
Abstract
Background:
Prolonged hypoglycemia leads to brain injury, despite treatment with 10% dextrose. Whether induction of hyperglycemia or ketonemia achieves better neuroprotection is unknown. Hyperglycemia is neuroprotective in other brain injuries during development; however, it worsens hypoglycemia-induced injury in the adult brain via poly(ADP-ribose)polymerase-1 (PARP-1) overactivation.
Methods:
Three-week-old rats were subjected to insulin-induced hypoglycemia and treated with 10% dextrose or 50% dextrose. Neuronal injury, PARP-1, and brain-derived neurotrophic factor (BDNF) III/TrkB/p75NTR expressions were determined. In the second experiment, ketonemia was induced by administering β-hydroxybutyrate during hypoglycemia and its effect on neuronal injury was compared with those conventionally treated using 10% dextrose.
Results:
Both 10 and 50% dextrose administration led to hyperglycemia (50% dextrose > 10% dextrose). Compared with the 10% dextrose group, neuronal injury was greater in the 50% dextrose group and was accompanied by PARP-1 overactivation. BDNF III and p75NTR, but not TrkBFL, mRNA expressions were upregulated. Neuronal injury was less severe in the rats subjected to ketonemia, compared with those conventionally treated using 10% dextrose.
Conclusion:
Hyperglycemia accentuated hypoglycemia-induced neuronal injury, likely via PARP-1 overactivation. Although BDNF was upregulated, it was not neuroprotective and potentially exaggerated injury by binding to p75NTR receptor. Conversely, ketonemia during hypoglycemia attenuated neuronal injury.
Similar content being viewed by others


Main
Hypoglycemia is a common metabolic problem during development. Infants with hyperinsulinism and children on insulin therapy for type 1 diabetes are particularly at risk. Inadequately treated hypoglycemia leads to cerebral energy failure and permanent brain injury. Therefore, early detection and effective treatment of hypoglycemia has considerable significance during development.
Currently, severe hypoglycemia in children is treated by parenteral administration of 10% dextrose in a 200–500 mg/kg dose (1,2). While this therapy rapidly achieves euglycemia, there is no evidence that it ensures neuroprotection. In hypoglycemic developing rats, cerebral energy production remains suboptimal for at least 50 min after 200 mg/kg of 10% dextrose administration (3). Neuronal injury, primarily in the anterior regions of the cerebral cortex, is seen in the brains harvested 24 h later (4,5). Thus, there is a need for exploring alternative treatment strategies for hypoglycemia.
Hyperglycemia to a blood glucose concentration of 450–720 mg/dl (25–40 mmol/l) decreases neuronal injury in other causes of cerebral energy failure, such as hypoxia–ischemia (HI) during development (6,7). Enhanced substrate availability, improved mitochondrial function, and attenuation of N-methyl-D-aspartate-mediated excitotoxicity are potentially responsible for this neuroprotection (8,9). Whether hyperglycemia provides similar neuroprotection during hypoglycemia is not known. In adult rats, hyperglycemia accentuates hypoglycemia-induced neuronal injury by leading to superoxide production (10) and poly(ADP-ribose)polymerase-1 (PARP-1) overactivation (11). PARP-1, a nuclear enzyme, normally participates in DNA repair by forming poly(ADP-ribose) (PAR) polymers in the vicinity of DNA breaks. However, PARP-1 overactivation leads to neuronal death by depleting cellular NAD+ and ATP stores and translocating apoptosis-inducing factor (AIF) from the mitochondria to the nucleus (12,13). As NAD+ is an essential cofactor for glycolysis, administration of excess glucose under these conditions worsens oxidant stress by generating superoxide via the hexose monophosphate shunt, instead of ATP production via glycolysis and oxidative phosphorylation (10). We have previously demonstrated that although baseline PARP-1 expression in the brain is higher during development (5), there is no additional upregulation after hypoglycemia treated with 10% dextrose. Whether this is also true when hypoglycemia is treated using hyperglycemia-inducing doses of dextrose, for example, by administering 50% dextrose is not known.
Similarly, whether ketonemia attenuates hypoglycemia-induced neuronal injury in the developing brain is not known. Although glucose is its primary energy substrate, the developing brain is capable of using ketone bodies (β-hydroxybutyrate and acetoacetate) for energy production, particularly in the preweaning period (14,15). Induction of ketonemia, for example, by feeding a ketogenic diet before and after hypoglycemia, reduces the severity of neuronal injury in 3-wk-old rats (16), likely by enhancing mitochondrial biogenesis and alternative energy stores (17). However, such preventive strategies are of limited clinical utility in acute hypoglycemia, which typically occurs without forewarning. Limited data suggest that induction of ketonemia after the onset of hypoglycemia also may be beneficial. In preweanling rats and mice, β-hydroxybutyrate administration reverses hypoglycemia-induced coma by correcting cerebral energy failure (18,19). Whether such treatment is also neuroprotective in the postweaning period, when cerebral ketone body uptake and utilization markedly decrease from the preweaning period (14) is unknown.
The aims of the study were to determine the neuroprotective effects of hyperglycemia and ketonemia in 3-wk-old rats. Rats of this age are used to model the neurological effects of hypoglycemia in human infants and young children due to similarities in brain development and substrate utilization (14,16). Glucose becomes the principal energy substrate to the brain at the corresponding ages in the two species (14). In the first experiment, we compared the severity of neuronal injury using Fluoro-jade B (FJB) histochemistry, and PARP-1 and AIF expressions in hypoglycemic rats treated with a hyperglycemia-inducing dose of dextrose with those treated with the conventional dose of dextrose. We also assessed the effect of the two treatments on the mRNA expression of brain-derived neurotrophic factor (BDNF) and its receptors, tropomyosin-related kinase B (TrkB) and p75 neurotrophin (p75NTR) receptors. BDNF expression is upregulated soon after hypoglycemia in the adult rat brain (20). In vitro studies demonstrate that BDNF provides neuroprotection during hypoglycemia by binding to the full-length isoform of TrkB (TrkBFL) receptor and stabilizing calcium homeostasis (21,22). Conversely, binding of BDNF to p75NTR receptor promotes neuronal injury (23,24). In the second experiment, we determined the neuroprotective efficacy of ketonemia by comparing the severity of neuronal injury in rats treated with β-hydroxybutyrate during hypoglycemia with those conventionally treated using 10% dextrose.
Results
Experiment 1: Effect of Hyperglycemia on Hypoglycemia-Induced Neuronal Injury
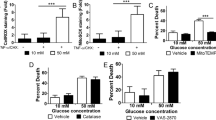
To determine whether hyperglycemia attenuates neuronal injury, P21 rats were subjected to insulin-induced hypoglycemia and treated with either 10% dextrose (HG-10% dextrose group) or 50% dextrose (HG-50% dextrose group) 240 min after the insulin administration. The blood glucose concentration during the 210 min of hypoglycemia was similar in the two groups (HG-10% dextrose group, 36.0 ± 4.2 mg/dl; HG-50% dextrose group, 36.2 ± 3.5 mg/dl, P = NS). Two hours after termination of hypoglycemia, the blood glucose concentration was higher in the HG-50% dextrose group, compared with the HG-10% dextrose group (HG-50% dextrose group, 582.0 ± 11.9 mg/dl; HG-10% dextrose group, 268.0 ± 53.9 mg/dl, P < 0.005, Figure 1a ).

Effect of 10% dextrose and 50% dextrose administration on blood glucose concentration (a), severity of neuronal injury (b), and poly(ADP-ribose)polymerase-1 (c) and apoptosis-inducing factor (d) mRNA expression in the brain of 3-wk-old rats. Rats were subjected to insulin-induced hypoglycemia and treated with either 10% dextrose or 50% dextrose 240 min after the insulin administration. The brain was collected 24 h later and processed for neuronal injury using Fluoro-jade B histochemistry and mRNA expression using quantitative RT-PCR method. Values are mean ± SEM, n = 6–8 per group. The mRNA data are normalized to the control group. *P < 0.05 vs. control group; **P < 0.01 vs. 10% dextrose group. AIF, apoptosis-inducing factor; D, dextrose; FJB+, Fluoro-jade B positive; PARP-1, poly(ADP-ribose)polymerase-1.
FJB-positive cells reflecting injured neurons were present in the brain sections collected 24 h posthypoglycemia in both hypoglycemia groups. FJB-positive cells were primarily seen in the orbital and cingulate regions of the prefrontal cortex and layers 2 and 3 of the parietal cortex. FJB-positive cells were absent in the other brain regions and in the control group. There was a main effect of treatment on the number of FJB-positive cells (P < 0.001). Compared with the HG-10% dextrose group, more FJB-positive cells were present in the HG-50% dextrose group (P < 0.05, Figure 1b ).
PARP-1 and AIF mRNA expressions were upregulated 24 h posthypoglycemia in the HG-50% dextrose group, relative to the control group (P < 0.05, Figure 1c , d ). This was accompanied by increased PAR (+132%) and AIF (+23%) protein expressions (not shown). NF-κB mRNA expression was upregulated 35% (P < 0.05), while Bcl2 and caspase 3 mRNA expressions were not altered in the HG-50% dextrose group. None of the mRNA and protein expressions in the PARP-1 pathway were altered in the HG-10% dextrose group.
There was a main effect of treatment on BDNF-III, TrkBTOTAL (TrkBFL + TrkBTRUNCATED) and p75NTR mRNA expressions 24 h posthypoglycemia (P < 0.05; Table 1 ). Relative to the control group, all three transcripts were upregulated in the HG-50% dextrose group (P < 0.05), but not in the HG-10% dextrose group. The mRNA expressions of TrkBFL and early growth response-2 (Egr-2), which is downstream of BDNF/TrkB signaling (25), were not altered in either hypoglycemia group.
Experiment 2: Effect of Ketonemia on Hypoglycemia-Induced Neuronal Injury
To determine whether ketonemia attenuates neuronal injury, P28 rats were subjected to insulin-induced hypoglycemia. β-hydroxybutyrate was administered in the ketone group, beginning at 120 min after the insulin administration, while the dextrose group was maintained without additional treatment until hypoglycemia was terminated 240 min after the insulin administration in both groups using 10% dextrose. The timing of β-hydroxybutyrate administration was based on the occurrence of neuronal injury after 120 min of hypoglycemia in P28 rats (4). The blood glucose concentration during the 210 min of hypoglycemia was comparable in the two groups (dextrose-group, 31.3 ± 4.3 mg/dl; ketone-group, 32.7 ± 2.3 mg/dl, P = NS). The ketone body concentration in the blood 120 min after the insulin administration (i.e., until the β-hydroxybutyrate administration in the ketone group) was comparable in the two groups (dextrose-group, 0.4 ± 0.1 mmol/l; ketone-group, 0.6 ± 0.2 mmol/l, P = NS). Beginning 30 min after the β-hydroxybutyrate administration and until termination of hypoglycemia at 240 min, the ketone body concentration was higher in the ketone-group (ketone group, 2.9 ± 0.3 mmol/l; dextrose group, 0.6 ± 0.1 mmol/l, P < 0.001).
Similar to Experiment 1, FJB-positive cells were absent in the control group and present in both hypoglycemia groups, primarily in the orbital and cingulate regions of the prefrontal cortex and the parietal cortex ( Figure 2 ). There was a main effect of treatment on the number of FJB-positive cells (P < 0.001). Compared with the dextrose-group, fewer FJB-positive cells were present in the ketone-group (FJB-positive/brain section: dextrose-group, 82 ± 19; ketone-group, 21 ± 12, P < 0.05).

Effect of ketonemia on hypoglycemia-induced neuronal injury in the parietal cortex (a–c) and prefrontal cortex (d–f) of developing rats. Twenty-eight-day-old rats were subjected to insulin-induced hypoglycemia. Ketonemia was induced in the ketone group by administering β-hydroxybutyrate, starting at 120 min after the insulin administration. Rats in the dextrose group were maintained without additional treatment until termination of hypoglycemia in both groups 240 min after the insulin administration using 10% dextrose. Neuronal injury was assessed 24 h later in the control (a and d), dextrose (b and e), and ketone (c and f) groups using Fluoro-jade B histochemistry. Arrows point to Fluoro-jade B-positive cells in the hypoglycemia groups. 20 µm coronal brain sections; scale bar in panel a = 100 µm, and applies to all the panels.
Discussion
Hypoglycemia led to neuronal injury in the developing rat brain. Similar to previous studies (4,5,16,26), neuronal injury was primarily seen in the anterior regions of the cerebral cortex, confirming the vulnerability of this brain region during moderate hypoglycemia. Contrary to our expectation, hyperglycemia accentuated the severity of injury, likely via glucose reperfusion and PARP-1 overactivation. Conversely, ketonemia during hypoglycemia was neuroprotective. Collectively, these data suggest the importance of avoiding extreme hyperglycemia and exploring alternative management strategies for hypoglycemia during development.
Both dextrose treatment protocols resulted in hyperglycemia, albeit of different severity. The blood glucose concentration achieved in the 50% dextrose group (582.0 ± 11.9 mg/dl) is comparable to those associated with neuroprotection in previous HI studies (450–720 mg/dl (6,7)). However, unlike those studies, hyperglycemia led to more severe injury in the present study. Several factors may explain the divergent results. The aforementioned HI studies were performed at P7. The brain is naturally resistant to injury at this age (4,27), secondary to lower energy demand, higher antioxidant concentrations, and greater ability to use alternative substrates (14,28). The glucose transporter expression at the blood brain barrier and neuronal membrane is low at P7 (29), which may limit excess glucose transport into the neurons at this age. Our experiments were performed at P21 when cerebral energy demand, glucose transport, ability to use alternative substrates and vulnerability to injury resemble those at adulthood (4,14,27). Further, HI causes intracellular acidosis, a known inhibitor of superoxide production (30), whereas hypoglycemia is not associated with acidosis. The timing of hyperglycemia in relation to the onset and duration of injury also may have influenced the results. The HI studies in P7 rats suggest that hyperglycemia induced prior to the onset of hypoxia or after a short duration (60 min) of hypoxia is neuroprotective (6,7). Conversely, mild hyperglycemia (blood glucose, 200–240 mg/dl) induced after prolonged (120 min) hypoxia is not protective and instead worsens the severity of injury (31). Hyperglycemia was induced 210 min after the onset of hypoglycemia in our study, when cerebral energy failure is likely to have already occurred (3). It is not known whether hyperglycemia early in the course of hypoglycemia would have altered the results.
As with our previous study in P14 rats (5), treatment with 10% dextrose led to mild hyperglycemia without altering PARP-1 expression. Conversely, treatment with 50% dextrose led to extreme hyperglycemia and PARP-1 upregulation, similar to a previous study in adult rats (10). The accompanying AIF upregulation suggests PARP-1 overactivation in this group (13). Although reactive oxygen species are produced during hypoglycemia (32), they are unlikely to explain the PARP-1 overactivation. A similar effect was not present in the HG-10% dextrose group, despite the equivalent severity and duration of hypoglycemia. The most likely explanation for the PARP-1 overactivation in the HG-50% dextrose group is oxidant stress due to superoxide production during glucose reperfusion with subsequent AIF-mediated cell death (5,10,13). The lack of Bcl2 upregulation, a powerful antiapoptotic protein that blocks PAR-mediated AIF release from mitochondria (12) supports this postulation. However, even if Bcl2 was upregulated, it may not have ensured neuroprotection. Addition of Bcl2 delays but does not prevent PARP-1-mediated apoptosis (13). The lack of caspase 3 upregulation confirms that PARP-1 overactivation promotes programmed cell death via caspase-independent pathway (5,12). Finally, NF-κB upregulation in the HG-50% dextrose group is not surprising, given that NF-κB is typically coexpressed with PARP-1. It is noteworthy that hyperglycemia activates NF-κB via reactive oxygen species production in astrocyte cultures (33) and suggests that NF-κB-mediated inflammation also may have contributed to the neuronal injury (34).
It was surprising that PARP-1 and AIF mRNA expressions remained upregulated 24 h posthypoglycemia. A previous study in adult rats demonstrated peak PAR expression in the hippocampus at 3 h posthypoglycemia with progressive decrease thereafter (11). The time course of PARP-1 and AIF expression was not reported in that study. Conversely, a biphasic PARP-1 and PAR expression in the cerebral cortex was demonstrated post HI in P7 rats, with an initial peak soon after the termination of hypoxia and a second peak 12 h later and lasting until 24 h (35). A biphasic pattern of neuronal injury slightly delayed in time, relative to the PARP-1 and PAR expression was present (35). Thus, persistent PARP-1 and AIF mRNA upregulation 24 h posthypoglycemia may portend the risk of additional neuronal injury. However, as our assessment was limited to one time point, this possibility remains speculative and requires confirmatory studies. Of note, our previous study comparing PARP-1 activation in P14 and adult rats also found concurrent expressions of PARP-1 and AIF mRNAs, PAR and AIF proteins, and the presence of neuronal injury at 24 h posthypoglycemia in the adult cerebral cortex (5).
Similar to a previous study in adult rats (20), BDNF-III mRNA expression was upregulated in the HG-50% dextrose group. Although there was concurrent TrkBTOTAL upregulation, it is unlikely to provide neuroprotection, since the functionally important TrkBFL isoform was not upregulated. The lack of Egr-2 upregulation, the downstream effector of BDNF/TrkB signaling, supports this postulation. In this setting, increased BDNF is likely to promote neuronal injury by binding to the upregulated p75NTR receptor. It is noteworthy that exposure to high glucose concentration upregulates p75NTR, but not TrkB receptor mRNA in pancreatic islet cells (36). Our data suggest a similar effect in the brain.
Unlike hyperglycemia, ketonemia attenuated hypoglycemia-induced injury. Compared with no treatment, maintaining ketonemia during hypoglycemia resulted in less severe injury. This is similar to the protective effects of ketogenic diet previously demonstrated in rats of similar age (16). Unlike the longer duration of ketonemia (3 d before and after hypoglycemia) in that study, ketonemia was of short duration and was induced after the onset of hypoglycemia in our study, yet was effective in reducing neuronal injury. The ketone body concentration in the blood was ~50% higher than the normal fasting values at this age (4). Participation of ketone bodies in cerebral energy production and protection against N-methyl-D-aspartate-mediated excitotoxicity and oxidant injury (37,38,39) were potentially responsible for the beneficial effects of ketonemia. Notably, the beneficial effects were seen at an age when cerebral ketone body transport and utilization has decreased markedly from the preweaning period (14,15). In this respect, our data are similar to the beneficial effects with α-ketoglutarate administration during hypoglycemia in adult rats (40). A previous study in P13 rats demonstrated that β-hydroxybutyrate administration corrects cerebral energy failure and leads to clinical improvement during hypoglycemia, but does not prevent neuronal injury (19). The inability of ketone bodies to correct deficiency of glycolytic substrates essential for anaplerosis and to support pentose phosphate pathway necessary for maintaining cellular oxidative defenses were considered responsible for the lack of sustained neuroprotection (19). This was not the case in the present study, potentially because hypoglycemia was terminated using dextrose, which can support anaplerosis and the pentose phosphate pathway. Thus, combined ketone body and dextrose administration appears to be more effective than administration of individual substrate. Further, relative to the Schutz study (19), ketonemia was induced earlier in the course of hypoglycemia in the present study, likely prior to the onset of cerebral energy failure (3). Whether ketonemia induced after the onset of cerebral energy failure also achieves neuroprotection was not evaluated.
In summary, extreme hyperglycemia does not protect the developing rat brain against hypoglycemia-induced neuronal injury and instead may accentuate it. Conversely, ketonemia combined with the conventional dextrose-based treatment appears to attenuate the severity of injury. Future studies are necessary to understand the mechanism of this neuroprotection. With additional studies, this knowledge may lead to novel management strategies for hypoglycemia in human infants and children.
Methods
Animals
Experiments were performed in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of the University of Minnesota. Pregnant Sprague-Dawley rats were purchased (Harlan Laboratories, Indianapolis, IN) and allowed to deliver spontaneously. The litter size was culled to 8 soon after birth and pups were weaned to standard rodent diet on P21. Animals were maintained under standard laboratory conditions and allowed to consume food and water ad libitum until the previous day of the experiment.
Induction of Hypoglycemia
Acute hypoglycemia was induced either on P21 (Experiment 1) or on P28 (Experiment 2) using previously published protocol (4,5). In brief, after an overnight fast, rats were injected with human regular insulin in a dose of 10 IU/kg i.p. Littermates in the control group were injected with equivalent volume of normal saline. Animals were maintained at an ambient temperature of 34 ± 1 °C and fasting was continued. In both experiments, the blood glucose concentration was maintained between 20 and 40 mg/dl by determining blood glucose every 30 min using commercial strips (Accu-Check Compact, Roche Laboratories, Indianapolis, IN) and administering 10% dextrose in a dose of 200 mg/kg s.c. when the blood glucose concentration was <20 mg/dl (4,5). In this model, animals become hypoglycemic (blood glucose <40 mg/dl) 30 min after the insulin administration and remain hypoglycemic until dextrose administration at 240 min (4,5). Cerebral energy failure typically occurs between 180 and 210 min after the insulin administration (3).
Treatment
In Experiment 1, hypoglycemia was terminated 240 min after the insulin administration by injecting either 10% dextrose (HG-10% dextrose group) or 50% dextrose (HG-50% dextrose group), in a volume of 0.2 ml i.p. This dose of 10% dextrose achieves euglycemia (blood glucose >50 mg/dl) and normalizes brain glucose concentration in developing rats (3). The dose of 50% dextrose is similar to that used for inducing hyperglycemia in previous HI studies in P7 rats (6,7,8,31).
In Experiment 2, 50% of the hypoglycemic rats were randomly assigned to receive β-hydroxybutyrate (ketone group). The remaining 50% was maintained without additional treatment (dextrose group). Rats in the ketone group were injected with DL-β-hydroxybutyrate in a loading dose of 250 mg/kg i.p., 120 min after the insulin administration. A previous study had demonstrated neuronal injury with 120 min of hypoglycemia in P28 rats (4). Ketone body concentration in the blood was measured every 30 min using a commercial blood ketone monitoring system (Precision Xtra, Abbott Laboratories, Oxon, UK). Additional doses of DL-β-hydroxybutyrate were administered to maintain plasma ketone concentration > 2 mmol/l, a value associated with protection against hypoglycemia-induced injury in P25 rats on ketogenic diet (16). Hypoglycemia was terminated 240 min after the insulin administration in both groups by injecting 10% dextrose in a dose of 200 mg/kg i.p. as in our previous studies (3,4,5).
After confirming euglycemia and normal activity, rats in both experiments were returned to their cage and food was reintroduced. They were killed 24 h later using sodium pentobarbital (120 mg/kg i.p.) and the entire brain was collected. Animals used for histochemistry underwent transcardial perfusion-fixation before brain removal as previously described (4,5). In animals used for mRNA and protein expression in Experiment 1, the brain was collected without perfusion-fixation and the cerebral cortex was dissected and processed.
Assessment of Neuronal Injury
FJB histochemistry was used to assess the distribution and severity of neuronal injury in both experiments as previously described (4,5) (n = 6–8/group). Briefly, 20 µm coronal sections were collected from the frozen brain using a cryostat and mounted on glass slides. Sections were stained for FJB, and all FJB-positive cells in the brain sections (four sections/rat) were counted and group means were determined.
Assessment of mRNA and Protein Expression
The mRNA expressions of the transcripts in the PARP-1 pathway (PARP-1, AIF, NF-κB, Bcl2, and caspase 3 (5,12)), and BDNF-III, TrkBTOTAL, TrkBFL, Egr-2, and p75NTR were determined using quantitative RT-PCR method, utilizing readymade primers (Life Technologies, Carlsbad, CA) detailed in Supplementary Table S1 (online) and previously described methods (5) (n = 6–8/group). PAR and AIF protein expressions were determined using western blot analysis, utilizing primary antibodies against each protein and β-actin as internal control as previously described (5) (n = 3/group). The mRNA and protein expression in the hypoglycemia groups were normalized against the control group.
Data Analysis
Data from the two experiments were analyzed separately. The effects of treatment (10% dextrose vs. 50% dextrose in Experiment 1, and dextrose vs. ketone in Experiment 2) on FJB-positive cells, and on mRNA and protein expressions in Experiment 1 were determined using ANOVA. Intergroup differences were determined using Bonferroni-adjusted unpaired t-tests. Data are presented as mean ± SEM. Significance was set at P < 0.05.
Statement of Financial Support
This study was funded by Viking Children’s Fund, Department of Pediatrics, University of Minnesota.
References
Adamkin DH . Postnatal glucose homeostasis in late-preterm and term infants. Pediatrics 2011;127:575–9.
Clarke W, Jones T, Rewers A, Dunger D, Klingensmith GJ . Assessment and management of hypoglycemia in children and adolescents with diabetes. Pediatr Diabetes 2009;10:Suppl 12:134–45.
Rao R, Ennis K, Long JD, Ugurbil K, Gruetter R, Tkac I . Neurochemical changes in the developing rat hippocampus during prolonged hypoglycemia. J Neurochem 2010;114:728–38.
Ennis K, Tran PV, Seaquist ER, Rao R . Postnatal age influences hypoglycemia-induced neuronal injury in the rat brain. Brain Res 2008;1224:119–26.
Rao R, Sperr D, Ennis K, Tran P . Postnatal age influences hypoglycemia-induced poly(ADP-ribose) polymerase-1 activation in the brain regions of rats. Pediatr Res 2009;66:642–7.
Vannucci RC, Mujsce DJ . Effect of glucose on perinatal hypoxic-ischemic brain damage. Biol Neonate 1992;62:215–24.
Hattori H, Wasterlain CG . Posthypoxic glucose supplement reduces hypoxic-ischemic brain damage in the neonatal rat. Ann Neurol 1990;28:122–8.
Vannucci RC, Brucklacher RM, Vannucci SJ . The effect of hyperglycemia on cerebral metabolism during hypoxia-ischemia in the immature rat. J Cereb Blood Flow Metab 1996;16:1026–33.
Seo SY, Kim EY, Kim H, Gwag BJ . Neuroprotective effect of high glucose against NMDA, free radical, and oxygen-glucose deprivation through enhanced mitochondrial potentials. J Neurosci 1999;19:8849–55.
Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA . Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest 2007;117:910–8.
Suh SW, Aoyama K, Chen Y, et al. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J Neurosci 2003;23:10681–90.
Chiarugi A, Moskowitz MA . Cell biology. PARP-1–a perpetrator of apoptotic cell death? Science 2002;297:200–1.
Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002;297:259–63.
Nehlig A . Cerebral energy metabolism, glucose transport and blood flow: changes with maturation and adaptation to hypoglycaemia. Diabetes Metab 1997;23:18–29.
Prins ML . Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab 2008;28:1–16.
Yamada KA, Rensing N, Thio LL . Ketogenic diet reduces hypoglycemia-induced neuronal death in young rats. Neurosci Lett 2005;385:210–4.
Bough KJ, Wetherington J, Hassel B, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol 2006;60:223–35.
Thurston JH, Hauhart RE, Schiro JA . Beta-hydroxybutyrate reverses insulin-induced hypoglycemic coma in suckling-weanling mice despite low blood and brain glucose levels. Metab Brain Dis 1986;1:63–82.
Schutz PW, Wong PK, O’Kusky J, Innis SM, Stockler S . Effects of d-3-hydroxybutyrate treatment on hypoglycemic coma in rat pups. Exp Neurol 2011;227:180–7.
Lindvall O, Ernfors P, Bengzon J, et al. Differential regulation of mRNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 in the adult rat brain following cerebral ischemia and hypoglycemic coma. Proc Natl Acad Sci USA 1992;89:648–52.
Nakao N, Kokaia Z, Odin P, Lindvall O . Protective effects of BDNF and NT-3 but not PDGF against hypoglycemic injury to cultured striatal neurons. Exp Neurol 1995;131:1–10.
Kokaia Z, Othberg A, Kokaia M, Lindvall O . BDNF makes cultured dentate granule cells more resistant to hypoglycaemic damage. Neuroreport 1994;5:1241–4.
Unsain N, Nuñez N, Anastasía A, Mascó DH . Status epilepticus induces a TrkB to p75 neurotrophin receptor switch and increases brain-derived neurotrophic factor interaction with p75 neurotrophin receptor: an initial event in neuronal injury induction. Neuroscience 2008;154:978–93.
Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ . Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci 2006;26:7756–66.
Tran PV, Fretham SJ, Carlson ES, Georgieff MK . Long-term reduction of hippocampal brain-derived neurotrophic factor activity after fetal-neonatal iron deficiency in adult rats. Pediatr Res 2009;65:493–8.
Moore H, Craft TK, Grimaldi LM, Babic B, Brunelli SA, Vannucci SJ . Moderate recurrent hypoglycemia during early development leads to persistent changes in affective behavior in the rat. Brain Behav Immun 2010;24:839–49.
Towfighi J, Mauger D, Vannucci RC, Vannucci SJ . Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Brain Res Dev Brain Res 1997;100:149–60.
Rao AR, Quach H, Smith E, Vatassery GT, Rao R . Changes in ascorbate, glutathione and α-tocopherol concentrations in the brain regions during normal development and moderate hypoglycemia in rats. Neurosci Lett 2014;568:67–71.
Vannucci SJ . Developmental expression of GLUT1 and GLUT3 glucose transporters in rat brain. J Neurochem 1994;62:240–6.
Brennan-Minnella AM, Won SJ, Swanson RA . NADPH oxidase-2: linking glucose, acidosis, and excitotoxicity in stroke. Antioxid Redox Signal 2014; e-pub ahead of print 14 March 2014.
Sheldon RA, Partridge JC, Ferriero DM . Postischemic hyperglycemia is not protective to the neonatal rat brain. Pediatr Res 1992;32:489–93.
McGowan JE, Chen L, Gao D, Trush M, Wei C . Increased mitochondrial reactive oxygen species production in newborn brain during hypoglycemia. Neurosci Lett 2006;399:111–4.
Hsieh HL, Lin CC, Hsiao LD, Yang CM . High glucose induces reactive oxygen species-dependent matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Mol Neurobiol 2013;48:601–14.
Kauppinen TM, Swanson RA . Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol 2005;174:2288–96.
Martin SS, Perez-Polo JR, Noppens KM, Grafe MR . Biphasic changes in the levels of poly(ADP-ribose) polymerase-1 and caspase 3 in the immature brain following hypoxia-ischemia. Int J Dev Neurosci 2005;23:673–86.
Raile K, Klammt J, Garten A, et al. Glucose regulates expression of the nerve growth factor (NGF) receptors TrkA and p75NTR in rat islets and INS-1E beta-cells. Regul Pept 2006;135:30–8.
Jiang L, Mason GF, Rothman DL, de Graaf RA, Behar KL . Cortical substrate oxidation during hyperketonemia in the fasted anesthetized rat in vivo. J Cereb Blood Flow Metab 2011;31:2313–23.
Haces ML, Hernández-Fonseca K, Medina-Campos ON, Montiel T, Pedraza-Chaverri J, Massieu L . Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp Neurol 2008;211:85–96.
Newman JC, Verdin E . Ketone bodies as signaling metabolites. Trends Endocrinol Metab 2014;25:42–52.
Suh SW, Aoyama K, Matsumori Y, Liu J, Swanson RA . Pyruvate administered after severe hypoglycemia reduces neuronal death and cognitive impairment. Diabetes 2005;54:1452–8.
Acknowledgements
We acknowledge the technical assistance of Kim Thao-Le and Kira Hinz. Presented as research abstract during the annual meetings of Pediatric Academic Societies (K.E., H.D., A.S., D.W., R.R. Treatment of hypoglycemia using 50% dextrose worsens neuronal injury in the developing rat brain, on 05 May 2013 at Washington, DC, USA) and Midwest Society for Pediatric Research (K.E., H.D., Le K-T, A.S., R.R. Hyperglycemia accentuates and beta-hydroxybutyrate attenuates hypoglycemia-induced neuronal injury in the young rat brain, on 10 October 2013 at Minneapolis, MN, USA).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(DOC 37 kb)
PowerPoint slides
Rights and permissions
About this article

Cite this article
Ennis, K., Dotterman, H., Stein, A. et al. Hyperglycemia accentuates and ketonemia attenuates hypoglycemia-induced neuronal injury in the developing rat brain. Pediatr Res 77, 84–90 (2015). https://doi.org/10.1038/pr.2014.146
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2014.146
This article is cited by
-
Prolonged transitional neonatal hypoglycaemia: characterisation of a clinical syndrome
Journal of Perinatology (2021)
-
Factors influencing glycaemic stability after neonatal hypoglycaemia and relationship to neurodevelopmental outcome
Scientific Reports (2019)
-
Continuous glucose monitoring in neonates: a review
Maternal Health, Neonatology and Perinatology (2017)
