
Abstract
Background:
Previous studies have shown that elosulfase alfa has a favorable efficacy/safety profile in Morquio A patients aged ≥5 y. This study evaluated safety and impact on urine keratan sulfate (uKS) levels and growth velocity in younger patients.
Methods:
Fifteen Morquio A patients aged <5 y received elosulfase alfa 2.0 mg/kg/week for 52 wk during the primary treatment phase of a phase II, open-label, multinational study. Primary endpoint was safety and tolerability; secondary endpoints were change in uKS and growth velocity over 52 wk.
Results:
All 15 patients completed the primary treatment phase. Six of 743 infusions (0.8%) administered led to adverse events (AEs) requiring infusion interruption and medical intervention. Eleven patients (73.3%) had ≥1 study drug-related AE, mostly infusion-associated reactions. Mean z-score growth rate per year numerically improved from −0.6 at baseline to −0.4 at week 52. Comparison to untreated subjects of similar age in the Morquio A Clinical Assessment Program study showed a smaller decrease in height z-scores for treated than for untreated patients. Mean percent change from baseline in uKS was −30.2% at 2 wk and −43.5% at 52 wk.
Conclusion:
Early intervention with elosulfase alfa is well-tolerated and produces a decrease in uKS and a trend toward improvement in growth.
Similar content being viewed by others

Main
Morquio A syndrome is a rare inherited disorder caused by mutations of the gene that codes for the lysosomal enzyme N-acetylgalactosamine-6-sulfatase (GALNS, EC 3.1.6.4), which degrades the glycosaminoglycans keratan sulfate (KS) and chondroitin sulfate (1,2). GALNS insufficiency leads to progressive accumulation of glycosaminoglycans in multiple tissues and organs. In turn, this accumulation leads to significant morbidities and multisystemic clinical impairments causing diminished functional capacity, decreased endurance, impaired quality of life, and early mortality (3). The most apparent and most commonly reported clinical features of patients with Morquio A syndrome are progressive skeletal dysplasia and musculoskeletal abnormalities, typically associated with disproportionate short stature, short neck and trunk, and joint abnormalities (3,4,5). Although growth impairment is a major manifestation of Morquio A syndrome, growth is generally normal in very young patients and starts to decrease between the ages of 2 and 5 y, resulting in a mean adult standing height around 120 cm in men and 110 cm in women (4,6,7). Morquio A patients also commonly present with nonskeletal manifestations such as respiratory dysfunction, cardiovascular abnormalities, cervical/thoracolumbar cord compression, vision and hearing impairment, dental abnormalities, and hepatosplenomegaly (3,5).
In 2014, recombinant human GALNS (elosulfase alfa, Vimizim; BioMarin Pharmaceutical, Novato) was approved as an enzyme replacement therapy (ERT) for patients with Morquio A syndrome in the United States, Europe, Canada, Australia, and Brazil (8) (http://investors.bmrn.com/releases.cfm). The pivotal randomized, double-blind, placebo-controlled phase III study showed significant improvement in the 6-min walk test distance (primary endpoint), a substantial reduction in urine KS (uKS), and numerical improvements in several exploratory endpoints, including respiratory function, height, and growth rate, with 2 mg/kg/week elosulfase alfa dosing (9,10). However, the phase III study, as well as a previous phase I/II dose-ranging study, only included patients of 5 y of age and older (9,11). This open-label study was designed to explore the safety and clinical activity of elosulfase alfa in Morquio A patients younger than 5 y of age. Growth was selected as the major clinical endpoint of the study in order to assess if elosulfase alfa could prevent or minimize the deviation from the normal growth curve that generally occurs during these early years.
Results
Patient Characteristics
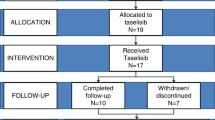
Fifteen patients aged 9 mo to 4.9 y were enrolled in the study. All patients completed the primary treatment phase, and none permanently discontinued elosulfase alfa. Demographic and baseline data are presented in Table 1 . Based on physical examination findings at screening and reported medical history, the majority of patients had abnormal musculoskeletal features (93.3%), with knee deformities (66.7%), pectus carinatum (66.7%), kyphosis (60.0%), and dysmorphism (60%) most frequently reported. Other findings were abnormal general appearance (60.0%), abnormal head, eyes, ears, nose, and throat (66.7%), corneal opacity (clouding) (33.3%), and deafness (53.3%). Cardiovascular abnormalities occurred in three (20%) patients and included mild to moderate mitral, pulmonary, and/or tricuspid valve regurgitation and mitral and/or aortic valve thickening. Standing height (in patients ≥ 2 y) was severely affected, with almost half of patients (46.7%) below the third percentile of normal for age. Normalized uKS (mean 35.9 μg/mg creatinine; Table 1 ) was elevated tenfold above the mean for a similar age control population (12).
Safety Results
All 15 enrolled patients were included in the safety analysis and completed 52 wk of treatment. The mean weekly dose received was 1.9 (±0.1) mg/kg/patient. Of 780 planned infusions, 37 (4.7%) were missed in 12 (80.0%) patients. All patients had ≥88% dosing compliance (100 × (actual dose/total planned dose)).
Table 2 summarizes the AEs reported during the 52 wk study. All 15 patients reported at least one AE. Eleven (73.3%) patients experienced at least one study drug-related AE. The most frequently reported drug-related AEs were pyrexia (40.0%) and vomiting (33.3%) ( Table 2 ). Most AEs were mild or moderate in severity. Only one patient experienced severe events, i.e., routine tonsillectomy for enlarged tonsils and cervical cord compression, which was unrelated to the tonsillectomy. Both events were not considered drug-related by the investigator. None of the children discontinued from the study due to AEs.
All 15 patients reported at least one infusion-associated reaction (IAR) (any AE occurring after infusion onset and within 1 d after infusion end, regardless of relation with study drug). All IARs were mild or moderate in severity and generally manageable with symptomatic treatment. Six of the 743 infusions (0.8%) were interrupted (three infusions) or discontinued (three infusions) due to an AE and required medical intervention in three patients (20%). AEs that resulted in infusion interruptions and/or discontinuations could be managed with symptomatic treatment (oxygen, IV steroids, IV antihistamines, or IV fluids) and infusion rate adjustment. In all cases, patients were successfully redosed and tolerated subsequent infusions.
There were no life-threatening AEs (grade 4) or deaths. Four patients (26.7%) reported a total of eight serious AEs (SAEs), with only one assessed as related to the study drug. Supplementary Appendix S1 online provides more details about this SAE. Eight hypersensitivity AEs occurred in four (26.7%) patients (more details are provided in Supplementary Appendix S1 online).
No clinically meaningful changes in vital signs, electrocardiograms, echocardiograms, clinical chemistry, hematology, or urinalysis results were observed. No new or unexpected safety signals were observed and no expedited safety reports were required.
Elosulfase alfa total antibodies were detected in all patients by week 4 and remained positive throughout the study. No patients tested positive for drug-specific IgE. The number of patients with hypersensitivity AEs was too low to identify a relationship with total antibody titers. No apparent relationship was observed between total antibody titers and height z-score measurements. Reductions in uKS occurred and were maintained despite the development of antidrug antibodies. Patients with total antibody titers greater than the mean had similar decreases in uKS as patients with total antibody titers below the mean.
Efficacy Results
All 15 patients were included in the efficacy analyses. Elosulfase alfa substantially decreased the mean normalized uKS levels within 2 wk, which was maintained over 52 wk ( Figure 1 ). The mean (±SD) percent change from baseline in uKS was −30.2% (±12.7) at 2 wk and −43.5% (±22.2) at 52 wk.

Normalized urine keratan sulfate (uKS): mean percent change from baseline (N = 15). Normalized uKS is calculated as uKS divided by urine creatinine.
The mean weight (all patients) increased by 1.7 (±0.8) kg from baseline to week 52; a mean percent change of 13.8% (±7.3). The mean height in patients ≥2 y of age (N = 12) increased by 5.3 (±2.4) cm from baseline to week 52; a mean percent change of 5.9% (±2.5). In patients <2 y (N = 3), the mean length increased by 6.0 (±2.7) cm from baseline to week 52; a mean percent change of 6.8% (±3.3). For all 15 patients, the mean height/length for age z-score was −1.6 (±1.6) at baseline and −1.9 (±1.6) after 52 wk of treatment ( Figure 2a ). The mean change from baseline at 52 wk was −0.4 (±0.5). The mean height z-score for the 12 patients ≥2 y of age at baseline was −2.0 (±1.5), as compared to −2.2 (±1.3) in 24 untreated subjects of similar age from the Morquio A Clinical Assessment Program (MorCAP) natural history study (1 of the 25 patients from the MorCAP study did not have a week 52 assessment). After 52 wk, height z-scores in both groups were −2.2 (±1.7) and −3.0 (±1.2), respectively ( Figure 2b ).

Normalized height/length and growth rate from baseline to week 52. (a) Mean standing height/lengtha z-score. Normalized standing height or length was computed using Centers for Disease Control normal values (17). (b) Mean standing height z-score at baseline and 52 wk for 12 patients ≥2 y of age included in this study (upper line) compared with 25 untreated subjects of 2–5 y of age included in the Morquio A Clinical Assessment Program (MorCAP) longitudinal natural history study at baseline and 52 wk (lower line). One of the 24 patients from the MorCAP study did not have a week 52 assessment and was excluded from this analysis. (c) Mean growth rate (per year) for standing height/lengtha z-score. Baseline rate calculation was based on observations up to 2 y prior to study entry. aLength was measured instead of standing height in subjects <2 y of age (N = 3).
The mean growth rate z-score per year was −0.6 (± 0.6; N = 8) at baseline and −0.4 (±0.5; N = 15) at week 52. Cumulative growth rate change was computed for the eight patients for whom prestudy growth rate data were available. The mean change in growth rate from baseline to week 52 in these patients was 0.2 (±1.0) ( Figure 2c ). In patients ≥2 y of age, the mean normalized growth rate z-score was −0.8 (±0.8; N = 5) at baseline and −0.3 (±0.5; N = 12) at week 52. Mean change in the patients with prestudy growth rate data was 0.6 (±1.1; N = 5).
Discussion
Elosulfase alfa is currently the only approved drug therapy for Morquio A syndrome that directly targets the underlying cause of the disease. Previous studies have assessed the efficacy and safety of elosulfase alfa in a wide age range, but not in patients younger than 5 y (9,10,11). The present study is the first to specifically examine the safety, tolerability, and pharmacodynamic responses, as demonstrated by reduction of uKS, and impact on growth of elosulfase alfa in this young patient group.
Overall, the safety analysis of this study showed that administration of elosulfase alfa is generally well tolerated in patients aged <5 y. The study did not reveal any new unexpected safety issues that were not reported in previous studies including older patients (9,10,11). Overall dosing compliance was high, i.e., ≥88% for all patients, with only 37 of 780 infusions missed (< 5%). No patients permanently discontinued treatment due to an AE and infusions requiring interruption or discontinuation due to an AE and requiring medical intervention, such as IV antihistamines or IV steroids, represented a small percentage of all 743 infusions (0.8%). This is comparable to the phase III study with 1.3% of infusions interrupted or discontinued (9). Only one patient experienced a SAE that was considered related to elosulfase alfa (moderate hypersensitivity). This patient continued to receive subsequent infusions and completed the 52 wk of treatment without experiencing additional hypersensitivity events. All patients experienced at least one IAR, but these were mild to moderate in severity and generally manageable with symptomatic treatment. The most frequently reported drug-related AEs (pyrexia, vomiting) were also IARs, similar to what was observed in the pivotal phase III study (9). Headache and nausea were less frequent in this study than in the phase III study (9). This may be related to the need to have appropriate language skills to verbalize and convey nausea and headache symptoms. Although all patients developed antidrug total antibodies by week 4, there was no apparent relationship between total antibody titers and uKS or height z-score measurements. The same has been reported for older patients included in the phase III study (9). Although promising, the favorable tolerability and safety data in this pediatric patient population should be interpreted cautiously due to certain limitations in the study design, including the open-label design and the limited sample size.
The efficacy analysis showed a substantial decrease in mean normalized uKS levels within 2 wk of treatment with elosulfase alfa, with the decreased levels being maintained over 52 wk. uKS is considered a pharmacodynamic biomarker for Morquio A syndrome with higher levels of uKS being associated with more severe clinical impairment (4,13). The observed decline in uKS of 43.5% was comparable to that reported for older children and adult patients in the elosulfase alfa phase I/II and phase III studies (9,11).
Skeletal abnormalities represent a major manifestation of Morquio A syndrome; however, young patients often do not possess, or have not yet developed, the severe abnormalities present in older patients. It is therefore postulated that early treatment may be particularly effective in changing the course of skeletal disease (4,6,7). Although there is currently limited data on younger patients, available data from natural history studies show that patients gradually fall off the anticipated growth trajectory by 2 y of age (4,6,7). In MorCAP, a natural history study including 325 untreated patients with Morquio A syndrome, height z-scores in patients older than 5 y were approximately 5–7 SDs below the normal range, while in younger patients, height was already decreasing by about 2 SDs below normal (4). These findings are consistent with the results of the current study in which the baseline mean normalized standing height z-score was −1.6. After 1 y of treatment, the mean z-score was only slightly lower (−1.9) and the mean normalized growth rate z-scores were numerically improved from baseline, indicating a closer to normal growth rate on treatment. Impact of treatment on growth may be more appropriately evaluated if disease-specific growth charts were used as they are currently available for other MPS disorders (14,15). Comparison of the current data with longitudinal growth data from the MorCAP study suggests that elosulfase alfa improves growth rates ( Figure 2b ). This finding is consistent with that of the phase III study where results suggested improved growth with treatment in individuals still growing (females <15 y and males <18 y) (10). However, as growth rates vary considerably during these years, a longer duration of follow-up will be needed to determine the significance of this early observation (16).
Corneal clouding, hearing loss/deafness, and cardiac valve abnormalities were common findings in the study population, even in the younger children. The prevalence of these findings was strikingly similar to that reported previously in the pediatric patients included in the MorCAP study (4), which highlights the importance of regular follow-up and the need for and potential benefit of early intervention. It should be noted that the effect of early ERT on corneal clouding and cardiac valves is unclear due to the poor vascularity at these sites, and based on the experience with ERT in other mucopolysaccharidoses.
In conclusion, the results of this study suggest a favorable benefit/risk profile for elosulfase alfa in pediatric Morquio A patients less than 5 y of age. Early intervention with elosulfase alfa produced a decrease in urinary KS and seemed to have improved growth. The extension phase of this study will provide more comprehensive data on growth effects and other long-term benefits in these patients.
Methods
Study Design
This study is an ongoing, phase II, open-label, multinational study consisting of an initial 52-wk primary treatment phase and an extension period of up to 156 wk plus 1 additional week for final study assessments. We report the results of the completed primary treatment phase, of which the primary objective was to evaluate safety and tolerability of infusions of elosulfase alfa 2.0 mg/kg/week in Morquio A patients <5 y of age. The secondary objectives were to evaluate its ability to reduce uKS levels and to assess its impact on growth velocity. The dose of 2.0 mg/kg/week was selected from the analysis of study data from a phase I/II dose escalation study (11).
The study is being conducted by four investigators at four study centers in three countries (USA, UK, and Italy) in accordance with the United States Code of Federal Regulations and/or other national and local regulations, the ICH Harmonized Tripartite Guideline (Guideline for Good Clinical Practice E6) and the ethical principles established by the Declaration of Helsinki.
Patient Selection
Individuals eligible to participate in this study were <5 y of age at first infusion, had a documented clinical diagnosis of Morquio A syndrome based on reduced fibroblast or leukocyte GALNS enzyme activity or genetic testing confirming the diagnosis. Each patient’s parent or other legally authorized representative provided written informed consent before study onset. Exclusion criteria were previous hematopoietic stem cell transplantation or treatment with elosulfase alfa, known hypersensitivity to any of the components of elosulfase alfa, major surgery within 3 mo prior to study entry or planned major surgery during the initial treatment period, use of any investigational product or investigational medical device within 30 d prior to screening or requirement for any investigational agent prior to study completion, concurrent disease or condition that would interfere with study participation or safety (symptomatic cervical spine instability, clinically significant spinal cord compression, severe cardiac disease), and any condition that placed the patient at high risk of poor treatment compliance or of not completing the study.
Study Drug Administration
All patients received pretreatment with an antihistamine within 1 h prior to each infusion. The choice of preinfusion medications was left to the discretion of the investigator. There was some minimal instruction to try nonsedating antihistamines first. Elosulfase alfa was diluted with sterile 0.9% sodium chloride (no standard source) at room temperature prior to administration and was infused IV over approximately 4 h, with vital signs monitored before, during, and after the infusion. Patients were also monitored by continuous pulse oximetry during and for at least 60 min after the infusion. Supplementary Appendix S2 online provides more detailed infusion procedures.
Safety Evaluation
Supplementary Appendix S3 online provides a schedule of the most important safety and efficacy assessments done during the primary treatment phase. Safety was assessed by examining incidence, severity, and relationship to study drug of treatment-emergent adverse events reported during the study. A treatment-emergent adverse event was defined as any adverse event (AE) that was new, increased in frequency, or worsened in severity after the first dose of study drug, including AEs with missing onset dates. An AE assessed by the investigator as possibly or probably related to study drug documented in the electronic case report form was counted as drug-related. In addition, vital signs, physical examinations, laboratory results, electrocardiograms, echocardiograms, cervical spine radiographs, concomitant medications, and immunogenicity were assessed per protocol. Machine types for making electrocardiograms and radiographs were not specified and varied from center to center, but a full set of instructions on how to collect the images was provided by BioClinica (Newtown, PA).
The investigator determined the severity of each event using the National Cancer Institute Common Terminology Criteria for Adverse Events v. 4.0. Hypersensitivity AEs were identified by utilizing the broad algorithmic Anaphylactic Reaction Standardized MedDRA Query (SMQ) v. 15.0 and the broad Angioedema SMQ. IARs were defined as all AEs (regardless of drug relationship) that occurred after infusion onset and within 1 d after infusion end.
Immunogenicity tests were performed using validated immunogenicity assays on blood samples and included a total antidrug antibody test (BioMarin Pharmaceutical), and drug-specific (anti-rhGALNS) IgE and total IgE tests (Viracor-IBT Laboratories, Lee’s Summit, MO).
Efficacy Evaluation
uKS levels (normalized to creatinine) were measured by liquid chromatography followed by tandem mass spectrometry (Pacific BioLabs, Hercules, CA) (see Supplementary Appendix S3 online for schedule of assessments) (12). Normalized standing height or length was computed using the Centers for Disease Control norms (see Supplementary Appendix S4 online for details on method) (17). Growth rate change was calculated as on-study growth rate (GR1S)–prestudy growth rate (GR0) for patients who had growth measurements within 2 y prior to study entry, with GR0 = (Zbaseline − Zprestudy)/T and GR1S = (ZS − Zbaseline)/T; where Zbaseline is the standing height/length z-score measured at baseline, Zprestudy the z-score closest to, but not greater than, 2 y prior to study entry, ZS the z-score measured at weeks 13, 26, 39, and 52, and T the time (in years) between corresponding measurements. All anthropometric measurements and growth rates were analyzed among all patients and for the subset of patients ≥2 y of age. Finally, mean height z-scores of the patients aged ≥2 y (Centers for Disease Control height measurements start at 2 y vs. length below this age) were compared to those of 25 untreated patients included in the MorCAP natural history study (4). These untreated patients were of similar age (2–5 y old) at baseline and at 52 wk. Baseline age, height, weight, and distribution of gender were similar between treated and untreated groups (Supplementary Appendix S5 online).
Statistical Methods
This paper reports the results of the completed 52-wk primary treatment phase. Safety and tolerability of elosulfase alfa was the primary endpoint. Secondary endpoints were change in uKS and growth velocity over 52 wk. The efficacy population consisted of all patients who received ≥1 dose of study drug and had ≥1 postbaseline efficacy measurement. The safety population consisted of all patients who received any study drug throughout the study duration. Compliance was derived from the total amount of study drug intake (actual dose) divided by the planned study drug intake during the study period (total planned dose), and multiplied by 100%.
Descriptive statistical summaries of continuous variables were done using SAS (r) Proprietary Software 9.2 (TS2M0) (SAS Institute, Cary, NC) and included the mean, SD, median, and range. Descriptive summaries of categorical variables included sample size and percent. Z-score height/length was compared to 25 patients of MorCAP who were aged 2–5 at entry to the MorCAP study. Patients in MorCAP had annual anthropometric assessments; however, there was some variability around the timing of the visit and the first annual follow-up score was linearly interpolated to precisely 365 d postbaseline.
Statement of Financial Support
This study was sponsored by BioMarin Pharmaceutical Inc., Novato, CA, USA and supported, in part, by the National Center for Advancing Translational Sciences, National Institutes of Health, Bethesda, MD, USA through UCSF-CTSI Grant Number UL1 TR000004 (P.H.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Disclosure
S.A.J., P.H., M.B., and R.P. are primary investigators for this trial. P.H. is a consultant and speaker for BioMarin and has received grant support from BioMarin. R.P. is a consultant and speaker for BioMarin. S.A.J. is a consultant/speaker for BioMarin and has received travel support. K.M., K.Y., and A.J.S. are employees and stock owners of BioMarin. H.W. is a consultant for BioMarin. The study design, collection, analysis and interpretation of data, the preparation of this manuscript, and the decision to submit the paper for publication were supported by BioMarin Pharmaceutical Inc.
References
Tomatsu S, Montaño AM, Nishioka T, et al. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum Mutat 2005;26:500–12.
Morrone A, Tylee KL, Al-Sayed M, et al. Molecular testing of 163 patients with Morquio A (Mucopolysaccharidosis IVA) identifies 39 novel GALNS mutations. Mol Genet Metab 2014;112:160–70.
Tomatsu S, Montaño AM, Oikawa H, et al. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol 2011;12:931–45.
Harmatz P, Mengel KE, Giugliani R, et al. The Morquio A Clinical Assessment Program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab 2013;109:54–61.
Hendriksz CJ, Al-Jawad M, Berger KI, et al. Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis 2013;36:309–22.
Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis 2007;30:165–74.
Montaño AM, Tomatsu S, Brusius A, Smith M, Orii T. Growth charts for patients affected with Morquio A disease. Am J Med Genet A 2008;146A:1286–95.
Sanford M, Lo JH. Elosulfase alfa: first global approval. Drugs 2014;74:713–8.
Hendriksz CJ, Burton B, Fleming TR, et al.; STRIVE Investigators. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis 2014;37:979–90.
Hendriksz CJ, Giugliani R, Harmatz P, et al.; STRIVE Investigators. Multi-domain impact of elosufase alfa in Morquio A syndrome in the pivotal phase III trial. Mol Genet Metab 2015;114:178–85.
Hendriksz C, Vellodi A, Jones S, et al. Long term outcomes of a phase 1/2, multicenter, open-label, dose-escalation study to evaluate the safety, tolerability, and efficacy of BMN 110 in patients with mucopolysaccharidosis IVA (Morquio A syndrome). Mol Genet Metab 2012;105:S35.
Martell LA, Cunico RL, Ohh J, Fulkerson W, Furneaux R, Foehr ED. Validation of an LC-MS/MS assay for detecting relevant disaccharides from keratan sulfate as a biomarker for Morquio A syndrome. Bioanalysis 2011;3:1855–66.
Dũng VC, Tomatsu S, Montaño AM, et al. Mucopolysaccharidosis IVA: correlation between genotype, phenotype and keratan sulfate levels. Mol Genet Metab 2013;110:129–38.
Quartel A, Hendriksz CJ, Parini R, Graham S, Lin P, Harmatz P. Growth Charts for Individuals with Mucopolysaccharidosis VI (Maroteaux-Lamy Syndrome). JIMD Rep 2015;18:1–11.
de Ruijter J, Broere L, Mulder MF, et al. Growth in patients with mucopolysaccharidosis type III (Sanfilippo disease). J Inherit Metab Dis 2014;37:447–54.
Tanner JM, Whitehouse RH. Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch Dis Child 1976;51:170–9.
Kuczmarski RJ, Ogden CL, Guo SS et al. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat 11 2002;1–190.
Acknowledgements
We acknowledge the participation of study patients and their families and the expert assistance of all study site coordinators and personnel. The authors are also grateful to Ismar Healthcare NV for support in the process of manuscript development, and to the Manchester National Institute for Health Research/Wellcome Trust Clinical research facility.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Appendixes
(DOC 73 kb)
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Jones, S., Bialer, M., Parini, R. et al. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) less than 5 y. Pediatr Res 78, 717–722 (2015). https://doi.org/10.1038/pr.2015.169
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.169
This article is cited by
-
Morquio A syndrome and effect of enzyme replacement therapy in different age groups of Turkish patients: a case series
Orphanet Journal of Rare Diseases (2021)
-
Elosulfase alfa in the treatment of mucopolysaccharidosis type IVA: insights from the first managed access agreement
Orphanet Journal of Rare Diseases (2021)
-
Diagnostic journey and impact of enzyme replacement therapy for mucopolysaccharidosis IVA: a sibling control study
Orphanet Journal of Rare Diseases (2020)
-
Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future
Journal of Human Genetics (2019)
-
Effect of enzyme replacement therapy on the growth of patients with Morquio A
Journal of Human Genetics (2019)
