb survivors. Although we only analysed data for three affected trios, the numbers of de novo variants and gross structural changes were comparable between children of radiation-exposed and non-exposed fathers, in line with previously published data [11]. All three exposed fathers suffered hair loss after the A-bomb explosion. Among the acute symptoms experienced following exposure to ionizing radiation, hair loss is considered to be a relatively clear sign of radiation effect, compared with symptoms related to infection, such as diarrhea or fever, which may have been caused by unsanitary conditions after the bombing. Hair loss was observed in about 30% of survivors exposed within 1.5 km of the hypocenter, mostly within 4 weeks [29]. The fact that the fathers in all the exposed trios mentioned that their hair loss started about 3 weeks after the explosion suggested that they had received a radiation dose of ≥2 Gy. The distance of the respective fathers from the hypocenter was 1.0–1.2 km, with some shields such as buildings or air-raid shelters. Their symptoms, and a calculated direct-exposure radiation dose of 8.6 Gy (as γ-rays) at 1.0 km [29] suggest that the fathers’ gonads would have been exposed to doses of radiation that could have affected the function of the spermatogonia. These factors indicate that the selected trios were appropriate subjects in which to address the study question.
We believe overall our filtering system is comparable to workflow in Kong’s paper except using QUAL when pick up the de novo variations. Difference from their workflow is the target genomic regions, i.e. we defined the target region with unique sequence because we must confirm almost all variants to count actual number in each trio. In Kong’s workflow, the target genomic regions are not defined, and “variant calls” are counted referring false positive rate. De novo variants in the segmentally duplicated region seem to be eliminated, but it is difficult to understand how many variants call in repetitive sequence were counted. Many of variants in repetitive sequences might be included, or might be excluded. Nonetheless, we think that our confirmed variants count and “variants calls” in readable region using strict filtering process in whole genome in Kong’s wokflow could be comparable. Because our target genomic region cover about 63% (eliminating 30.11% of “blacklist region” and 7.7% of unreadable genomic unique sequences), should the same mutation rate be applied to the repetitive sequence, 1.5 times de novo variants in trio shown in Table 3 could be found in whole genome.
This study focused on de novo SNVs, gross structural changes and copy number alterations in the genomes of A-bomb survivors’ children. However, the accuracy of the Illumina HiSeq 2500 short-read sequencer was limited, and short indels (1 bp to several kbp long) were not investigated in this study. Our conclusions concerning genomic alterations are therefore limited to SNVs, gross structural changes and copy number alterations including chromosomal abnormality. We set the window size 10 kb in cn.MOPS analyses to screen the relatively large deletion/insertion precisely and we never found large deletion or insertion. We did not intended to find small insertion/deletion (1 bp ~ several kb) in the current study, because the detection accuracy for small insertion/deletion is limited in the present short read NGS analyses. Now the only genomic changes that are not analyzed in three A-bomb survivor trio is small insertion/deletion (1 bp ~ several kb). Because short indels are the most common type of structural genomic change induced in irradiated cells, and novel sequencing technologies, such as the use of one-molecule sequencers for long reads instead of short-read sequencers, are needed to investigate these genomic alterations. Furthermore, we did not investigate transcriptome or epigenomic factors, such as DNA methylation or histone acetylation/methylation, and further studies are needed to determine the genome/epigenome status in offspring of atomic-bomb survivors. Despite these limitations, our data on de novo SNV variants in bomb-survivors’ children were compatible with the numbers in previous reports in the whole genome [11], thus supporting the idea that the incidences of de novo SNVs and gross structural changes are not increased in the offspring of atomic-bomb survivors. Of course, de novo SNVs information is now limited from the only three trios, many A-bomb survivor trios should be analysed along with control trios by confirming variants not by estimation.
Our investigation was also limited to trios in which only the father had been exposed to A-bomb radiation, and additional studies of the effects of maternal, and maternal/paternal exposure are needed to allow final conclusions to be drawn regarding de novo SNVs, gross structural changes and copy number alterations. The paternal survivors in this study were exposed to radiation at 6, 9, and 16 years old, respectively (before or during the development of secondary sexual characteristics and during non-dividing periods for spermatogonia), and the number of de novo SNV variants in radiation-exposed mothers of similar ages would thus not be expected to be increased. However, this possibility should be investigated. More genetic/epigenetic, as well as experimental and epidemiological studies, are required to further our understanding of the heritable effects of atomic-bomb radiation.
References
Kamiya K, Ozasa K, Akiba S, Niwa O, Kodama K, Takamura N, et al. Long-term effects of radiation exposure on health. Lancet. 2015;386:469–78.
Hsu WL, Preston DL, Soda M, Sugiyama H, Funamoto S, Kodama K, et al. The incidence of leukemia, lymphoma and multiple myeloma among atomic bomb survivors: 1950–2001. Radiat Res. 2013;179:361–82.
Preston DL, Ron E, Tokuoka S, Funamoto S, Nishi N, Soda M, et al. Solid cancer incidence in atomic bomb survivors: 1958–98. Radiat Res. 2007;168:1–64.
Muller HJ. Artificial transmission in the gene. Science. 1927;66:84–87.
Russell WL, Russell LB, Kelly EM. Radiation dose rate and mutation frequency. Science. 1958;128:1546–50.
Otake M, Schull WJ, Neel JV. Congenital malformations, stillbirths, and early mortality among the children of atomic bomb survivors: a reanalysis. Radiat Res. 1990;122:1–11.
Neel JV, Satoh C, Goriki K, et al. Search for mutations altering protein charge and/or function in children of atomic bomb survivors: final report. Am J Hum Genet. 1988;42:663–76.
Kodaira M, Izumi S, Takahashi N, Nakamura N. No evidence of radiation effect on mutation rates at hypervariable minisatellite loci in the germ cells of atomic bomb survivors. Radiat Res. 2004;162:350–6.
Kodaira M, Ryo H, Kamada N, Furukawa K, Takahashi N, Nakajima H, et al. No evidence of increased mutation rates at microsatellite loci in offspring of A-bomb survivors. Radiat Res. 2010;173:205–13.
Grant EJ, Furukawa K, Sakata R, Sugiyama H, Sadakane A, Takahashi I, et al. Risk of death among children of atomic bomb survivors after 62 years of follow-up: a cohort study. Lancet Oncol. 2015;16:1316–23.
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–5.
Kent WJ. BLAT-The BLAST-Like Alignment Tool. Genome Res. 2002;12:656–64.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:391–8.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinforma. 2013;43:10.1–33.
Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0. http://www.repeatmasker.org. 1996–2010.
Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–7.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–74.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. http://www.R-project.org/.
Wickham H. ggplot2: Elegant graphics for data analyses. New York: Springer-Verlag; 2009. http://ggplot2.org/.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
Yang L, Luquette LJ, Gehlenborg N, Xi R, Haseley PS, Hsieh C-H, et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell. 2013;153:919–29.
Batzer MA, Deininger PL, Hellmann Blumberg U, Jurka J, Labuda D, Rubin CM, et al. Standardized nomenclature for Alu repeats. J Mol Evol. 1996;42:3RepeatMasker Open-4.0, 2013-2015.6.
Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0, 2013–2015. http://repeatmasker.org.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3 -- new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006.
Gilbert ES, Ohara JL. Analysis of the DS86 atomic bomb radiation dosimetry methods using data on severe epilation. Radiat Res. 1984;100:124–38.
Young RW, Kerr GD, editors. Reassessment of the atomic bomb radiation dosimetry for Hiroshima and Nagasaki-Dosimetry System 2002. The Radiation Effect Research Foundation, Hiroshima 732-0815, Japan, 2005. http://www.rerf.or.jp/shared/ds02/index.html.
Acknowledgements
This work was partly supported by the Program of the Network-type Joint Usage/Research Centre for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University and Fukushima Medical University, and Takeda Science Foundation.
Author contributions
MH, YN, TM, and TH approached subjects, and collected data and samples. CH, AK, and K-iY performed sample preparation. MH, SS, DI, YI, KT, KY, and YM analysed the laboratory data. HM, AK, and K-iY analysed the genome data. YM and K-iY planned and organised the project. YM and K-iY wrote the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Horai, M., Mishima, H., Hayashida, C. et al. Detection of de novo single nucleotide variants in offspring of atomic-bomb survivors close to the hypocenter by whole-genome sequencing. J Hum Genet 63, 357–363 (2018). https://doi.org/10.1038/s10038-017-0392-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0392-9
This article is cited by
-
Heterozygous missense variant of the proteasome subunit β-type 9 causes neonatal-onset autoinflammation and immunodeficiency
Nature Communications (2021)
-
Multisite de novo mutations in human offspring after paternal exposure to ionizing radiation
Scientific Reports (2018)
