
Abstract
Background:
Only 15 point mutations in NFIX gene have been reported so far, nine of them cause the Marshall-Smith syndrome (MSS) and the remaining mutations lead to an overgrowth disorder with a less severe phenotype, defined as Sotos-like.
Methods:
The clinical findings in three patients with MSS and two patients with a Sotos-like phenotype are presented. Analysis of the NFIX gene was performed both by conventional or next-generation sequencing.
Results:
Five de novo mutations in NFIX gene were identified, four of them not previously reported. Two frameshift mutations and a donor-splice one caused MSS, while two missense mutations in the DNA binding/dimerisation domain entailed an overgrowth syndrome with some clinical features resembling Sotos syndrome, accompanied by a marfanoid habitus, very low BMI, long narrow face, or arachnodactyly.
Conclusion:
Marshall-Smith mutations are scattered through exons 6–10 of NFIX gene, while most point mutations causing an overgrowth syndrome are clustered in exon 2. Clinical features of this overgrowth syndrome may well be considered an intermediate phenotype between Sotos and Marfan syndromes.
Similar content being viewed by others

Main
Marshall-Smith syndrome (MSS; MIM 602535) is a genetic disorder characterized by accelerated skeletal maturation, relative failure to thrive, respiratory difficulties often leading to neonatal death, intellectual disability and unusual faces, including prominent forehead, shallow orbits, blue sclerae, depressed nasal bridge, and micrognathia (1). The only known causes for this syndrome are de novo mutations in the NFIX gene, recently reported by Malan et al. (2). In addition, some mutations in NFIX gene can cause a Sotos-like phenotype, in NSD1 mutation-negative patients, an overgrowth disorder with a less severe phenotype than MSS. So far only six different point mutations that cause Sotos-like syndrome and nine mutations resulting in MSS have been reported (2,3,4,5). Consequently, the range of genetic variants in the NFIX gene that gives rise to these clinical entities is scarce. To better define the phenotypic and genotypic spectrum associated with NFIX mutations, here we present the clinical and genetic findings in five further patients with de novo mutations in the NFIX gene associated to Sotos-like phenotype or MSS.
Results
We identified five NFIX mutations, each on a different patient, four of them not previously reported (see Table 1 ). Patient 1 showed a heterozygous de novo deletion of one thymidine in exon 10 (c.1496delT). This deletion causes two different effects depending of the isoform of NFIX messenger RNA. In the short isoforms (lacking the alternatively spliced exon 9), the deletion gives rise to the loss of the stop codon and the addition of 20 extra residues to the whole polypeptide sequence in these short isoforms. Conversely, long isoforms become more affected by the substitution of the last 12 residues (highly conserved even in paralogs) by 100 new aminoacids. Patient 2 carries another novel deletion of two base pairs in exon 7 leading to a frameshift mutation that originates a loss of the C-terminal domain. Patient 3 harbors a de novo variant affecting the donor-splice site of intron 6, previously reported in other MSS patient as causing a partial inclusion of intron 6, what represents a similar consequence to a frameshift mutation (2).
Patient 4 is a carrier of the de novo mutation c.136C>T, which causes the aminoacid change arginine to cysteine at position 46 of the protein. Patient 5 showed the de novo variant c.185G>C, which is predicted to substitute the arginine at position 62 by a proline. Both substitutions, present in exon 2, affect highly conserved aminoacids in both orthologs and paralogs of NFIX protein that are part of the DNA-binding and dimerisation domain (see Figure 1 ). In silico predictions of both variants confirm that they are probably damaging (see Table 1 ) which, together with the fact of occurring de novo in patients, are highly suggestive of their pathogenic condition.

Partial electropherograms showing the five mutations detected by Sanger sequencing. (a) Deletion c.1496delT in Patient 1; (b) deletion c.1074_1075delGC in Patient 2; (c) splicing mutation IVS6+1G>T in Patient 3; (d) nucleotide change c.136C>T in Patient 4; (e) nucleotide change c.185G>C detected in Patient 5; (f) multiple sequence alignment of the N-terminus of NFI family members obtained from the UniProt consortium/European Bioinformatics Institute (Hinxton, UK). In blue are represented hydrophobic aminoacids, red negatively charged, and green positively charged. Note the evolutionary conservation of the aminoacids altered by the two nucleotide changes.
Discussion
Five mutations in the NFIX gene are here described, four of them for the first time: two missense variants, two frameshift and one splice donor variant (see Table 1 ). Including the present study, a total of 20 NFIX point mutations have been reported so far (see Figure 2 ). All these mutations are unique, except for the first base of intron 6, mutated in three unrelated patients (2,3,4,5).

Distribution of point mutations along NFIX gene. (a) Schematic representation of NFIX gene in its different isoforms, where each exon is represented by vertical bars joined by a line (introns) with the arrows indicating the transcription sense (taken from the UCSC genome browser, hg19). (b) Distribution of the published mutations (below the diagram) and the novel mutations reported in this work (above the diagram). Each exon is represented by a rectangle (approximately to scale) joined by a continuous line (canonical isoform 1; NM_001271043.1) or by discontinuous lines to indicate alternatively spliced exons (1, 7 and 9). Mutations giving rise to Malan syndrome cluster to exons 2 and 3, while mutations causing the more severe Marshall-Smith syndrome are distributed along exons 6 to 10.
The clinical features of Patients 1–3 fit well with those previously reported associated to MSS. This one is a well characterized syndrome that can be suspected when facing a moderate to severe developmental delay, respiratory compromise during early infancy (secondary to upper airway obstruction), accelerated skeletal maturation, failure to thrive and the characteristic dysmorphic facial features. Any patient with these features should be referred to a clinical geneticist to make the correct diagnosis.
By contrast, Patients 4 and 5 present a Sotos-like phenotype, recently proposed to be referred as Malan syndrome (5). In spite of sharing several clinical characteristics with Sotos syndrome (overgrowth, advanced bone age, macrocephaly, and some dysmorphic craniofacial features in addition to a moderate developmental delay), both patients lack the facial gestalt and, in fact, the genetic study of NSD1 gene was negative. However, the most striking feature in both patients was a marfanoid habitus, with long and slender morphotype, a very low BMI, long narrow face, and arachnodactyly. These signs are also present in other Malan patients with previously reported NFIX mutation (patients A–C in ref. (2)). Consequently we propose that the study of this gene should be the first diagnostic option for any patient with an intermediate phenotype between Sotos and Marfan syndromes, sharing characteristics of both, such as accelerated linear growth, skeletal anomalies included bone maturation, intellectual disability with autistic traits or anxiety, hypotonia, long and narrow face with high forehead and small mouth (see Table 2 ). It is worth noting that patients at younger ages do not manifest this marfanoid habitus (Patients 2–6 in ref. (5)), suggesting that marphanoid signs develop with age.
Most mutations causing MSS are clustered in exon 7, which can be considered a mutational hotspot for microdeletions/microinsertions in this gene. Conversely, nearly all the mutations causing a Sotos-like phenotype cluster in the first two coding exons, affecting evolutionary conserved aminoacids in the DNA binding/dimerisation domain. Malan et al. (2) argued that the different phenotypic consequences are due to a different fate of the messenger. The overgrowth phenotype (Malan syndrome) would be due to pure NFIX haploinsufficiency because of the lack of one dosage of functional protein. By contrast, the MSS would be due to the dominant-negative effect of those mutations that give rise to truncated NFIX proteins. The presence of NFIX proteins lacking their C-terminal end could exert a deleterious effect due to dimerization with products of the normal allele (homodimer) or even with other members of the family (heterodimer). This hypothesis was supported by the presence of both normal and mutated messenger RNAs in MSS patients’ samples, but not in a Malan patient (2). However, here we report two missense mutations, associated to a Malan phenotype, that not necessarily lead to a decrease of the corresponding messenger RNA. Consequently, the haploinsufficiency of NFIX factor should be understood as a dosage decrease of NFIX functional protein with ability to bind DNA. Therefore deletions or missense mutations, but also some frameshifts or nonsense mutations, that lead to a loss of the ability to bind DNA will be less deleterious than those mutations that give rise to an anomalous NFIX factor with preservation of the DNA-binding domain. Both missense mutations are in the N-terminal DNA-binding and dimerisation domain comprising residuals 9–202 (6), but only c.136C>T (p.R46C) affects the highly conserved domain corresponding to the CTF/NF-I DNA-binding domain signature (R-K-R-K-Y-F-K-K-H-E-K-R), whose function is not known.
The functional importance of the multiple NFIX splice variants remains largely unknown, although they presumably fulfill very specific biological functions (7). Mutations on the different isoforms encoded by NFIX might conceivably have variable phenotypic consequences. In this sense, mutation of patient 1 (c.1496delT) is probably the most interesting one because it alters the stop codon of the short isoforms. Consequently, the polypeptidic sequence of the short isoforms remains preserved except for the addition of 20 extra aminoacids. By contrast, the C-terminal end of the long isoforms becomes much more modified. These long isoforms are generated by the inclusion of exon 9 (see Figure 2 , upper side), which introduces a shift of the reading frame in exons 10 and 11, giving rise to a different peptide sequence with a long proline-rich transcriptional activation domain. This domain, fully conserved in primates, is the target of gene expression regulatory pathways induced by growth factors and it interacts with basal transcription factors and with histone H3. Furthermore, the expression of long isoforms, the minority in other tissues, has been demonstrated to be strongly upregulated during the differentiation of neural progenitors (8), suggesting a relevant role on central nervous system development. It is not unreasonable to suppose that any mutation affecting exclusively the short isoforms would have rather benign neurologic consequences.
In summary, here we present one recurrent and four novel mutations of NFIX, a gene where only 15 mutations have been so far reported. We confirm that the missense mutations causing a Sotos-like syndrome cluster in the first exons affecting the N-terminal DNA-binding and dimerisation domain. We propose that patients with a phenotype intermediate between Sotos and Marfan syndromes should be preferentially tested for this gene. Conversely, the mutations associated to the more severe MSS cluster in the last exons, presumably giving rise to a truncated protein with a dominant negative effect. Additionally, the mutation c.1496delT, which affects the stop codon of the short isoforms, lets us point to the importance of the longer isoforms in this syndrome.
Methods
Subjects
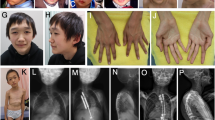
Patient 1. This patient is a 5-y-old girl, born from the second pregnancy of unrelated healthy parents of 31 and 29 y of age. The family history is significant for a distant paternal relative with Fragile X syndrome and a distant maternal relative with molecularly-confirmed Cornelia de Lange syndrome, which do not affect the parents of the patient. She was born at 39 wk of gestation by natural delivery after uneventful pregnancy except for arrest of fetal growth noted in the eighth month. Apgar scores were 9/10. At birth, weight was 2,840 g (10–25th percentile), length 48.5 cm (10–25th percentile), and occipitofrontal circumference (OFC) 33.5 cm (50th percentile). Since the first month, vomiting, poor weight gain, laryngomalacia with progressive afonic stridor, continuous drooling, and serious feeding problems were evidenced. During lactation, she suffered three episodes of aspiration pneumonia. At 6 mo old, she began to be fed by tube and at the 18th month of life she required gastrostomy. Bilateral conductive hearing loss was confirmed, and coincident with intubation maneuvers for placement of grommet tubes she suffered a difficult to recover cardiac arrest. At 10 mo, she showed a weight of 7,100 g (−2.82 SD), length 70.3 cm (−1.76 SD) and head circumference 45.5 cm (10th percentile). Abnormal phenotype included dolichocephaly, prominent occiput, frontal bossing and triangular facies, arched eyebrows, proptosis, with blue sclera, depressed nasal bridge, anteverted nares, microretrognathia, highly arched palate with gum hypertrophy and hypoplastic teeth. Also, pectum excavatum with anterior protrusion of left rib cage was evidenced. Limbs were slender with very little muscle mass, wrists in flexion, long thin fingers, and toes with limitation on the size of the phalanges of the third and fourth fingers, clinodactyly of the fifth finger on both hands and bilateral hallux valgus. Hair was sparse and brittle. At 25 mo, she presents thoracic scoliosis and persisting malnutrition (weigh −2.6 SD, length −2.1 SD and head circumference 10th percentile). Carpal radiograph at 2 y and 9 mo shows a bone age of 7 y. Significant psychomotor delay was evidenced since 3–6 mo with axial hypotonia, flexion contracture of hands, limited abduction of the hips and bending of femurs. She was able to sit at 10 mo, to walk with aid at 30 mo and autonomous walking and first words were achieved at 3 y old. She has a friendly demeanor and very good relationship with his family. Currently, at 5 y, she weights 12,100 g (−2.4 SD), a height of 96 cm (−3.1 SD), and an OFC of 50 cm (50 centile). She shows moderate intellectual and language disability and a general physical aspect of aged. Brain MRI, karyotype, 22q11 and arrayCGH studies were normal. Advancement of the bone age and other radiographic findings (presence of Wormian bones, shallowness of the orbits, thinning of long tubular bones, etc.) were consistent with clinical suspicion of MSS. Other supportive findings included bilateral short ulna, hallux valgus and coxa vara, ischia thinning, dolicocephaly, prominent forehead, proptosis, depressed nasal bridge, anterverted nares, and micrognathia ( Figure 3a – c ).

Overview of Patient 1 at 2 mo (a), 7 mo (b), and 5 y old (c). Notice the facial features of Marshall-Smith syndrome: frontal bossing, triangular faces, arched eyebrows, proptosis with blue sclera, depressed nasal bridge, anteverted nares, retrognathia, and hypoplastic teeth. Patient 4 (d) and Patient 5 (e) show generalized longilinear appearance.
Patient 2. This patient is a 19-y-old boy, third son of unrelated healthy parents. After a pregnancy without significant incidents, he was born in the 40th week by induced labor and cephalic presentation. Born with 3,250 g weight (25–50th centile), length 54.5 cm (>90th centile), OFC 37.5 cm (>90th centile), and Apgar 8–9. He was admitted at the Neonatal Unit of our Hospital 41 h after birth due to respiratory distress. He was discharged from hospital at 22 d of life with the diagnoses of dysplasia of aortic valve with mild insufficiency, congenital hydrocephalus, and dysmorphic syndrome. During his development, he frequently suffered upper respiratory infections without respiratory obstruction.
His first words were at 3 y, although his vocabulary is very limited with less than 10 words in total. When he was 7 y old, tympanostomy tube placement due to bilateral cholesteatoma was performed. He developed severe bilateral hypoacusia needing hearing amplification.
He began to walk at 4 y old. He had bilateral hip dysplasia and instability of the cranio-cervical junction. Brain MRI showed ventriculomegaly with septum pellucidum agenesis. He underwent surgery for bilateral cryptorchidism at 3 y of age. Since the age of 11 y, he needed follow up by Dermatologist due to multiple acquired melanocytic nevi.
He always had typical facial features of MSS, with accelerated bone age, wide forehead, proptosis, blue sclerae, anteverted nares, ears with multiple folds, and underdeveloped midface. Both height and OFC were higher than 99th percentile until 4 y of life, when he began to loss height due to severe kyphosis. His weight was always below the 10th percentile.
He has a severe cognitive deficit, although with a very sociable and happy disposition.
Patient 3. This patient is the second daughter of unrelated healthy parents. At 20th week of pregnancy, the size of cerebral ventricles was on 90th percentile. No other abnormalities were observed on fetal MRI. She was born at 41 wk of gestation by vaginal delivery. The Apgar scores were 3/3/6 at the first, fifth, and tenth minutes respectively. She needed intubation at the delivery room due to hypotonia and absence of respiratory effort. The body weight at birth was 2,830 g (10th to 25th percentile), length 50 cm (50th to 75th percentile) and OFC 37 cm (>98th percentile). She was extubated at the second day of life, but she needed noninvasive ventilation with nasopharyngeal tube.
In cranial TC midface hypoplasia, hypoplasia of external ear canals and choanal estenosis were observed. Full skeletal survey showed an advanced bone age, corresponding 18–24 mo and bullet-shaped phalanges. Other characteristic craniofacial features suggestive of MSS were high forehead with proptosis, short nose, anteverted nares, underdeveloped midface, retrognathia, and gum hypertrophy. She had large hands and feet and hypertrichosis. At neurologic examination, central hypotonia and hyperreflexia were detected.
She died at the 17th day of live due to respiratory failure.
Patient 4. This patient is the second son of unrelated healthy parents. During the pregnancy, cerebral ventriculomegaly was suspected, but not confirmed after birth. He was born at the 38th week of gestation by cesarean section, with Apgar 8/10. The birth weight was 3,510 g (75th–90th percentile), height 50.5 cm (75th–90th percentile), and OFC 39 cm (>98th percentile). He was referred to the Neuropediatric clinic at 4 mo of life due to hypotonia and motor developmental delay. No brain anomalies were recognized by cerebral MRI. Metabolic studies, cardiological and oftalmological examination were uneventful, except for a mild strabismus. At 6 y old, his weight was 25.5 kg (90th percentile), height 135 cm (>98th percentile), OFC 59.5 cm (>98th percentile). The facial appearance showed triangular shape with frontal bossing, down-slanting palpebral fissures, small mouth, prominent, thin mandible, and everted lower lips. Slender habitus, dolichostenomielia, large hands and fingers and moderate joint hyperextensibility were also noted ( Figure 3d ). Bone age was estimated 8 y at 6 y of age. He shows mild intellectual disability and attention deficit. Karyotype, fragile-X and NSD1 (sequenciation and MLPA) testing were negative.
Patient 5. This patient is the first daughter of unrelated healthy parents. She was born after 39 wk of uneventful gestation by natural delivery. Body weight at birth was 2,850 g (25th percentile), height 49 cm (50th percentile), and OFC 34 cm (50th percentile). Apgar scores were 9 and 10 at the first and the fifth minutes respectively. At 5 mo of life, she was referred to the Neuropediatric clinic due to hypotonia. No brain abnormalities were observed at cerebral MRI. Due to a systolic murmur, echocardiographical study was performed with normal result. At our first examination, she was 12 y old. The following features were noticed: weight 33.5 kg (10th percentile), height 164 cm (>97th percentile), OFC 55 cm (>97th percentile). She showed slender habitus, with dolichostenomielia, arachnodactily, large hands and fingers, triangular face with wide forehead, prominent mandible, small mouth, scoliosis, and strabismus ( Figure 3e ). She has mild intellectual disability, with attention deficit and hypotonia with poor motor skills. Although no advanced bone age was noticed, Sotos syndrome was suspected due to the overgrowth. NSD1 gene analysis by sequencing and gene deletion testing were normal.
Molecular Analyses
Genomic DNA was extracted from peripheral blood lymphocytes of patients and parents following standard methods. Informed consent, approved by the Hospital Universitario y Politecnico La Fe Biomedical Research Ethics Committee, was obtained from all the participants or from their parents, when appropriate. The whole NFIX (sequence of reference Genbank NM_ 001271043.1) coding regions and splice sites were searched for mutations by two different strategies. Patient 1 underwent next-generation sequencing targeted to a panel of 179 genes with known or putative implication on syndromic intellectual disability. Sequences capture was carried out with the HaloPlex Target Enrichment System (Agilent, Santa Clara, CA), following the specifications of the manufacturer, and run on an Illumina 150 bp paired-end MiSeq platform (Illumina, San Diego, CA). Anotation of nucleotide variants was performed with DNAnexus platform (DNAnexus, Mountain View, CA), and validated by Sanger sequencing as detailed below.
In Patients 2–5, bidirectional sequencing was performed following the Big Dye Terminator kit and an ABI PRISM 3130xl automated sequencer (Life Technologies, Carlsbad, CA). Primers and PCR conditions are available on request.
In Silico Analyses
The effect of the missense changes was evaluated by the web-based prediction tool PolyPhen-2 (9). Conservation scores and any putative correspondence with variants integrated in the dbSNP and 1000-genomes databases were obtained through UCSC Genome Browser (10).
Statement of Financial Support
This work was supported by the grant PI11/00389 (ISCIII -Subdirección General de Evaluación y Fomento de la Investigación, PN de I+D+I 2008–2011), FEDER (Fondo Europeo de Desarrollo Regional, EU) (Madrid, Spain).
Disclosure
There are no financial ties to products in the study or potential/perceived conflicts of interest.
References
Adam MP, Hennekam RC, Keppen LD, et al. Marshall-Smith syndrome: natural history and evidence of an osteochondrodysplasia with connective tissue abnormalities. Am J Med Genet A 2005;137:117–24.
Malan V, Rajan D, Thomas S, et al. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a Sotos-like or a Marshall-Smith syndrome. Am J Hum Genet 2010;87:189–98.
Yoneda Y, Saitsu H, Touyama M, et al. Missense mutations in the DNA-binding/dimerization domain of NFIX cause Sotos-like features. J Hum Genet 2012;57:207–11.
Priolo M, Grosso E, Mammì C, et al. A peculiar mutation in the DNA-binding/dimerization domain of NFIX causes Sotos-like overgrowth syndrome: a new case. Gene 2012;511:103–5.
Klaassens M, Morrogh D, Rosser EM, et al. Malan syndrome: Sotos-like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. Eur J Hum Genet 2015;23:610–5.
Stefancsik R, Sarkar S. Relationship between the DNA binding domains of SMAD and NFI/CTF transcription factors defines a new superfamily of genes. DNA Seq 2003;14:233–9.
Gründer A, Qian F, Ebel TT, et al. Genomic organization, splice products and mouse chromosomal localization of genes for transcription factor Nuclear Factor One. Gene 2003;304:171–81.
Singh SK, Wilczynska KM, Grzybowski A, et al. The unique transcriptional activation domain of nuclear factor-I-X3 is critical to specifically induce marker gene expression in astrocytes. J Biol Chem 2011;286:7315–26.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9.
Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006.
Acknowledgements
The authors appreciate the collaboration of the patients and their families for participating in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Martinez, F., Marín-Reina, P., Sanchis-Calvo, A. et al. Novel mutations of NFIX gene causing Marshall-Smith syndrome or Sotos-like syndrome: one gene, two phenotypes. Pediatr Res 78, 533–539 (2015). https://doi.org/10.1038/pr.2015.135
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.135
This article is cited by
-
Erythroid lineage chromatin accessibility maps facilitate identification and validation of NFIX as a fetal hemoglobin repressor
Communications Biology (2023)
-
A deep phenotyping experience: up to date in management and diagnosis of Malan syndrome in a single center surveillance report
Orphanet Journal of Rare Diseases (2022)
-
Fronto-orbital advancement in a patient with Marshall-Smith syndrome: a case report and review of the literature
Child's Nervous System (2021)
-
Overgrowth syndromes — clinical and molecular aspects and tumour risk
Nature Reviews Endocrinology (2019)
-
Sindromi da iperaccrescimento: aspetti clinici e genetici
L'Endocrinologo (2019)
