
Abstract
Background:
We describe childhood growth patterns in a series of well-characterized patients with congenital hypogonadotropic hypogonadism (CHH) with special emphasis on genotype–phenotype correlation.
Methods:
We retrospectively evaluated the growth charts of 36 males with CHH (27 from Finland and 9 from Denmark). Fifteen patients (42%) had representative growth measurements during the first year of life. Genetically verified diagnosis of CHH was made in 15 (42%) patients (KAL1, FGFR1, GNRHR, or PROK2).
Results:
We found a deceleration of growth rate during early childhood. The mean (SD) length standard deviation score (SDS) at birth (0.2 (1.6) SDS) decreased significantly during the first 3 (to −0.9 (1.2) SDS) and 6 mo of life (to −0.7 (1.3) SDS). At the average age of 3 y, mean height SDS (−0.2 (1.3) SDS) did not differ from mid-parental target height (MPH). Mean height SDS reached its nadir (−1.7 (1.4) SDS) at an average age of 15.8 (0.8) years reflecting pubertal failure. Final heights did not differ from MPH. No clear genotype-growth associations emerged.
Conclusion:
Moderate postnatal length deflection is a novel feature of CHH and may reflect early androgen deficiency. Childhood growth patterns are not of clinical value in targeting molecular genetic diagnosis of CHH.
Similar content being viewed by others
Main
In boys, the postnatal activity of the hypothalamic–pituitary–gonadal (HPG) axis displays fascinating temporal variation. During the first 24 h of life, there is a transient peak in LH secretion, which subsides rapidly (1). The next wave of HPG activation starts approximately at the age of 1 wk, and lasts for a few months (2,3,4,5,6). During this minipuberty of infancy, gonadotropin, and sex steroid levels may reach adult levels. Thereafter, the HPG axis remains relatively quiescent until the late prepubertal years when hypothalamic GnRH secretion is gradually reactivated. One of the apparent physiological correlates of the HPG axis activity is somatic growth. During puberty, for example, there is complex crosstalk between the HPG axis activity and GH/IGF-1 axis, which leads to pubertal growth spurt and attainment of final height (7). However, it is unclear whether sex steroids within or below the normal range regulate length and weight gain in infants and children (8).
Congenital hypogonadotropic hypogonadism (CHH) is a rare genetic disorder with impaired GnRH secretion or action resulting in sex steroid deficiency, absent puberty, and infertility. Subjects with CHH typically suffer from low sex steroid levels during minipuberty (9,10), and thereby CHH provides a meaningful model to study the relationship between early sex steroid deficiency and growth. Although somatic growth and final heights of patients with CHH have been characterized before (11,12,13,14,15,16), these studies stem from the era when no comprehensive knowledge on molecular genetics of CHH was available. Previous reports show, however, that subjects with CHH appear to grow relatively normally during childhood (11,12,13,14,15,16), and expectedly display a decrease in growth rate during adolescence due to the absence of pubertal growth spurt. On the other hand, previous series included very limited growth data during early childhood (12,13).
Herein, we investigated growth during infancy, childhood, and adolescence in a clinically and genetically well-characterized series of CHH males.
Results
Growth in CHH Patients During Early Infancy
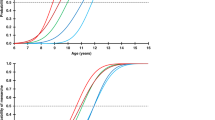
We first examined growth patterns in 15 male CHH patients with available auxological data during the first 6 mo of life. Their average length standard deviation score (SDS) decreased significantly from birth (0.2 (1.6) SDS) to 3 mo (−0.9 (1.2) SDS, P < 0.01) and 6 mo of age (−0.7 (1.3) SDS, P < 0.05) ( Figure 1a ). The mean length SDSs at 3 and 6 mo of age were lower than their mid-parental target height (MPH) (−0.02 (0.9) SDS; P < 0.05). To further test if the postnatal growth deflection exists in CHH patients with birth length within the normal range (±2 SD), we excluded three Danes and one Finn from first year growth analysis. Even within this subgroup (n = 11), the average length SDS decreased significantly from birth (−0.3 (1.3) SDS) to 3 mo (to −1.0 (1.3) SDS, P < 0.01).

Growth during early infancy. (a) Length SDS of patients with CHH during the first 6 mo of life. (b) Weight-for-length in patients (DW%) with CHH. *P < 0.05 and **P < 0.01. CHH, congenital hypogonadotropic hypogonadism; SDS, standard deviation score.
One boy had received i.m. testosterone treatment for micropenis during the first 6 mo of life, which may accelerate growth (17); exclusion of this subject did not change these results. Analysis of weight gain in infancy showed a significant reduction in weight-for-length in the CHH patients from birth (2 (10) %) to 6 mo of age (−4 (10) %, P < 0.05) ( Figure 1b ).
Growth During Childhood, Adolescence, and Final Height
After the first 6 mo, growth rate appeared to increase, as the mean height SDS reached −0.1 (1.9) SDS (n = 21) by the age of 3 y. However, the change in height SDS between 6 mo and 3 y of age in patients with measurements available at both time points (n = 13) failed to reach statistical significance (mean change 0.7 SDS, P = NS). At the average age of 3 y, the respective mean height SDS was similar as compared to the general population and MPH SDSs (n = 21) (P = NS). At the mean age of 7 y, the boys with CHH (n = 29) were shorter (mean height, −0.7 (1.2) SDS, P < 0.01) than the general population, and their mean height SDS was below their MPH SDS (P < 0.05). Similar results were found in boys with CHH and birth length within the normal range (± 2 SD). Height SDS reached its nadir (mean = −1.7 (1.4) SDS) at an average age of 15.8 (0.8) y and increased thereafter due to the accelerated growth rate after induction of puberty ( Figure 2 ). The mean final height of the patients (n = 33) did not differ from the general population (mean final height, −0.4 (1.3) SDS, P = NS) or MPH (mean difference = −0.3 (1.1) SDS, n = 20, P = NS; Figure 2 ). We found no correlation between the age at the induction of puberty and final height (r = 0.12, n = 31, P = NS). However, the cumulative dose of androgens used during the first year of pubertal induction correlated negatively with final height (r = −0.7, n = 14, P < 0.05) and with final height–MPH (r = −0.6, n = 10, P < 0.05).

Height SDSs of 27 Finnish (gray line) and 9 Danish (dashed line) males with CHH. Final heights (FH) (crosses, n = 31) and MPH (box plot, n = 22) are shown. Horizontal line in the FH plot indicates the median. CHH, congenital hypogonadotropic hypogonadism; SDS, standard deviation score.
During childhood and adolescence, the majority of the patients were of normal weight with mean age- and sex-adjusted BMI (ISO-BMIs) of 21.5 (4.1) kg/m2 (n = 24), 22.1 (4.8) kg/m2 (n = 24), and 22.3 (4.4) kg/m2 (n = 30) at the mean ages of 5, 10, and 16 years, respectively. During childhood, five boys were overweight (ISO-BMI > 25 kg/m2) and two boys were obese (ISO-BMI > 30 kg/m2).
Genotype–Phenotype Correlations
For genotype–phenotype analyses, patients with a molecular genetic diagnosis were divided to three groups: those with mutations in KAL1 (seven patients), FGFR1 (five patients), and to those (three patients) with mutations in either GNRHR or PROK2. The frequencies of micropenis and/or cryptorchidism were not different across these groups (57, 80, and 33%, respectively, P = NS). At 3 mo of age, CHH patients harboring mutations in these genes (n = 7) were shorter than the general population (−1.0 (0.9) SDS, P < 0.05), and their length SDS deflected strongly from birth (0.1 (1.2) SDS) to 3 mo of age (−1.0 (0.9) SDS, P < 0.01). Patients with KAL1 (n = 7) mutations had significantly lower birth length SDS as compared to patients with FGFR1 (n = 4) mutations (−0.7 (0.8) and 0.8 (0.8) SDS, respectively, P < 0.05). During adolescence, growth rate and attained final heights (mean = −0.8 (1.1) SDS) did not differ between the three groups of patients with different molecular genetic diagnoses, or between those with or without a molecular genetic diagnosis (P = NS).
Age at Induction of Puberty in Finnish Patients With CHH
In the Finnish CHH patients, clinical data were available from the 1970s onwards, and, during that time, the age at induction of puberty had declined (r = −0.4, P < 0.05). The age at the induction of puberty did not correlate with final height or MPH subtracted from final height (r = 0.1, P = NS and r = −0.3, P = NS, respectively).
Discussion
In the current work, we describe growth patterns in a clinically and genetically well-characterized population of males with CHH. We found that patients with CHH (i) display decreased growth rate during the minipuberty of infancy, (ii) exhibit normal childhood growth, and (iii) attain normal adult height following induction of puberty. Although patients with KAL1 mutations were shorter than those with FGFR1 mutations at birth, no postnatal genotype-specific growth patterns emerged.
During infancy, growth is modulated by thyroid hormone, GH/IGF-1 axis, insulin, and nutrition. Carbohydrate and amino acid-containing diet increases the secretion of IGF-1 (18), which enhances growth rate and correlates positively with length gain at 3 to 12 mo of age (18,19,20,21,22,23). The stimulatory influence of sex steroids on the GH/IGF-I axis in boys during puberty has been well established (7), while little evidence exists about the interactions between sex steroids and the GH/IGF-I axis during infancy. Thus, sex steroids have been considered to have little, if any, influence on growth during prenatal and immediate postnatal periods of life. Our results challenge this view, as patients with CHH displayed decreased growth rate at the time of minipuberty. Indeed, androgens may promote growth through the GH/IGF-I axis, since IGF-I levels correlate with growth velocity during minipuberty and puberty (7,20,23). Further, one cannot exclude a direct growth-promoting effect of sex steroids on the developing growth plate in mediating the growth-promoting effects of androgens in infant boys (24,25). The exact reason for the difference in birth lengths between patients with KAL1 or FGFR1 mutations remains speculative. Typically mutations in KAL1 cause very severe gonadotropin and sex steroid deficiency, and there is evidence suggesting that intrauterine sex steroid exposure may modulate birth length (26). Thus, it is tempting to assume that differences in intrauterine gonadotropin and sex steroid levels have contributed to this finding. Unfortunately, this genotype–phenotype correlation cannot be studied further in genetically modified mouse models, as a murine ortholog for KAL1 has not yet been identified.
After infancy, the nutritional component of IGF-1–dependent growth is gradually replaced by GH stimulation on IGF-1 synthesis (20,27). During childhood, boys with CHH experienced relatively normal childhood growth. Similarly, previous studies have described unremarkable childhood growth in patients with CHH (11,12,13,14,15,16). This growth phase is mainly GH/IGF axis dependent, and our results support the concept that growth during this period of life is not significantly modified by testicular androgens, although some gonadotropin secretion is detected already in 5-y-old-boys (28). In contrast, during adolescence, the influence of androgens on the regulation of linear growth increases, and boys with CHH suffer from absent pubertal growth spurt due to sex steroid deficiency. Consequently, their height reaches its nadir at adolescence as described in our series. Despite the fact that a single-nucleotide polymorphism rs7012413*T in FGRF1 is associated with obesity (29), we found normal ISO-BMI trajectories in patients with FGFR1-mutations. Thus, FGFR1 does not seem to directly regulate body composition in childhood.
Whereas previous reports have suggested that due to delayed fusion of the growth plates, the final heights of CHH patients typically exceed their MPH or the final height of general population (11,15,16), we found no difference between the average final heights and MPHs. This discrepancy is potentially explained by the apparently younger age at the age of pubertal induction in our CHH patient series (15,16). In line with this, Uriarte et al. (16) reported a positive correlation between the age at initiation of sex hormone replacement therapy and final height. Interestingly, we showed that the cumulative dose of androgens during the first year of pubertal induction associated negatively with final height. Some patients, however, were treated in the 1970s and received directly injections of high-dose testosterone, which is not recommended at present. We found a downward trend in the age of pubertal induction during the last five decades, which is assuring, as we recently showed that delayed diagnosis and treatment in CHH patient associates with impaired health-related quality of life (30).
This study has limitations that merit consideration. First, growth data was collected retrospectively. This was inevitable, because approximately only one patient with Kallmann syndrome is born in Finland annually, and thus, a prospective study design would have been practically impossible to conduct (31). Second, it may be argued that our patient series was heterogeneous. Clinical and genetic heterogeneity is, however, characteristic for CHH (32), and the absence of genotype–phenotype correlation in postnatal growth reported herein suggests that this is not a major concern. Finally, we anticipate that deficient growth in length during minipuberty will facilitate the early diagnosis of CHH, but this hypothesis needs to be prospectively tested.
In conclusion, our finding suggest that decreased growth rate during the first months of life is a novel nonreproductive clinical feature of males with CHH. During childhood, growth appears to be normal, and following the decrease in height SDS due to sex steroid deficiency during adolescence, a final height consistent with MPH is reached. Childhood growth patterns appear to provide no clinical value in targeting molecular genetic diagnostics of CHH.
Methods
This retrospective study included growth charts of 36 patients, 27 from Finland and 9 from Denmark, taking part in a nationwide study of CHH in both countries. Clinical features and genetic data of the patients have been partly published before (31,33,34). In brief, the diagnosis of CHH in the majority of patients was based on (i) absent or incomplete puberty (as judged by testis growth) at the age of 18 y, (ii) low sex steroid levels accompanied by normal or subnormal gonadotropin levels, (iii) otherwise normal anterior pituitary function, and (iv) no organic or syndromic cause for their condition. Six Finnish and two Danish patients were diagnosed earlier based on the history of micropenis and/or cryptorchidism and no spontaneous testicular growth during the clinical follow-up (n = 7) (35). In addition, one Dane had a micropenis and very low LH (<0.05 IU/l), inhibin B (53 pg/ml), and testosterone (<0.23 nmol/l) levels during the minipuberty. It is important to note that induction of puberty occurred (and should occur) clearly earlier than 18 y of age (i.e., prior to the definitive diagnosis of the condition) to diminish the psychological suffering caused by sexual infantilism (35).
Kallmann syndrome was diagnosed in 21 (78%) Finnish and in 5 (56%) Danish patients. During infancy or early childhood, 10 (37%) Finnish and 8 (89%) Danish subjects were diagnosed with micropenis and/or cryptorchidism. Two subjects with a micropenis were hormonally treated; the Danish patient received i.m. testosterone three times at 7, 15, and 24 mo, respectively, and the Finnish patient received testosterone starting at the age of 2 mo. In 14 patients, we were able to calculate the cumulative dose (mg/kg/y and mg/m2/y) of androgens (testosterone enanthate, undecanoate, and propionate) used during the first year of pubertal induction. Twenty-seven subjects (75%) were born after full-term pregnancies, and all subjects were born at gestational age of 36 wk or more (data not available in nine patients). Gestational diabetes was not reported in any of the pregnancies. The clinical features and molecular genetic diagnoses are described in Table 1 (31,33,34). The coding exons and exon–intron boundaries of the known CHH genes were PCR-amplified from the genomic DNA from peripheral blood lymphocytes of the patients (31,33,34), and bi-directionally sequenced. The study protocol was approved by the Ethics Committee of the Helsinki University Central Hospital and Danish regional ethical committee. A written informed consent was obtained from all subjects.
Growth Data
The growth data of the participating patients were collected from medical records. Fifteen of the 36 CHH patients had representative length measurements during early infancy. Length deflection was calculated from the change in length SDS from birth to 3 and 6 mo of age. If the length measurement was not available at 3 or 6 mo of age, the nearest available measurement (respective ranges, 2–4 mo and 5–7 mo) was used. Final heights were available from 23 Finnish and 8 Danish patients, and both parental heights from 16 and 6 patients, respectively. MPH SDS was calculated using the equation: 0.886 × ((father’s height + mother’s height)/2 + 6.8 − 178.9066)/6.6784 − 0.071 (36). Mid-parental target height data were available for 11 of the 15 patients with growth data available from early infancy.
Between birth and 2 y of age, the relative weight was analyzed using weight-for-length, i.e., the percentage deviation of weight from the median weight for length and sex (%DW) (37). After the age of 2 y, ISO-BMI was used for describing weight progression (36). The following ISO-BMI categories were used: severe underweight (less than 16), underweight (16 to 17), normal weight (17 to 25), overweight (more than 25), and obesity (more than 30 kg/m2) (36). Height/length and weight measurements were carried out by trained nurses in maternity hospitals, child welfare clinics during infancy, and early childhood and in school health care or university hospital endocrinology clinics during childhood and adolescence. To calculate height and length SDS for each patient, we used the most recent national reference data (36,38).
Statistical Analyses
Values are mean (SD) unless otherwise stated. Analyses were carried out with the SPSS statistical software for Windows, release 22.0 (SPSS, Chicago, IL). Paired samples t-test was used for analyzing longitudinal changes in serial measurements of height (SDS) and weight (%DW) during the first 6 mo of life. One sample t-test was used for analyzing length/height SDS and weight-for-length deviation from the general population (0 SDS and 0 %DW). Comparisons between two subgroups of CHH patients were carried out with independent samples t-test. Phenotype–genotype associations were analyzed using one-way ANOVA and Fisher’s exact test. Correlations were determined with Spearman’s rank correlation. All statistical tests were two sided. The statistically significant level was set to P < 0.05.
Statement of Financial Support
This study was supported by the Finnish Foundation for Pediatric Research (7495), the Academy of Finland (268356), the Helsinki University Central Hospital Research Funds (2010307), and the Sigrid Juselius Foundation, Helsinki, Finland (2613).
Disclosure
The authors have nothing to disclose. No one received payment to produce this manuscript.
References
Corbier P, Dehennin L, Castanier M, Mebazaa A, Edwards DA, Roffi J. Sex differences in serum luteinizing hormone and testosterone in the human neonate during the first few hours after birth. J Clin Endocrinol Metab 1990;71:1344–8.
Forest MG, Sizonenko PC, Cathiard AM, Bertrand J. Hypophyso-gonadal function in humans during the first year of life. 1. Evidence for testicular activity in early infancy. J Clin Invest 1974;53:819–28.
Winter JS, Hughes IA, Reyes FI, Faiman C. Pituitary-gonadal relations in infancy: 2. Patterns of serum gonadal steroid concentrations in man from birth to two years of age. J Clin Endocrinol Metab 1976;42:679–86.
Bolton NJ, Tapanainen J, Koivisto M, Vihko R. Circulating sex hormone-binding globulin and testosterone in newborns and infants. Clin Endocrinol (Oxf) 1989;31:201–7.
Andersson AM, Toppari J, Haavisto AM, et al. Longitudinal reproductive hormone profiles in infants: peak of inhibin B levels in infant boys exceeds levels in adult men. J Clin Endocrinol Metab 1998;83:675–81.
Bergadá I, Milani C, Bedecarrás P, et al. Time course of the serum gonadotropin surge, inhibins, and anti-Müllerian hormone in normal newborn males during the first month of life. J Clin Endocrinol Metab 2006;91:4092–8.
Murray PG, Clayton PE. Endocrine control of growth. Am J Med Genet C Semin Med Genet 2013;163C:76–85.
Lampit M, Golander A, Guttmann H, Hochberg Z. Estrogen mini-dose replacement during GnRH agonist therapy in central precocious puberty: a pilot study. J Clin Endocrinol Metab 2002;87:687–90.
Main KM, Schmidt IM, Skakkebaek NE. A possible role for reproductive hormones in newborn boys: progressive hypogonadism without the postnatal testosterone peak. J Clin Endocrinol Metab 2000;85:4905–7.
Grumbach MM. A window of opportunity: the diagnosis of gonadotropin deficiency in the male infant. J Clin Endocrinol Metab 2005;90:3122–7.
Raboch J, Reisenauer R. Analysis of body height in 829 patients with different forms of testicular pathology. Andrologia 1976;8:265–8.
Kaushanski A, Laron Z. Growth pattern of boys with isolated gonadotropin deficiency. Isr J Med Sci 1979;15:518–21.
Van Dop C, Burstein S, Conte FA, Grumbach MM. Isolated gonadotropin deficiency in boys: clinical characteristics and growth. J Pediatr 1987;111:684–92.
Moorthy B, Papadopolou M, Shaw DG, Grant DB. Depot testosterone in boys with anorchia or gonadotrophin deficiency: effect on growth rate and adult height. Arch Dis Child 1991;66:197–9.
Dickerman Z, Cohen A, Laron Z. Growth in patients with isolated gonadotrophin deficiency. Arch Dis Child 1992;67:513–6.
Uriarte MM, Baron J, Garcia HB, Barnes KM, Loriaux DL, Cutler GB Jr . The effect of pubertal delay on adult height in men with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab 1992;74:436–40.
Landier F, Chaussain JL, Job JC. [Early treatment of congenital hypoplasia of the penis with intramuscular delayed-action testosterone]. Arch Fr Pediatr 1984;41:467–71.
Socha P, Grote V, Gruszfeld D, et al.; European Childhood Obesity Trial Study Group. Milk protein intake, the metabolic-endocrine response, and growth in infancy: data from a randomized clinical trial. Am J Clin Nutr 2011;94:Suppl 6:1776S–84S.
Hill DJ, Hogg J. Growth factors and the regulation of pre- and postnatal growth. Baillieres Clin Endocrinol Metab 1989;3:579–625.
Low LC, Tam SY, Kwan EY, Tsang AM, Karlberg J. Onset of significant GH dependence of serum IGF-I and IGF-binding protein 3 concentrations in early life. Pediatr Res 2001;50:737–42.
Chellakooty M, Juul A, Boisen KA, et al. A prospective study of serum insulin-like growth factor I (IGF-I) and IGF-binding protein-3 in 942 healthy infants: associations with birth weight, gender, growth velocity, and breastfeeding. J Clin Endocrinol Metab 2006;91:820–6.
Ong KK, Langkamp M, Ranke MB, et al. Insulin-like growth factor I concentrations in infancy predict differential gains in body length and adiposity: the Cambridge Baby Growth Study. Am J Clin Nutr 2009;90:156–61.
Wang X, Xing KH, Qi J, Guan Y, Zhang J. Analysis of the relationship of insulin-like growth factor-1 to the growth velocity and feeding of healthy infants. Growth Horm IGF Res 2013;23:215–9.
Kusec V, Virdi AS, Prince R, Triffitt JT. Localization of estrogen receptor-alpha in human and rabbit skeletal tissues. J Clin Endocrinol Metab 1998;83:2421–8.
Hero M, Norjavaara E, Dunkel L. Inhibition of estrogen biosynthesis with a potent aromatase inhibitor increases predicted adult height in boys with idiopathic short stature: a randomized controlled trial. J Clin Endocrinol Metab 2005;90:6396–402.
Balsamo A, Wasniewska M, Di Pasquale G, et al. Birth length and weight in congenital adrenal hyperplasia according to the different phenotypes. Eur J Pediatr 2006;165:380–3.
Huet F, Carel JC, Nivelon JL, Chaussain JL. Long-term results of GH therapy in GH-deficient children treated before 1 year of age. Eur J Endocrinol 1999;140:29–34.
Mitamura R, Yano K, Suzuki N, Ito Y, Makita Y, Okuno A. Diurnal rhythms of luteinizing hormone, follicle-stimulating hormone, and testosterone secretion before the onset of male puberty. J Clin Endocrinol Metab 1999;84:29–37.
Jiao H, Arner P, Dickson SL, et al. Genetic association and gene expression analysis identify FGFR1 as a new susceptibility gene for human obesity. J Clin Endocrinol Metab 2011;96:E962–6.
Varimo T, Hero M, Laitinen EM, Sintonen H, Raivio T. Health-related quality of life in male patients with congenital hypogonadotropic hypogonadism. Clin Endocrinol (Oxf) 2015;83:141–3.
Laitinen EM, Vaaralahti K, Tommiska J, et al. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J Rare Dis 2011;6:41.
Valdes-Socin H, Rubio Almanza M, Tomé Fernández-Ladreda M, Debray FG, Bours V, Beckers A. Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front Endocrinol (Lausanne) 2014;5:109.
Laitinen EM, Tommiska J, Sane T, Vaaralahti K, Toppari J, Raivio T. Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS One 2012;7:e39450.
Tommiska J, Känsäkoski J, Christiansen P, et al. Genetics of congenital hypogonadotropic hypogonadism in Denmark. Eur J Med Genet 2014;57:345–8.
Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2015;11:547–64.
Saari A, Sankilampi U, Hannila ML, Kiviniemi V, Kesseli K, Dunkel L. New Finnish growth references for children and adolescents aged 0 to 20 years: length/height-for-age, weight-for-length/height, and body mass index-for-age. Ann Med 2011;43:235–48.
Sorva R, Tolppanen EM, Perheentupa J. Variation of growth in length and weight of children. I. Years 1 and 2. Acta Paediatr Scand 1990;79:490–7.
Tinggaard J, Aksglaede L, Sørensen K, et al. The 2014 Danish references from birth to 20 years for height, weight and body mass index. Acta Paediatr 2014;103:214–24.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Varimo, T., Hero, M., Laitinen, EM. et al. Childhood growth in boys with congenital hypogonadotropic hypogonadism. Pediatr Res 79, 705–709 (2016). https://doi.org/10.1038/pr.2015.278
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.278
This article is cited by
-
Maternal Cortisol and Paternal Testosterone Correlated with Infant Growth via Mini Puberty
Adaptive Human Behavior and Physiology (2021)
-
Serum inhibin B for differentiating between congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty: a systematic review and meta-analysis
Endocrine (2021)
-
La statura nell’adolescente con ipogonadismo
L'Endocrinologo (2019)
