
Abstract
Background:
Therapeutic hypothermia (HT) is the only intervention that improves outcomes in neonatal hypoxic-ischemic encephalopathy (HIE). However, the multifactorial mechanisms by which HT impacts HIE are incompletely understood. The complement system plays a major role in the pathogenesis of ischemia-reperfusion injuries such as HIE. We have previously demonstrated that HT modulates complement activity in vitro.
Methods:
Term equivalent rat pups were subjected to unilateral carotid ligation followed by hypoxia (8% O2) for 45 min to simulate HIE. A subset of animals was subjected to HT (31–32°C for 6 h). Plasma and brain levels of C3a and C5a were measured. Receptors for C3a (C3aR) and C5a (C5aR) along with C1q, C3, and C9 were characterized in neurons, astrocytes, and microglia.
Results:
We found that HT increased systemic expression of C3a and decreased expression of C5a after HIE. In the brain, C3aR and C5aR are predominantly expressed on microglia after HIE. HT increased local expression of C3aR and decreased expression on C5aR after HIE. Furthermore, HT decreased local expression of C1q, C3-products, and C9 in the brain.
Conclusion:
HT is associated with significant alteration of complement effectors and their cognate receptors. Complement modulation may improve outcomes in neonatal HIE.
Similar content being viewed by others


Main
Brain injury in newborn infants resulting from perinatal asphyxia (hypoxic ischemic encephalopathy, HIE; hypoxia ischemia, HI) affects 1–3/1,000 live term births in the developed world (1). The incidence of HIE is much higher in developing countries, with estimates as high as 8/1,000 live term births (2). Therapeutic hypothermia (HT) is the only known intervention to improve short-term outcomes in HIE, with a 25% reduction in death or major neurodevelopmental disability at 18 mo of age (3,4). However, there is no significant difference in the composite primary outcome of death or IQ < 70 at 6– 7 y of age between the hypothermia treated and untreated controls (5). The incidence of death and disability remains unacceptably high at 40% even after HT (4), highlighting the urgent need for adjunct therapies to further improve outcomes in HIE. An important step in developing new therapies is to first elucidate the mechanisms by which HT manifests its beneficial effects.
The neuropathology of ischemia-reperfusion injuries (IRI) such as HIE is characterized by primary energy failure and cellular dysfunction due to oxygen deprivation during the ischemic phase, and augmentation of tissue damage during the reperfusion phase due to oxidative stress and inflammatory mediators (6). The human complement system, comprised of numerous membrane-bound and plasma proteins, acts as a bridge between the innate and adaptive immune response (7). In IRI, damaged hypoxic cells express neoepitopes that bind to circulating IgM antibodies, thus activating the inflammatory complement cascade via the classical or lectin pathways (8,9,10,11). Antibody-initiated complement activation appears to play a role in HIE by mediating inflammatory brain damage through deposition of C1q and C3 in the brain and generation of proinflammatory mediators such as C5a (12,13). Complement activation also leads to downstream effects such as neutrophil and macrophage aggregation, worsening the injury through the release of lysosomal enzymes, oxygen free radicals, and proinflammatory cytokines (14).
Experimental studies in rats have shown that C1q is highly expressed in the brain following ischemia (15), and that classical complement pathway activation via C1 generates proinflammatory mediators such as C5a, which are associated with hypoxic-ischemic brain injury (16). Additionally, deletion of C1q in mice not only reduces brain infarction and neurofunctional deficit but also results in protection of mitochondrial respiration, indicating a role for classical complement activation and brain oxidative stress (17,18). In the brain, C5a receptor (C5aR) is expressed on astrocytes, microglia, and neurons (19). Engagement of C5aR with C5a induces neuronal apoptosis, and inhibition of the C5a–C5aR interaction significantly blocks ischemia-induced apoptosis (20). Recent evidence has shown that C3a can induce either pro- or anti-inflammatory effects and in contrast to C5a, C3a prevents mobilization of neutrophils limiting their accumulation into tissues (21). Interestingly, signaling through C3a receptor (C3aR) positively regulates in vivo neurogenesis in adult mouse brain (22). Importantly, the anti-inflammatory effects of C3a dominate the acute phase of inflammation (21). C3a–C3aR interaction appears to ameliorate behavioral deficits after HIE, and C3aR has been suggested as a novel therapeutic target for the treatment of neonatal HIE (23).
In vitro data from our research group has demonstrated that HT temperatures modulate complement activation (12). We hypothesized that HT alters complement effectors in neonatal HIE. We therefore evaluated the extent to which HT after HIE altered systemic and local expression of C3a, C5a, and their receptors in an established animal model.
Methods
Antibodies and Reagents
Primary antibodies used for assays: Goat anti-rat C3 IgG (MP Biomedicals, Santa Ana, CA), Chicken anti-C3/C3a (Abcam, Cambridge, MA), Rabbit anti-rat C9 (generously provided by Professor Paul Morgan, Cardiff, UK), Mouse anti-C3aR (Thermo Fisher Scientific, Rockford, IL), Rabbit anti-C3aR (Bioss, Woburn, MA), Mouse anti-C5aR (Hycult Biotech., Plymouth Meeting, PA), goat anti-human C1q (Complement Technology, Tyler, Texas) mouse anti-NeuN (EMD Millipore, Temecula, CA), chicken anti-NeuN (EMD Millipore), mouse anti-GFAP (Sigma-Aldrich, St. Louis, MO), chicken anti-GFAP (Abcam), goat anti-Iba1 (Abcam) and mouse anti-GAPDH (Abcam). Secondary antibodies included goat antimouse horseradish peroxidase (HRP) (Sigma-Aldrich), goat antichicken IgY HRP (Genway Biotech, San Diego, CA), goat antirabbit HRP (Sigma-Aldrich), rabbit antigoat HRP (Thermo Fisher Scientific, Grand Island, NY), biotinylated goat antimouse IgG (H+L), biotinylated goat antichicken IgG (H+L) (Vector Laboratories, Burlingame, CA), Biotinylated goat antirabbit IgG (H+L) (Bioss), donkey antigoat IgG (H+L) Alexa Fluor (AF) 488/568, goat antichicken IgG (H+L) AF 488, goat antimouse IgG (H+L) AF 568, goat antirabbit IgG (H+L) AF 405/488 (Life Technologies, Grand Island, NY).
Animal Model of Unilateral Hypoxia-ischemia
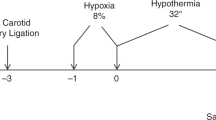
All animal experiments were performed under approved protocols by the Eastern Virginia Medical School Institutional Animal Care and Use Committee. Timed pregnant Wistar rats (Charles River Laboratories, Wilmington, MA) were purchased at embryonic (E) day 19, housed individually, and allowed to deliver spontaneously. Pups were randomized on the day of birth to control for litter effects (10/litter). P10 is considered equivalent to a human term newborn (24). At P10, HI brain injury was induced using the Vannucci method of unilateral carotid ligation (25). Briefly, the right carotid artery was isolated and ligated twice with 4-0 silk suture under 2% isoflurane anesthesia. After recovery with the dam for 60 min, pups were subjected to 8% O2/balanced nitrogen for 45 min at 37°C. Pups were then randomly assigned to normothermia (NT) and hypothermia (HT) groups. Pups in the HT group were placed in open jars in a temperature controlled chamber set to 28–30°C to maintain a target rectal temperature of 31–32°C for 6 h, while pups in the NT group were kept in a separate chamber at 37°C. Pups were rewarmed and placed back with the dam, and harvested at different time points (30 min – 5 d).
Tissue Harvest and Processing
Animals were deeply anesthetized with a fatal dose of pentobarbital (FatalPlus). After drawing blood, animals were perfused with ice-cold PBS (followed by 10% neutral buffered formalin for perfusion fixation if being harvested for histopathology). Harvested brains were separated into right and left hemisphere and stored in liquid nitrogen until use. Complete mini EDTA-free protease inhibitor cocktail (Roche) was dissolved in homogenization buffer (300 mmol/l sucrose, 0.05 mmol/l CaCl2, 0.1 mmol/l MgCl2, 0.1 mmol/l NaHCO3, 1 mmol/l Na3VO4) immediately before use, and each brain lobe was homogenized on ice. Whole cell and membrane protein fractions were extracted using a series of centrifugation steps, aliquoted and stored at −80oC until use. Protein concentration was determined using a bicinchoninic acid assay (BCA) according to the manufacturer’s recommendations (Thermo Fisher Scientific). For histopathology, 10% neutral buffered formalin was used for perfusion-fixation, brains were removed, processed and paraffin embedded (Excalibur Pathology, Norman, OK) and 5 µm coronal sections were cut using a RM2125 rotary microtome (Leica Microsystems, Wetzlar, Germany). To isolate plasma, blood collected from cardiac puncture in EDTA tubes was incubated at room temperature for 45 min, incubated on ice for 45 min, centrifuged and supernatant collected, aliquoted, and stored at –80oC.
Fluorescent Western Blotting
40 µg of whole cell lysate in 1× Tris-buffered saline (TBS) with 0.1% Tween 20 (TBST) were separated on a 4–20% Mini-PROTEAN TGX precast gradient gel (Bio-Rad) under reducing conditions and transferred to a 0.2 µm Immun-Blot PVDF membrane (Bio-Rad, Hercules, CA). After washing, the membrane was blocked overnight with 5% normal donkey serum (NDS) in TBST, followed by probing with rabbit anti-C3aR, followed by a 1 : 500 goat anti-rabbit IRDye 680 secondary antibody. The membrane was then blocked with 5% normal goat serum (NGS), followed by probing with mouse antiglyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1 : 500), followed by a 1 : 500 goat anti-mouse IRDye 800 secondary antibody. The membrane was imaged using a LICOR Odyssey imaging system using both 700 and 800 nm fluorescent channels.
Enzyme-Linked Immunosorbent Assay
The C3a and C5a levels from serum samples were measured according to the manufacturer’s instructions (rat C3a ELISA kit (TSZ ELISA, Waltham, MA) and rat C5a ELISA kit SEA388Ra (Cloud-Clone; Houston, TX)) using a 1 : 10 dilution of each serum sample or 100 µg of whole cell lysate in 1× PBS from the processed right hemisphere brain tissues.
Immunohistochemistry
About 5 μm paraffin-embedded coronal brain sections on slides were incubated (1 h; 60oC), deparaffinized with xylene, and rehydrated with ethanol (100, 95, and 70%; RT). Endogenous peroxidase activity was blocked (0.3% H2O2/MeOH; 10 min; RT), and heat induced antigen retrieval was performed using 10 mmol/l sodium citrate, pH 6.0. 10% normal serum (from the same species as the host of the secondary antibody) was used to block and to dilute antibodies. Sections were incubated with primary antibody (overnight, 4oC), washed, and then incubated with biotinylated secondary antibody (2 h; RT), followed by incubation with avidin-biotin complex (Vectastain ABC Elite kit, Vector) (45 min, RT). Sections were developed with diaminobenzidine (DAB substrate kit, Vector), counterstained in 20% Harris modified hematoxylin solution (Sigma), dehydrated with ethanol and cleared in xylene before mounting (Cytoseal XYL, Thermo Fisher Scientific), and coverslipping.
Fluorescent Immunohistochemistry
After hydration, antigen retrieval, and blocking as described, sections were incubated in both the primary antibodies together (overnight, 4oC). Sections were washed, incubated with one Alexa Fluor-conjugated secondary antibody (from the same host species as the blocking serum, targeting the host species of one of the primary antibodies) (2 h, RT), washed, incubated with the second Alexa Fluor-conjugated secondary antibody (targeting the host species of the other primary antibody), mounted with aqueous mounting media with 4′,6-diamidino-2-phenylindole (Vectashield antifade mounting medium with DAPI, Vector) and coverslipped.
Laboratory Methods
See the Supplementary Methods online section for other detailed experimental procedures, including Fluorescent ELISA, Tetrazolium chloride (TTC) Staining, Fluoro-Jade Staining, Hematoxylin and eosin (H&E), and Cresyl Violet Staining.
Imaging
For histopathology, a digital camera (DP70, Olympus Center, Valley Forge, PA), mounted on a BX50, Olympus microscope was used. Adobe photoshop CS5 was used to merge fluorescent IHC images and Image J (National Institutes of Health) was used for analysis.
Statistical Analysis
Means and SEMs were calculated from independent experiments. Statistical comparisons were made using the paired t-test and ANOVA where appropriate. Statistical analysis was performed with OpenEpi (Emory University) and SAS V9.3 (Cary, NC).
Results
HT is Neuroprotective in Neonatal HIE
In the Vannucci rat model, we assessed whether HT (decreasing body temperature to 31–32°C) after HIE is neuroprotective as demonstrated by decreased brain infarct size on TTC staining. HT animals displayed a decrease in infarct size by an average of 40% (±3.2% SEM) compared with normothermia (NT) animals (P = 0.01) ( Figure 1a ). H&E and Cresyl violet staining were performed for qualitative assessment of brain injury. H&E staining demonstrated a greater degree of neuronal necrosis, pyknosis, and karyorrhexis in the cortex of NT animals when compared HT animals ( Figure 1b – d ). Cresyl violet staining shows profound neuronal loss in NT animals, with relative preservation of Nissl substance in the cortex of the HT group ( Figure 1e – g ). Quantitation of Fluorojade B staining demonstrated greater numbers of degenerating neurons in the cortex, hippocampus, and thalamus of NT animals compared with HT animals (P = 0.04) ( Figure 1h – j ). Neuroprotection was consistently reproducible in HT animals.

Therapeutic hypothermia (HT) is neuroprotective in neonatal hypoxic-ischemic encephalopathy (HIE). (a) Tetrazolium chloride (TTC) staining shows that HT decreased brain infarct size by an average of 40% (±3.2% SEM) compared with normothermia (NT) animals (n = 5 per group, representative example shown). (b-d) H&E staining of the ipsilateral cortex reveals a greater degree of neuronal necrosis, pyknosis and karyorrhexis (arrows) in NT animals compared with HT animals (40X magnification). (e-g) Cresyl Violet staining of the ipsilateral cortex shows significantly greater preservation of neuronal architecture and Nissl substance in the HT brain compared with NT animals. (h–j) Fluorojade B staining of the ipsilateral cortex, hippocampus, and thalamus demonstrated a 2.5-fold decrease in the number of degenerating neurons (arrows) in the HT animals compared with the NT animals. (10X magnification, n = 3 per group, P = 0.04). (Control - b, e, h; NT – c, f, i; HT – d, g, j).
HT Modulates Systemic and Brain Levels of C3a and C5a
C3a has been shown to have an anti-inflammatory role, especially in the acute phase of the inflammatory processes such as IRI (26). We assessed C3a levels in the plasma of both NT and HT animals exposed to HIE by ELISA. As previously reported in rats (27), C3a is present systemically in no intervention controls. In this model of HIE, systemic C3a expression in the HT group was increased at 1, 8, and 16 h after the hypoxic insult, when compared with time-matched NT controls, reaching a maximum twofold difference at the 16 h time point (P = 0.03) ( Figure 2a ). C3a levels decreased in both groups by 48 h after the hypoxic insult ( Figure 2a ). In contrast to C3a, C5a is known to exacerbate IRI such as HIE by enhancing the release of inflammatory cytokines from activated microglia and astrocytes (28). To assess systemic C5a levels in NT and HT animals, plasma samples at various time points after intervention were analyzed by ELISA. Systemic C5a expression increased in NT animals at 8 and 16 h relative to early time points after brain hypoxia ( Figure 2b ). However, hypothermia significantly decreased C5a levels at 4, 8, and 16 h after the hypoxic insult ( Figure 2b ). C5a levels in NT animals increased 1.8-fold compared with HT at 8 h and 1.6-fold compared with HT at 16 h (P = 0.04) ( Figure 2b ). Measurement of C3a levels in brain homogenates by ELISA showed a sixfold increase in C3a levels in the HT group 48 h after the hypoxic insult when compared with NT animals (P = 0.001) ( Figure 2c ). Relative quantification of C5a in brain homogenates by fluorescent ELISA showed a significant decrease in C5a levels at 48 h after hypoxic insult in the HT animals when compared with NT (P = 0.001) ( Figure 2d ). These data demonstrate that HT after brain hypoxia increases systemic and brain C3a levels compared with normothermia controls, suggesting that C3a may contribute to hypothermia-mediated neuroprotection. Systemic C5a levels are decreased by hypothermia after brain hypoxia, suggesting that hypothermia moderates complement-mediated inflammation in this setting.

Therapeutic hypothermia modulates systemic and brain levels of C3a and C5a. (a) Systemic plasma C3a levels in normothermia (NT) and hypothermia (HT) animals were quantified by enzyme-linked immunosorbent assay (ELISA) at the indicated time points (n = 4–7 per time point). P < 0.05 at 1, 8, and 16 h. (b) Systemic plasma C5a levels in NT and HT animals were quantified by ELISA at the indicated time points (n = 4–7 per time point). P < 0.05 at 4, 8, and 16 h. (c) C3a levels in ipsilateral whole brain homogenates of NT and HT animals were quantified by ELISA at 24 and 48 h (n = 3). P = 0.001 at 48 h. (d) Relative quantification of C5a in ipsilateral whole brain homogenates of NT and HT animals at 24 and 48 h by fluorescent ELISA normalized to no-intervention controls (n = 3). P = 0.002 at 48 h. Asterisks represent statistically significant results; error bars represent SEM. Measurements in ng/mg brain protein. Solid lines/black bars represent NT and dotted line/white bars represent HT.
Microglia are the Predominant Cell Type Expressing Receptors for C3a and C5a in the Neonatal Rat Brain Cerebral Cortex
Receptors for C3a (C3aR) and C5a (C5aR) are known to be expressed on neurons, astrocytes, and microglia in most areas of the central nervous system including the cerebral cortex, hippocampus, thalamus, and cerebellum (13,29,30). In the neonatal rat brain subjected to HIE, we observed widespread reactive astrocytosis, neuronal necrosis, and microglial activation, as described in the literature (31). Given the changes in plasma and brain C3a and C5a levels in NT and HT animals subject to HIE, we were interested in identifying the specific CNS cell types expressing their cognate receptors C3aR and C5aR. Using multicolor fluorescent immunohistochemistry, at 48 h postintervention, the C3aR and C5aR were assessed for colocalization with activated microglia in the cerebral cortex. Baseline C3aR expression and ramified microglia with scant colocalization in no intervention controls is shown in Figure 3a – c . After hypoxic insult, colocalized staining for C3aR and microglia is observed, less in the NT animals ( Figure 3d , e ) when compared with HT animals ( Figure 3g – i ). Low baseline C5aR expression and ramified microglia are shown in Figure 3j – l . While robust colocalized staining for C5aR and microglia is seen in the NT cortex ( Figure 3m – o ), low levels of C5aR in the HT brain leads to scant colocalization with microglia ( Figure 3p – r ). Thus, C3aR and C5aR show robust colocalization with microglia after HIE. In contrast, there was relatively scant colocalization with astrocytes and neurons in areas of reactive gliosis (shown below, quantitation in Supplementary Figure S1 online). There was no significant difference in the relative number of NeuN, GFAP, and Iba1 positive cells in the measured fields between the NT and HT groups (Supplementary Figure S2 online)

Microglia express receptors for C3a (C3aR) and C5a (C5aR) in the infant rat brain. Coronal brain sections for normothermia (NT) and Therapeutic hypothermia (HT) animals (ipsilateral cortex) harvested at 48 h, were stained for microglia with antibody to microglial marker Iba-1, C3aR, and C5aR for analysis by fluorescent immunohistochemistry at 40X magnification. (a–c) show cortical brain tissue stained with Iba-I and C3aR (no intervention control), (d–f) show cortical brain tissue stained with Iba-I and C3aR (right side, NT), (g–i) show cortical brain tissue stained with Iba-I and C3aR (right side, HT), (j–l) show cortical brain tissue stained with Iba-I and C5aR (no intervention control), (m–o) show cortical brain tissue stained with Iba-I and C5aR (right side, NT), (p–r) show cortical brain tissue stained with Iba-I and C5aR (right side, HT). (C3aR – a,d,g; C5aR – j,m,p; Iba1 – b,e,h,k,n,q; Merged – c,f,i,l,o,r). Arrows indicate colocalization of C3aR or C5aR with activated microglia.
Hypothermia Increases C3a and C3aR Expression in the Brain
Next, we sought to evaluate whether changes in C3a plasma levels corresponded with changes in C3aR levels in the brain. As demonstrated by western blotting, C3aR is expressed in the normal neonatal rat brain with increased levels of C3aR signal detected in HT treated animals at 24 and 48 h compared with NT animals ( Figure 4a ). To quantify C3aR from the fluorescent IHCs (C3aR/GFAP, C3aR/Iba1, and C3aR/NeuN), 10 random fields per animal per assay (n = 3–4 animals per intervention per time point) at 40X magnification were photographed from the cortex, hippocampus and thalamus on the right side. Image J was used to threshold these images and the integrated density was compared between groups, and plotted as a graph. Quantification of fluorescent IHC images showed that expression of C3aR was 2.5-fold higher in HT brains compared with NT brains (P = 0.01) ( Figure 4b ). There was scant colocalization of C3aR with astrocytes ( Figure 4c – e ), and almost no colocalization with neurons ( Figure 4f – h ). These data demonstrate that HT increased C3aR expression after brain hypoxia compared with NT. Given that C3a–C3aR interactions are believed to play a positive role in neurogenesis, these data suggest that increased C3a–C3aR interactions may be one of the mechanisms by which hypothermia mediates neuroprotection (32).

Hypothermia increases C3a receptor (C3aR) expression in the brain. (a) Brain C3aR levels in normothermia (NT) and Therapeutic hypothermia (HT) animals were analyzed by western blot in ipsilateral brain homogenates at the indicated time points. A no intervention control (No Int) was included to provide a baseline level of C3aR. (b) Brain C3aR expression at 48 h: Quantification of C3aR in fluorescent IHCs (n = 3–4 per group per assay, C3aR/Iba-1, C3aR/GFAP, C3aR/NeuN, 10 fields per animal including the ipsilateral cortex, hippocampus, and thalamus at 40X magnification (P = 0.01). (c–e) Fluorescent IHC simultaneously staining for astrocytes (GFAP) and C3aR in the ipsilateral cortex in no-intervention controls (c), 48 h after NT (d) and HT (e). Arrows indicate C3aR expression on astrocytes in HT animals (f–h) Fluorescent IHC simultaneously staining for neurons (NeuN) and C3aR in the ipsilateral cortex in no-intervention controls (f), 48 h after NT (g) and HT (h). Black bars represent NT and white bars represent HT.
Hypothermia Decreases C5a Receptor Expression in the Brain
In order to test, if the significantly increased systemic C5a expression in the NT group translated into increased C5aR expression in the brain, we performed a series of IHC experiments probing for C5aR. A qualitatively greater C5aR expression in the NT group 48 h after intervention when compared with the HT group was observed by IHC ( Figure 5a – c ). Fluorescent IHCs were quantified as described above for C3aR assays. Quantification of fluorescent IHC showed that expression of C5aR was 2.5-fold higher in NT brains compared with HT brains (P = 0.04) ( Figure 5d ). There was scant C5aR expression in the no-intervention neonatal brain ( Figure 5a , e , h ). Increased C5aR expression in the NT group was primarily seen in microglia ( Figure 3o ) and, to a lesser extent, astrocytes ( Figure 5e – g ). There was almost no colocalization of C5aR with neurons ( Figure 5h – j ). C5aR signaling has been shown to regulate astrocyte proliferation, and subsequent scar formation (33). These data show that HT may mediate neuroprotection by decreasing C5aR expression after HIE.

Hypothermia decreases C5a receptor (C5aR) expression in the brain. (a–c) Qualitative IHC staining of the ipsilateral cortex for C5aR in no-intervention controls (a), 48 h after normothermia (NT) (b) and Therapeutic hypothermia (HT) (c). Arrows represent C5aR expression (20X magnification). (d) Brain C5aR expression at 48 h: Quantification of C5aR in fluorescent IHCs (n = 3–4 per group per assay, C5aR/Iba-1, C5aR/GFAP, C5aR/NeuN, 10 fields per animal including the ipsilateral cortex, hippocampus, and thalamus at 40X magnification (P = 0.04). Error bars represent SEM. (e–g) Fluorescent IHC simultaneously staining for astrocytes (GFAP) and C5aR in the ipsilateral cortex in no-intervention controls (e), 48 h after NT (f) and HT (g). Arrows indicate C5aR expression on astrocytes in NT animals (h–j) Fluorescent IHC simultaneously staining for neurons (NeuN) and C5aR in the ipsilateral cortex in no-intervention controls (h), 48 h after NT (i) and HT (j).
Hypothermia Decreases Total C1q, C3, and C9 Expression in the Brain Cortex
The classical complement pathway initiator molecule C1q has been shown to enhance apoptosis in IRI through microglia, and enhance release of MCP-1 and IL-6 (34). Additionally, increased C3 and C9 expression in the brain after HIE has been previously described (35). In order to determine the relative levels of these complement factors in NT and HT animals subject to HIE, brain sections were analyzed by IHC 48 h after intervention. The level of staining for total C3/C3-fragment expression in NT cortex was much greater than that observed in HT animals ( Figure 6a – c ). Quantification of fluorescent IHCs (C3/Neun, C3/GFAP, C3/Iba1, as described above) showed a trend toward decreased C3/C3-fragment staining in the HT group compared with NT (P = 0.06) ( Figure 6d ) Total C9 expression in brain cortex was greater in NT compared with HT animals ( Figure 6e – g ). Quantification of fluorescent IHCs showed that C9 expression decreased by twofold in the HT group compared with NT (P = 0.04) ( Figure 6h ). For C1q, there appeared to be greater expression of C1q in the NT brain cortex compared with the HT group ( Figure 6i – k ). C1q levels were measured in whole brain lysates by fluorescent ELISA, and showed that C1q expression decreased by twofold in the HT group compared with NT (P = 0.04) ( Figure 6l ). C3 and C9 were expressed on microglia and neurons (Supplementary Figure S3 online), whereas C1q was predominantly expressed on microglia (data not shown). These data demonstrate that hypothermia after brain hypoxia decreased expression of critical complement effectors C3-fragments (i.e., opsonins), C9 (i.e., membrane attack complex), and C1q (i.e., apoptotic cell clearance) compared with normothermia. Increased C1q binding together with increased C3-fragment and C9 expression suggests that classical pathway activation may be a major contributor to the generation of these effectors.

Hypothermia decreases total C1q, C3, and C9 expression in the brain. (a–c) Qualitative IHC staining for C3/C3-fragments in the ipsilateral cortex of no-intervention controls (a), 48 h after normothermia (NT) (b) and Therapeutic hypothermia (HT) (c) (20X magnification). (d) Quantification of C3 IHC (P = 0.06) (e–g) Qualitative IHC staining for C9 fragments in the ipsilateral cortex of no-intervention controls (e), 48 h after NT (f) and HT (g) (20X magnification) (h) Quantification of C9 IHC (*P = 0.04). For C3 and C9 quantification, in fluorescent IHCs (n = 3–4 per group per assay, C3 (or C9)/Iba-1, C3 (or C9)/GFAP, C3 (or C9)/NeuN, 10 fields per animal including the ipsilateral cortex, hippocampus, and thalamus at 40X magnification (i–k) IHC staining for C1q fragments in the ipsilateral cortex of no-intervention controls (i), 48 h after NT (j) and HT (k). (l). Relative quantification of C1q in ipsilateral whole brain homogenates of NT and HT animals at 48 h by fluorescent enzyme-linked immunosorbent assay (ELISA) normalized to no-intervention controls (n = 3). P = 0.04. Asterisks represent statistically significant results and error bars represent SEM.
Discussion
This study modeling HIE in full-term neonates demonstrates previously unknown changes in C3a and C5a and their respective receptors in the pathophysiology of ischemia-reperfusion. HIE increased systemic expression of C5a and the local (microglia and astrocytes) expression of C5aR in the brain. Activation of the C5a–C5aR axis has been shown to increase astrogliosis and glial scar formation, inducing chemotaxis and activation of granulocytes, with subsequent chemokine and cytokine production, release of reactive oxygen species and enhancement of apoptosis (33). Hypothermia appears to mediate its anti-inflammatory action, in part, by inhibiting the C5a–C5aR axis. Hypothermia also increases systemic C3a expression and local (microglia and astrocytes) expression of C3aR in the brain. To our knowledge, this is the first study describing the complex changes in complement effectors in neonatal HIE.
There is evidence demonstrating that C3a regulates neurogenesis by directly affecting the properties of neural progenitor cells, and thus has a role in repair and regeneration of the brain (22). Another study showed that prolonged exposure to C3a reduced HI-induced hippocampal tissue loss and that single dose C3a treatment of wild-type mice 1 h after hypoxic ischemia ameliorated hypoxic ischemia-induced memory impairment by acting through its canonical receptor C3aR (23). In a mouse model of intestinal ischemia-reperfusion, the elimination of C3aR led to greater numbers of neutrophils infiltrating the intestine, thus exacerbating tissue damage (26). Our data suggest that increased C3aR expression and increased C3a–C3aR interactions during hypothermia after brain hypoxia may contribute to decreased inflammation and tissue damage.
In our model of neonatal HIE, C3aR, and C5aR in the brain were predominantly expressed on microglia. Previous investigators have shown that neuroinflammation after kainite administration led to microglial activation in the brain by regulating the expression of the complement C5a receptor genes in microglia (36). C5a and C5aR have been shown to be upregulated in other microglia mediated inflammatory processes (37). Additionally, blockade of C5aR by the antagonist PMX205 has been shown to be beneficial in different models of neurodegeneration and neuroinfammation (38). Immunotherapy with a C5a peptide vaccine has been shown to reduce microglial activation, and thus neuroinflammation (39). Our data suggest that hypothermia decreases C5a generation and C5aR expression after brain hypoxia potentially contributing to decreased reperfusion injury.
Additionally, hypothermia inhibited the expression of C1q (i.e., clearance of apoptotic cells and classical pathway activation), C3-fragments (i.e., opsonins) and C9 (i.e., membrane attack complex) in brain tissue after hypoxia, suggesting that inhibition of these complement effectors plays a role in hypothermia neuroprotection. We show decreased C3 expression with simultaneous significant elevation of C3a presence in the HT group. Since C3 is a converging point for classical, mannose binding protein and alternative pathways, this may be secondary to C3 consumption due to activation of all three pathways. There are conflicting data regarding the role of C9 in HIE (18,40). We show decreased C9 deposition in the brain in HT treated animals, but this does not prove that decreased C9 deposition is neuroprotective.
This study demonstrates significant and consistent changes in C3a, C5a, and their respective receptors, but these novel associations do not prove causality. Moreover, one could argue that the changes seen are not a direct consequence of HT, but are a result of its nonspecific neuroprotective effects. The role of complement in ischemia-reperfusion is an emerging field with rapidly evolving understanding. Recent studies have shown that the previously understood roles of many complement effectors have been incomplete. For example, pharmacological blockade of C5aR during the first 7 d after spinal cord injury improved recovery, but continued administration of the antagonist beyond the acute phase of injury resulted in worse outcomes, when compared with vehicle controls (33). Our study helps define role of complement in the current medical therapy (HT), and provides important information for future intervention studies. Further investigation is needed to refine our understanding of the myriad complement-meditated effects of this single intervention (hypothermia) on HIE. Future studies will use specific modulators of the complement pathway that will target C3a, C5a, and their receptors selectively and in combination to further understand these associations. Of critical importance is determining the roles of complement in HIE in terms of how the timing of various effectors impact inflammation, apoptosis, and healing.
Statement of Financial Support
This study was supported by a grant from the Commonwealth Health Research Board, Richmond, Virginia.
Disclosure
The authors have no conflicts of interest or financial disclosures to report with regard to this manuscript.
References
Rocha-Ferreira E, Hristova M. Antimicrobial peptides and complement in neonatal hypoxia-ischemia induced brain damage. Front Immunol 2015;6:56.
Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 2010;86:329–38.
Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev 2013;1:Cd003311.
Papile LA, Baley JE, Benitz W, et al. Hypothermia and neonatal encephalopathy. Pediatrics 2014;133:1146–50.
Shankaran S, Pappas A, McDonald SA, et al.; Eunice Kennedy Shriver NICHD Neonatal Research Network. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 2012;366:2085–92.
Lai MC, Yang SN. Perinatal hypoxic-ischemic encephalopathy. J Biomed Biotechnol 2011;2011:609813.
Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res 2010;20:34–50.
Arumugam TV, Magnus T, Woodruff TM, Proctor LM, Shiels IA, Taylor SM. Complement mediators in ischemia-reperfusion injury. Clin Chim Acta 2006;374:33–45.
Elvington A, Atkinson C, Kulik L, et al. Pathogenic natural antibodies propagate cerebral injury following ischemic stroke in mice. J Immunol 2012;188:1460–8.
Chan RK, Ibrahim SI, Verna N, Carroll M, Moore FD Jr, Hechtman HB. Ischaemia-reperfusion is an event triggered by immune complexes and complement. Br J Surg 2003;90:1470–8.
Zhang M, Austen WG Jr, Chiu I, et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci USA 2004;101:3886–91.
Shah TA, Mauriello CT, Hair PS, et al. Clinical hypothermia temperatures increase complement activation and cell destruction via the classical pathway. J Transl Med 2014;12:181.
Van Beek J, Bernaudin M, Petit E, et al. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol 2000;161:373–82.
Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 2012;298:229–317.
Schäfer MK, Schwaeble WJ, Post C, et al. Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J Immunol 2000;164:5446–52.
Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol 2007;171:715–27.
Ten VS, Sosunov SA, Mazer SP, et al. C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke 2005;36:2244–50.
Ten VS, Yao J, Ratner V, et al. Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J Neurosci 2010;30:2077–87.
Crane JW, Baiquni GP, Sullivan RK, et al. The C5a anaphylatoxin receptor CD88 is expressed in presynaptic terminals of hippocampal mossy fibres. J Neuroinflammation 2009;6:34.
Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J 2012;26:3680–90.
Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immunol 2015;194:3542–8.
Shinjyo N, Ståhlberg A, Dragunow M, Pekny M, Pekna M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells 2009;27:2824–32.
Järlestedt K, Rousset CI, Ståhlberg A, et al. Receptor for complement peptide C3a: a therapeutic target for neonatal hypoxic-ischemic brain injury. FASEB J 2013;27:3797–804.
Patel SD, Pierce L, Ciardiello A, et al. Therapeutic hypothermia and hypoxia-ischemia in the term-equivalent neonatal rat: characterization of a translational preclinical model. Pediatr Res 2015;78:264–71.
Vannucci SJ, Seaman LB, Vannucci RC. Effects of hypoxia-ischemia on GLUT1 and GLUT3 glucose transporters in immature rat brain. J Cereb Blood Flow Metab 1996;16:77–81.
Wu MC, Brennan FH, Lynch JP, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci USA 2013;110:9439–44.
Lillegard KE, Loeks-Johnson AC, Opacich JW, et al. Differential effects of complement activation products c3a and c5a on cardiovascular function in hypertensive pregnant rats. J Pharmacol Exp Ther 2014;351:344–51.
O’Barr S, Cooper NR. The C5a complement activation peptide increases IL-1beta and IL-6 release from amyloid-beta primed human monocytes: implications for Alzheimer’s disease. J Neuroimmunol 2000;109:87–94.
Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Fang S, Parsa AT. Complement and the central nervous system: emerging roles in development, protection and regeneration. Immunol Cell Biol 2010;88:781–6.
Bénard M, Gonzalez BJ, Schouft MT, et al. Characterization of C3a and C5a receptors in rat cerebellar granule neurons during maturation. Neuroprotective effect of C5a against apoptotic cell death. J Biol Chem 2004;279:43487–96.
Liu F, McCullough LD. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol Sin 2013;34:1121–30.
Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol 2011;48:1592–603.
Brennan FH, Gordon R, Lao HW, et al. The complement receptor C5aR controls acute inflammation and astrogliosis following spinal cord injury. J Neurosci 2015;35:6517–31.
Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni MG. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci 2014;8:380.
Cowell RM, Plane JM, Silverstein FS. Complement activation contributes to hypoxic-ischemic brain injury in neonatal rats. J Neurosci 2003;23:9459–68.
Nomaru H, Sakumi K, Katogi A, et al. Fosb gene products contribute to excitotoxic microglial activation by regulating the expression of complement C5a receptors in microglia. Glia 2014;62:1284–98.
Ager RR, Fonseca MI, Chu SH, et al. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer’s disease. J Neurochem 2010;113:389–401.
Woodruff TM, Crane JW, Proctor LM, et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J 2006;20:1407–17.
Landlinger C, Oberleitner L, Gruber P, et al. Active immunization against complement factor C5a: a new therapeutic approach for Alzheimer’s disease. J Neuroinflammation 2015;12:150.
Aly H, Khashaba MT, Nada A, et al. The role of complement in neurodevelopmental impairment following neonatal hypoxic-ischemic encephalopathy. Am J Perinatol 2009;26:659–65.
Acknowledgements
The authors thank Susan Vannucci, Shyama Patel, Jiyong Zhang, Amber Ciardiello, Trent Woodruff, and C.W. Gowen for their support and guidance.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Material
(DOCX 18 kb)
Supplementary Figures
(PDF 558 kb)
Rights and permissions
About this article

Cite this article
Shah, T., Nejad, J., Pallera, H. et al. Therapeutic hypothermia modulates complement factor C3a and C5a levels in a rat model of hypoxic ischemic encephalopathy. Pediatr Res 81, 654–662 (2017). https://doi.org/10.1038/pr.2016.271
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.271
This article is cited by
-
Mild Hypothermia Alleviates Complement C5a-Induced Neuronal Autophagy During Brain Ischemia–Reperfusion Injury After Cardiac Arrest
Cellular and Molecular Neurobiology (2023)
-
Targeting Complement C3a Receptor to Improve Outcome After Ischemic Brain Injury
Neurochemical Research (2021)
-
Identification of novel biomarkers for neonatal hypoxic-ischemic encephalopathy using iTRAQ
Italian Journal of Pediatrics (2020)
