
Abstract
Episodic ataxias (EAs) are rare channelopathies characterized by recurrent ataxia and vertigo, having eight subtypes. Mutated genes were found in four of these eight subtypes (EA1, EA2, EA5, and EA6). To date, only four missense mutations in the Solute Carrier Family 1 Member 3 gene (SLC1A3) have been reported to cause EA6. SLC1A3 encodes excitatory amino-acid transporter 1, which is a trimeric transmembrane protein responsible for glutamate transport in the synaptic cleft. In this study, we found a novel missense mutation, c.383T>G (p.Met128Arg) in SLC1A3, in an EA patient by whole-exome sequencing. The modeled structural analysis suggested that p.Met128Arg may affect the hydrophobic transmembrane environment and protein function. Analysis of the pathogenicity of all mutations found in SLC1A3 to date using multiple prediction tools showed some advantage of using the Mendelian Clinically Applicable Pathogenicity (M-CAP) score. Various types of SLC1A3 variants, including nonsense mutations and indels, in the ExAC database suggest that the loss-of-function mechanism by SLC1A3 mutations is unlikely in EA6. The current mutation (p.Med128Arg) presumably has a gain-of-function effect as described in a previous report.
Similar content being viewed by others
Introduction
The most common and recognizable causes of pediatric acute ataxia are post-infectious cerebellar processes, from which recovery occurs naturally [1]. Chronic or recurrent spells of vertigo and ataxia may suggest episodic ataxias (EAs), which are rare inherited disorders of ion channel function (channelopathies) [2]. EAs include eight subtypes defined by the responsible mutated genes/loci and clinical features [3]. Mutated genes were found in four of eight subtypes, exhibiting autosomal dominant expression. EA1 and EA2 are the most common subtypes, caused by mutations in the Potassium Voltage-Gated Channel Subfamily A Member 1 gene (KCNA1) and the Calcium Voltage-Gated Channel Subunit Alpha1 A gene (CACNA1A) [2]. EA1 is clinically characterized by brief attacks of ataxia with myokymia. The episodes are commonly triggered by physical and emotional stress in early childhood [4]. EA2 is usually characterized by more prolonged attacks of ataxia and nystagmus lasting minutes to hours (or even days). Acetazolamide and 4-aminopyridine have been effective in treating EA2 [5]. In addition, mutations in Calcium Voltage-Gated Channel Auxiliary Subunit Beta 4 (CACNB4) and Solute Carrier Family 1 Member 3 (SLC1A3) cause EA5 and EA6, respectively [6, 7]. EA6 shows similar features to EA2, but without CACNA1A mutations. The other four subtypes (EA3, EA4, EA7, and EA8) have been linked to different chromosomal loci [2, 3].
SLC1A3 encodes excitatory amino-acid transporter 1 (EAAT1), consisting of 542 amino acids. EAAT1 clears glutamate from the synaptic cleft and regulates neurotransmitter concentrations at the excitatory glutamatergic synapses in mammals [7]. Three missense mutations in SLC1A3 have been registered as disease-causing mutations in the Human Gene Mutation Database (HGMD, Qiagen) [7,8,9]. In addition, very recently, Choi et al. [10] also reported one missense mutation. However, the clinical features of EA6 have not been fully elucidated.
In this paper, we report on a patient with EA harboring a novel missense mutation in SLC1A3. Clinical features are compared with those of previously reported cases and the pathogenicity associated with all of the reported mutations is evaluated.
Materials and methods
Patient
A patient with EA was focused on in this study. Detailed clinical information was obtained after careful examination by the clinician seeing this patient. The Institutional Review Board of Yokohama City University of Medicine approved the experimental protocols. Informed consent was obtained, in agreement with Japanese regulations.
Sample preparation and whole-exome sequencing
Genomic DNA was isolated from peripheral blood leukocytes using QuickGene 610L (Wako, Osaka, Japan). Genomic DNA was sheared and captured using the SureSelect Human All Exon v6 Kit (60 Mb; Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq2500 (Illumina, San Diego, CA, USA) with 101-bp paired-end reads. Exome data processing, variant calling, and variant annotation were performed as previously described [11]. The average read depth of protein-coding regions was 67.2× and at least 97% of the target bases were sequenced by 10 or more reads for the patient. Common single-nucleotide polymorphisms (SNPs) with minor allele frequencies ≥1% in dbSNP 137, and variants observed in more than five of our 575 in-house ethnically matched controls as an exome database were filtered out. Among the remaining rare variants, we focused on amino acid-altering or splicing-affecting variants. Particular attention was given to the mutations in previously reported causative genes associated with ataxia, cerebellar atrophy, and other neurodegenerative diseases. The variant in SLC1A3 was confirmed by Sanger sequencing with an ABI PRISM 3500×l autosequencer (Life Technologies, Carlsbad, CA, USA) using genomic DNA from the patient and her parents as a template.
Structural consideration
We constructed a modeled structure of human EAAT1 from an archaeal homolog using Protein Homology/analogY Recognition Engine V 2.0 (Phyre2) [12] with mapping of the mutation site.
Results
Clinical features
The patient was a 10-year-old girl born to healthy non-consanguineous parents and the family history was unremarkable for neurological disorders. She showed truncal ataxia and crossed eyes at the age of 11 months. Initial brain magnetic resonance imaging (MRI) and cerebrospinal fluid examination performed at this time were normal. She recovered spontaneously in a few days. Although her development was normal, she showed repeated episodes of cerebellar ataxia, such as truncal ataxia, intentional tremor, and slurred speech, at 8–9 years of age. Brain single-photon emission computed tomography (SPECT) indicated mild hypoperfusion of her cerebellum and brainstem at 8 years of age. Her symptoms resolved spontaneously in ~2 weeks and were absent for periods between the episodes. A similar episode occurred again at 10 years of age. Acetazolamide was effective for her symptoms. Most of her episodes arose in winter.
Genetic analysis
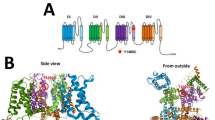
We performed whole-exome sequencing in the patient and found a missense mutation in SLC1A3, NM_004172.4:c.383T>G (p.Met128Arg). Sanger sequencing of samples from the patient and her parents revealed that c.383T>G had occurred de novo in the patient (Fig. 1a). Met128 is conserved in the eukaryotic EAAT1 protein (Fig. 1b) and located in transmembrane domain 3 (TM3) (Fig 1c) [13].

The SLC1A3 mutation found in this family. a Familial pedigree and electropherograms of the mutation. c.383T>G (p.Met128Arg) occurred de novo in the patient. b Met128 is evolutionarily highly conserved in vertebrates. c Schematic representation of the EAAT1 protein together with previously reported and our mutations. EAAT1 contains eight transmembrane domains and helical hairpins (HP1 and HP2). Black (reported) or red (this case) circles with lines indicate mutations. p.Met128Arg is located at transmembrane domain 3
c.383T>G is absent from dbSNP 137, our ethnically matched control database (n = 575), the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) database, and the Tohoku Medical Megabank Organization (ToMMo, https://ijgvd.megabank.tohoku.ac.jp/) database. Web-based prediction tools, SIFT (http://sift.jcvi.org/), MutationTaster (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and M-CAP, indicated that the mutation was pathogenic (in the first two) and possibly damaging (in the latter two).
Structural consideration
EAAT1 is a trimeric transmembrane protein (Fig. 2a) and the identified mutation falls within the third transmembrane α-helix (TM3), which comprises hydrophobic amino-acid residues (Fig. 2b). The methionine residue at the mutation position is located at the center of TM3 and would be exposed to lipids of the membrane. This might be related to the pathogenic role of Met128Arg.

Structural consideration of the SLC1A3 mutation. A modeled structure of human EAAT1 constructed from the crystal structure of a ligand-free trimeric glutamate transporter homolog from Thermococcus kodakarensis (PDB code 4ky0) [18] is viewed from the extracellular side (a) and parallel to the membrane (b). Each monomer of the trimer is colored cyan, magenta, or green. The methionine residue at the mutation site is shown as red van der Waals spheres in one monomer. b A magnified view around the mutation site is also shown. Hydrophobic side chains in the third transmembrane α-helix (TM3) are shown as cyan van der Waals spheres. Red and blue faces depicted as series of slits represent the extracellular and cytoplasmic surfaces of the lipid bilayers, respectively. The orientation of the protein in the membrane was obtained from the Orientations of Proteins in Membranes (OPM) database [19]
Discussion
To date, a total of five missense mutations (including our mutation) in SLC1A3 have been found in cases of ataxic disorders [7,8,9,10]. A summary of the comparative data of the clinical and mutational features in all cases is shown in Supplementary Table 1. SLC1A3 mutations are associated with a wide range of clinical phenotypes. Our case showed repetitive ataxic episode triggered physical stress, effective response to acetazolamide and normal MRI finding. Jen et al. [7] reported exceptionally the most severe case of c.869C>G (p.Pro290Arg) showing EA, mild cerebellar atrophy, severe intellectual disability, and seizures. The other four patients (three reported and the one in this study) harboring c.383T>G (p.Met128Arg), c.556T>A (p.Cys186Ser), c.1496G>A (p.Arg499Gln), or c.1177 G>A (p.Val393Ile) consistently showed episodic cerebellar ataxia but with normal MRI findings [8,9,10]. In addition, Adamczyk et al. [14] reported another case with c.657G>C (p.Glu219Asp), which has been registered as a polymorphism (rs2032892) associated with Tourette’s syndrome.
The modeled structure study suggested that Met128Arg would perturb the hydrophobic status of the membrane by the strong positive charge of the replaced arginine and affect EAAT1’s function. The reported mutations (Pro290Arg and Cys186Ser) reside in the transmembrane domain of EAAT1 and lead to decreased glutamate uptake [7, 8]. Met128 is located at the center of the TM3 domain and might similarly affect glutamate uptake. On the basis of the pathogenicity classification of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines [15], c.383T>G is pathogenic (PS2, PM1, PM2, PP3, and PP4).
The six missense mutations (including the current mutation) were evaluated using multiple computational predictive tools, but none of the prediction tools (such as Polyphen-2 and CADD) could consistently predict their pathogenicity (Supplementary Table 2). M-CAP is a machine learning method combining nine established pathogenicity likelihood scores for only rare missense variants [16]. M-CAP outperforms other tools with the evaluation of receiver operating characteristic curves [16]. M-CAP indicated that four of the six missense mutations are possibly pathogenic and the M-CAP scores were consistent with the age of onset or severity of the patients, indicating that M-CAP is the most useful prediction tool, especially in the evaluation of SLC1A3 variants.
The ExAC database contains 153 non-synonymous missense variants, 2 nonsense, 2 indels (leading to frameshift), and 2 canonical splice site substitutions in SLC1A3, indicating that the loss-of-function mechanism by SLC1A3 mutations is unlikely in EA6. The previous report described that p.Pro290Arg in SLC1A3 led to the gain-of-function of the EAAT1 transporter and increases anion currents in whole-cell patch clamp analyses [17]. Other mutations (including our p.Met128Arg) may have similar gain-of-function effects. M-CAP classified 58 (of 153) missense variants in ExAC as possibly pathogenic (Supplementary Table 3). Even pathogenic SLC1A3 variants might be included in ExAC, considering possible low penetrance and late onset in EA6 patients. c.1496G>A is indeed a pathogenic variant found in an EA6 patient [9], but also in ExAC. We should thus be very cautious in evaluating EA-related variants registered in ExAC.
In conclusion, this paper presents a novel pathogenic SLC1A3 mutation: c.383T>G (p.Met128Arg). The structural consideration suggests that Met128Arg may disturb the hydrophobic transmembrane environment and possibly affect protein function. The assessment of missense variants in SLC1A3 using M-CAP was relatively useful, but the evaluation of variants in control databases should be undertaken with caution, like in EA6.
References
Caffarelli M, Kimia AA, Torres AR. Acute ataxia in children: a review of the differential diagnosis and evaluation in the emergency department. Pediatr Neurol. 2016;65:14–30.
Jen JC. Hereditary episodic ataxias. Ann N Y Acad Sci. 2008;1142:250–3.
Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, McCoy B, et al. A novel locus for episodic ataxia: UBR4 the likely candidate. EJHG. 2014;22:505–10.
Mestre TA, Manole A, MacDonald H, Riazi S, Kraeva N, Hanna MG, et al. A novel KCNA1 mutation in a family with episodic ataxia and malignant hyperthermia. Neurogenetics. 2016;17:245–9.
Maksemous N, Roy B, Smith RA, Griffiths LR. Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol Genet Genomic Med. 2016;4:211–22.
Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66:1531–9.
Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529–34.
de Vries B, Mamsa H, Stam AH, Wan JJ, Bakker SLM, Vanmolkot KRJ, et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. 2009;66:97–101.
Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain. 2015;138:276–83.
Choi KD, Jen JC, Choi SY, Shin JH, Kim HS, Kim HJ, et al. Late-onset episodic ataxia associated with SLC1A3 mutation. J Hum Genet. 2017;62:443–6.
Iwama K, Sasaki M, Hirabayashi S, Ohba C, Iwabuchi E, Miyatake S, et al. Milder progressive cerebellar atrophy caused by biallelic SEPSECS mutations. J Hum Genet. 2016;61:527–31.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–58.
Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–8.
Adamczyk A, Gause CD, Sattler R, Vidensky S, Rothstein JD, Singer H, et al. Genetic and functional studies of a missense variant in a glutamate transporter, SLC1A3, in Tourette syndrome. Psychiatr Genet. 2011;21:90–97.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–6.
Winter N, Kovermann P, Fahlke C. A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents. Brain. 2012;135:3416–25.
Jensen S, Guskov A, Rempel S, Hanelt I, Slotboom DJ. Crystal structure of a substrate-free aspartate transporter. Nat Struct Mol Biol. 2013;20:1224–6.
Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–5.
Acknowledgements
We thank the individuals and their families for their participation in this study. We also thank Nobuko Watanabe and Mai Sato for technical assistance. We are also grateful to Tom Buckle, MSc, from Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript. This work was supported by grants from Research on Measures for Intractable Diseases; Comprehensive Research on Disability Health and Welfare; the Strategic Research Program for Brain Science (SRPBS); the Practical Research Project for Rare/Intractable Diseases; the Initiative on Rare and Undiagnosed Diseases in Pediatrics; the Initiative on Rare and Undiagnosed Diseases in Adults from the Japan Agency for Medical Research and Development; a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Grants-in-Aid for Scientific Research (A and B); Grant-in-Aid for Young Scientists (B); Challenging Exploratory Research from the Japan Society for the Promotion of Science; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency; grants from the Ministry of Health, Labor and Welfare; and the Takeda Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Iwama, K., Iwata, A., Shiina, M. et al. A novel mutation in SLC1A3 causes episodic ataxia. J Hum Genet 63, 207–211 (2018). https://doi.org/10.1038/s10038-017-0365-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0365-z
This article is cited by
-
Sex-Stratified Single-Cell RNA-Seq Analysis Identifies Sex-Specific and Cell Type-Specific Transcriptional Responses in Alzheimer’s Disease Across Two Brain Regions
Molecular Neurobiology (2022)
-
DNA methylation and gene expression signatures are associated with ataxia-telangiectasia phenotype
Scientific Reports (2020)
-
Haploinsufficiency of A20 caused by a novel nonsense variant or entire deletion of TNFAIP3 is clinically distinct from Behçet’s disease
Arthritis Research & Therapy (2019)
-
Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction
Communications Biology (2018)
