
Abstract
Background:
The propensity for sustained inflammation after bacterial infection in neonates, resulting in inflammatory sequelae such as bronchopulmonary dysplasia and periventricular leucomalacia, is well known, but its molecular mechanisms remain elusive. Termination of inflammatory reactions physiologically occurs early after removal of bacteria by phagocytosis-induced cell death (PICD) of immune effector cells such as monocytes. PICD from cord blood monocytes (CBMOs) was shown to be reduced as compared with that of peripheral blood monocytes (PBMOs) from adult donors in vitro.
Methods:
PBMOs, CBMOs, and Fas (CD95)-deficient (lpr) mouse monocytes were analyzed in an in vitro infection model using green fluorescence protein–labeled Escherichia coli (E. coli-GFP). Phagocytosis and apoptosis were quantified by flow cytometry and CD95L secretion was quantified by enzyme-linked immunosorbent assay.
Results:
We demonstrate the involvement of the CD95/CD95 ligand pathway (CD95/CD95L) in PICD and provide evidence that diminished CD95L secretion by CBMOs may result in prolonged activation of neonatal immune effector cells.
Conclusion:
These in vitro results offer for the first time a molecular mechanism accounting for sustained inflammation seen in neonates.
Similar content being viewed by others


Main
Neonatal sepsis is a major cause of mortality and morbidity in term and preterm infants (1,2). Although acute bacterial infection can be controlled in many cases, the accompanying inflammatory reaction, sometimes starting before birth, may be excessive and hardly controllable (3). Inflammation may even persist after resolution of infection, because sepsis predisposes to inflammatory diseases exclusively manifesting during the neonatal period, i.e., bronchopulmonary dysplasia and periventricular leukomalacia (2,4,5,6,7). As for this, Dammann et al. coined the term “sustained inflammation” to describe a prolonged inflammatory reaction after an initial activation of the neonatal immune system, with an inability to resolve the resultant inflammation (8,9). Although the clinical consequences of excessive inflammation in developing organs have been well recognized, a conclusive pathophysiological paradigm for the “sustained inflammation” observed during the neonatal period is still missing (8,9).
Monocytes and macrophages represent one of the key players during bacterial infection. As phagocytes, they are essential for the rapid elimination of invading pathogens and the activation of other immune cells by secretion of proinflammatory cytokines. As antigen-presenting cells, they can further transmit signals to cells of the adaptive immune system, launching a profound inflammatory response (10).
Under physiological conditions, activity of monocytes at sites of infection is terminated by apoptosis (11), mainly induced by a process called phagocytosis-induced cell death (PICD), leading to elimination of activated monocytes (12). PICD seems to be a common event in monocyte–bacteria interaction, given that it has been shown for a huge variety of bacteria, although the signals required to induce apoptosis in this process have not yet been entirely identified (12). PICD of monocytes, as well as apoptosis of other immune cells, is important during the course of sepsis, limiting inflammation (13). Reduced monocyte apoptosis has been associated with poor outcome in adult septic patients (14).
Apoptosis can be induced by two major pathways: the external pathway, initiated by ligation of death ligands to their receptors, and the internal pathway, initiated by a variety of stimuli, leading to alteration of the mitochondrial membrane potential. Both end up in the activation of effector caspases (13).
Fas-ligand (CD95L) is one of the major death ligands belonging to the tumor necrosis factor family. It is expressed by different immune effector cells (15). In monocytes, CD95L is stored in the intracellular compartment and can be externalized and secreted as soluble ligand (sCD95L) upon stimulation (16).
In previous studies, we analyzed different steps of the interaction of cord blood monocytes (CBMOs) with common bacterial pathogens causative for neonatal sepsis, i.e., Escherichia coli (E. coli) and group-B streptococci. Although phagocytic activity and intracellular degradation of bacteria did not differ in comparison with those of monocytes from peripheral blood of adults (PBMOs) (17,18), PICD was greatly diminished in CBMOs (19). The remaining monocytes were capable of producing inflammatory signals (17). We could show that expression of Fas-receptor (CD95) was not different between PBMOs and CBMOs but that CD95L-mRNA was diminished in CBMOs (19).
In the current study, we analyzed the involvement of the CD95/CD95L pathway in PICD of monocytes exposed to E. coli and hypothesized that this pathway would be altered in CBMOs.
We show that (i) the CD95/CD95L pathway is crucial for induction of PICD; (ii) secretion of CD95L leads to apoptosis of nonphagocytizing bystander cells, and (iii) CBMOs exhibit a diminished CD95L secretion, which leads to reduced effector cell apoptosis resulting in remnant activated monocytes. These results provide an initial molecular explanation for the sustained inflammation seen in neonates.
Results
Apoptosis Upon Infection With E. coli-GFP Is Diminished in CBMOs and Mediated by Soluble Factors
Infection with E. coli-green fluorescent protein (GFP) induced significant apoptosis in PBMOs, whereas apoptosis of CBMOs was greatly diminished ( Figure 1a ) as described earlier (17,19).

Apoptosis upon infection with E. coli-GFP is diminished in CBMOs and mediated by soluble factors. CBMOs (white boxes) and PBMOs (gray boxes) were infected with E. coli-GFP for 1 h as described in the Methods. (a) After incubation for an additional 24 h, percentage of hypodiploid nuclei was assessed by flow cytometry. (b) After 3 h, conditioned media were filtered and applied to freshly isolated allogeneic PBMOs and apoptosis was assessed after 24 h. PBMOs cultivated in fresh medium only (media only) and PBMOs cultivated in medium conditioned by E. coli-GFP in the absence of cells (supernatant E. coli) served as controls. The box represents the median and 25th and 75th percentiles; n = 8. CBMC, cord blood mononuclear cell; CBMO, cord blood monocyte; E. coli, Escherichia coli; GFP, green fluorescent protein; PBMC, peripheral blood mononuclear cell; PBMO, peripheral blood monocyte.
To address whether apoptosis was mediated by soluble factors, we used a media conditioned by infected monocytes. Media from peripheral blood mononuclear cells (PBMCs) given on freshly isolated allogeneic PBMOs were capable apoptosis inductors, with median (minimum–maximum) apoptosis rates after 3 h of 46% (20–67%) as compared with 5% (3–12%) in control PBMOs cultured in fresh medium ( Figure 1b ). However, when incubating PBMOs in media conditioned by cord blood mononuclear cells (CBMCs), apoptosis occurred in only 23% (7–41%) of target PBMOs ( Figure 1b ). Neither media conditioned by PBMCs nor that conditioned by CBMCs induced apoptosis in freshly isolated, allegeneic CBMOs (data not shown). Medium conditioned by E. coli-GFP alone was also capable of inducing apoptosis in a relevant percentage of PBMOs 20% (19–28%). ANOVA revealed a significant main effect for unconditioned media (P value < 0.001). Post hoc pair-wise comparisons showed significant differences between (i)unconditioned media and all other conditions (all P values < 0.001) and (ii) media conditioned by infected PBMCs and that conditioned by infected CBMCs (P value = 0.041). Thus, conditioning by infected PBMCs generated significant apoptosis-inducing capacity in cell culture media, whereas this effect was only marginally detectable for infected CBMCs.
PBMOs and CBMOs Are Sensitive to CD95L-Induced Apoptosis
We next analyzed the impact of CD95L on PBMOs and CBMOs. Addition of CD95L induced apoptosis in both naive PBMOs and CBMOs in a concentration-dependent manner. Apoptosis occurred in up to 61% (44–77%) of CBMOs and 86% (71–93%) of PBMOs ( Figure 2a ). ANOVA showed only one significant main effect (CD95L; P value <0.001), i.e., cell type (CBMO or PBMO) and the interaction term between cell type and CD95L were not significant. Post hoc pair-wise comparisons showed significant differences only for medium (100 ng/ml; P value <0.001) and high (1,000 ng/ml; P value <0.001) CD95L concentrations as compared with incubation without external CD95L. These results indicated that CD95L is potent to induce apoptosis, showing similar effects in CBMOs and PBMOs.

PBMOs and CBMOs are sensitive to CD95L-induced apoptosis. (a) Naive and (b) E. coli-GFP–infected CBMOs (white boxes) and PBMOs (gray boxes) were incubated with external CD95L with concentrations indicated. After 24 h, the percentage of hypodiploid nuclei was assessed by flow cytometry. The box represents the median and 25th and 75th percentiles; open circles indicate outliers; n = 6. CBMO, cord blood monocyte; E. coli, Escherichia coli; GFP, green fluorescent protein; PBMO, peripheral blood monocyte.
When incubating PBMOs and CBMOs after infection with E. coli-GFP, low concentrations of external CD95L (10 ng/ml) increased the low apoptosis rates seen in infected CBMOs from 20% (16–21%) to 36% (26–47%), comparable with those seen in infected PBMOs (43% (37–58%), Figure 2b ), whereas CD95L in this concentration had no effect on infected PBMOs (43% (32–61%)). External CD95L in higher concentrations led to further rising of apoptosis rates in infected PBMOs and CBMOs. ANOVA revealed significant main effects for cell type (CBMO or PBMO) and CD95L (all P values <0.001), as well as for the interaction term of both (P value =0.032). Again, post hoc pair-wise comparisons showed significant differences only for medium (100 ng/ml; P value <0.001) and high (1,000 ng/ml; P value <0.001) CD95L concentrations as compared with incubation without external CD95L. In contrast to uninfected cells, these data indicated different effects of CD95L in infected CBMOs as compared with PBMOs, with CBMOs tending to be more susceptible to apoptosis induction.
Infected PBMOs showed significantly elevated apoptosis rates as compared with naive PBMOs exposed to 10 and 100 ng/ml CD95L, whereas addition of 1,000 ng/ml CD95L resulted in higher apoptosis rates in noninfected PBMOs ( Figure 2a,b ). However, infection had only little effect in CBMOs (data not shown).
CD95/CD95L Signaling Is Crucial for E. coli-GFP–Induced Apoptosis
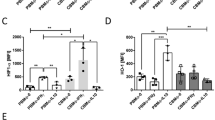
To demonstrate the relevance of CD95/CD95L signaling for E. coli-GFP–mediated apoptosis, we chose two different approaches. In a first series of experiments, a blocking monoclonal antibody against CD95 (αCD95), the receptor of CD95L, was used to inhibit CD95L signaling. Incubation of PBMOs with αCD95 before, during, and after infection with E. coli-GFP resulted in a dramatic decrease in apoptosis rates to nearly 50% of controls (52% (28–74%) vs. 31% (17–46%), Figure 3a ). Incubation of CBMOs with αCD95 did not further diminish the low apoptosis rate after infection (22% (15–25%) vs. 19% (11–27%), Figure 3a ). However, ANOVA did not show a significant main effect of blocking antibody incubation (P value =0.072), nor was the interaction term between cell type (CBMO or PBMO) and antibody incubation significant (P value =0.195). In one post hoc pair-wise comparison, the mean individual differences (SD) in apoptosis rate between incubation with isotype antibody and incubation with αCD95 were significant (−2% (7%) in CBMOs vs. −21% (12%) in PBMOs; unpaired t-test: P value =0.006). This supported a relevant role for CD95/CD95L signaling in E. coli-GFP–mediated apoptosis induction. Control staining with fluorescent labeled anti-CD95 monoclonal antibodies (mAbs) for quantifying the blocking effect of αCD95 revealed that 60% of CD95 receptors were blocked (data not shown).

CD95/CD95L signaling is crucial for E. coli-GFP–induced apoptosis. (a) CBMOs (white boxes) and PBMOs (gray boxes) were incubated with blocking αCD95L mAb (ZB4, 4 μg/ml) or isotype control and then infected with E. coli-GFP for 1 h and free bacteria removed as described in the Methods. After 24 h, the percentage of hypodiploid nuclei was assessed by flow cytometry, n = 3. (b,c) PBMCs from wild-type (WT) and MRL-Faslpr mice were infected for 1 h with E. coli-GFP and free bacteria removed as described in Methods. Cells were either stained with CD14-FITC and (b) percentage of GFP+ cells of CD14+ cells was quantified or cultured for another 24 h and (c) percentage of hypodiploid nuclei was quantified by flow cytometry. The box represents the median and 25th and 75th percentiles; open circles indicate outliers; n = 3. CBMO, cord blood monocyte; E. coli, Escherichia coli; FITC, fluorescein isothiocyanate; GFP, green fluorescent protein; mAb, monoclonal antibody; PBMC, peripheral blood mononuclear cell; PBMO, peripheral blood monocyte.
In a second approach, a transgenic mouse model helped to confirm the relevance of CD95/CD95L signaling in PICD, using isolated CD95-deficient MRL-Faslpr mouse monocytes. MRL-Faslpr monocytes showed slightly elevated phagocytosis activity as compared with wild-type mice (21% (21–26%) vs. 17% (13–18%), Figure 3b ) after in vitro infection with E. coli-GFP. However, E. coli-GFP–induced apoptosis rates were diminished in MRL-Faslpr as compared with monocytes from wild-type mice (10% (9–30%) vs. 39% (38–66%), Figure 3c ).
sCD95L Secretion Is Diminished in CBMCs
Having shown that media conditioned by CBMCs exhibited a weaker potency to induce apoptosis, we quantified sCD95L in media conditioned by PBMCs and CBMCs. CD95L content in media of naive CBMCs and PBMCs was low (8 (7–10) pg/ml and 10 (2–23) pg/ml, Figure 4 ). Twenty-four hours after infection with E. coli-GFP, CD95L concentration was increased to 52 (21–88) pg/ml in media from PBMCs, whereas in media from CBMCs, CD95L content remained low (10 (5–18) pg/ml, Figure 4 ). ANOVA showed significant main effects for cell type (CBMO or PBMO; P value =0.001) and infection (P value <0.001), as well as for the interaction term of the two (P value =0.001). These data indicate a strongly diminished secretion of CD95L by CBMCs upon infection with E. coli-GFP.

sCD95L secretion is diminished in CBMCs. CBMCs (white boxes) and PBMCs (black bars) were infected for 1 h with E. coli-GFP and free bacteria removed as described in Methods. After 24 h, media were removed and analyzed for sCD95L by ELISA. Uninfected PBMCs and CBMCs served as controls. The box represents the median and 25th and 75th percentiles; open circle indicate outliers; n = 6. CBMC, cord blood mononuclear cell; E. coli, Escherichia coli; ELISA, enzyme-linked immunosorbent assay; GFP, green fluorescent protein; PBMC, peripheral blood mononuclear cell.
Remaining CBMOs Are Capable of Inducing T-Cell Proliferation
Finally, the functional consequences related to the observed differences in monocyte PICD of PBMOs and the functionality of the remaining CBMOs were assessed. Therefore, we analyzed an obligatory monocyte-dependent function and chose αCD3 mAb–stimulated T-cell proliferation, which depends on monocyte capacity to provide costimulatory signals (20,21). PBMOs and CBMOs were infected with E. coli-GFP and, after removal of free bacteria, T cells were stimulated with αCD3 for 48 h and analyzed by flow cytometry. Noninfected cells served as controls. Representative histograms are presented in Figure 5a .

Apoptosis-resistant CBMOs are capable of inducing T-cell proliferation. CFSE-labeled PBMCs and CBMCs were infected for 1 h with E. coli and free bacteria removed as described in Methods. Cells were stimulated with αCD3 (OKT3, 5 μg/ml) for 48 h, and T-cell proliferation was analyzed by flow cytometry. Uninfected cells and cells not treated with αCD3 served as controls. (a) Representative histogram overlays of αCD3-treated PBMCs (above) and CBMCs (below) infected with E. coli (black lined graph) or uninfected controls (filled gray graph) are given. (b) Proliferating T cells in CBMCs (white boxes) and PBMCs (gray boxes) are depicted. The box represents the median and 25th and 75th percentiles; n = 11. CBMC, cord blood mononuclear cell; CBMO, cord blood monocyte; CFSE, 5-carboxyfluorescein diacetate succinimidyl ester; E. coli, Escherichia coli; PBMC, peripheral blood mononuclear cell.
In the absence of αCD3 mAb, T-cell proliferation was negligible, both in uninfected and infected CBMCs vs. PBMCs (9% (4–16%) vs. 2% (1–6%)) and 15% (4–25%) vs. 3% (1–22%) ( Figure 5b ), respectively. As shown earlier (20,22), αCD3 mAb-mediated T-cell proliferation in uninfected CBMCs was diminished as compared with that of PBMCs (28% (15–80%) vs. 64% (40–85%), Figure 5b ). Infection of monocytes resulted in a greatly diminished αCD3-mediated T-cell proliferation in PBMCs as compared with their uninfected counterparts (21% (12–82%), Figure 5b ), whereas T-cell proliferation in CBMCs was increased (82% (46–89%), Figure 5b ). In ANOVA, the interaction term between cell type (CBMC or PBMC) and infection with E. coli-GFP was not significant in the absence of αCD3 stimulation (P value =0.614) but was significant in the presence of αCD3 (P value <0.001). This indicated that infection of PBMCs leads to diminished capacity to stimulate T cells, whereas in CBMCs, stimulatory capacity was strongly enhanced.
Discussion
In this study, we analyzed signals involved in inducing PICD in monocytes and contributing to diminished PICD in CBMOs and found that (i) media conditioned by infected monocytes contained apoptosis-inducing capacity, (ii) blocking CD95 and using CD95-knockout monocytes diminished PICD, and (iii) CD95L was secreted during PICD. Furthermore, we showed that diminished PICD of neonatal monocytes was associated with a diminished secretion of CD95L and provided evidence that the remaining CBMOs were capable costimulators and may thus be causing continuing inflammation.
Signals inducing apoptosis in monocytes during their interaction with bacterial pathogens have mostly been linked to direct binding and/or phagocytosis, leading to the term “phagocytosis-induced cell death” (12). In addition, phagocytizing monocytes were shown to be capable of inducing apoptosis in bystander cells (15).
Our data show that cell culture media conditioned by E. coli-infected monocytes was capable of inducing apoptosis in bystander cells ( Figure 1 ). This effect has already been shown for in vitro infection models of monocytes/macrophage phagocytosis with a variety of other pathogens such as Chlamydia trachomatis (23), Streptococcus pneumoniae (24), or yeast wall–prepared zymosan (15). Moreover, two studies with sera from patients suffering from severe infections, i.e., sepsis, pneumonia, or urinary tract infection, also exhibited apoptosis-inducing capacity when tested in an in vitro cell culture system (25,26). Of note, in the experiments presented here, even cell-culture medium inoculated with E. coli only and conditioned in the absence of cells was capable of inducing apoptosis, although no bacteria could be cultivated from it ( Figure 1 ). We therefore think that bacterial cell wall components that cannot be detected by flow cytometric analysis might be ingested as subcellular fragments thereby inducing apoptosis in monocytes. On the whole, these data underscore the role of soluble factors involved in PICD and document the occurrence of bystander apoptosis.
Apoptosis-inducing capacity was associated with dramatically elevated CD95L content in media from PBMOs ( Figure 4 ). This is in line with data showing elevated CD95L mRNA (19) and protein levels (27) after infection of PBMOs, and with data from adult patients showing elevated CD95L levels during the course of sepsis (26). The relevance of CD95-signaling for termination of inflammation demonstrated here ( Figure 3 ) has been demonstrated in a mouse model of bacteria-induced acute lung injury. Monocytes invading the lung during the acute phase of inflammation were effectively eliminated by CD95L-induced apoptosis and cleared by local immune cells. Apoptosis could be prevented by the addition of neutralizing antibodies against CD95 or CD95L, leading to a delayed resolution of inflammation and pronounced organ damage (28).
CBMOs exhibited diminished CD95L secretion ( Figure 4 ), leading to remnant and activated immune effector cells ( Figure 5 ). Former reports on serum sCD95L predominantly revealed decreased levels in cord blood and during the neonatal period as compared with adults and pregnant women (29,30). In view of balancing inflammatory reactions in neonates, we therefore speculate that reduced levels of external death ligands such as CD95L might shift the balance toward prolonged inflammation by insufficient downregulation of immune effector cells, possibly leading to tissue destruction.
Recent data indicate that concentrations of inflammation-related proteins were elevated peripartally and slowly decreased in an “inverse developmental” pattern (8). Moreover, clinical, histological, and experimental data on diseases exclusively manifesting during the neonatal period, such as bronchopulmonary dysplasia (3,6,31) and periventricular leukomalacia (4,7,32), are all linked to prolonged inflammation leading to tissue destruction. Thus, the data presented here might contribute to a molecular explanation for the “sustained inflammation” seen in neonates.
However, because the data presented here are in vitro data, the relevance of CD95/CD95L signaling during inflammation in neonates has to be evaluated in clinical studies and data from preterm infants should be included, as we analyzed only cord blood from term neonates.
In view of regulating apoptosis not only in immune cells, but in the developing organism at large, the role of the CD95/CD95L system might be a double-edged one. For CD95L to become operant, its receptor, CD95, has to be expressed on target cells, rendering these cells sensitive toward CD95L-mediated apoptosis (33). Although CD95 is constitutively expressed on monocytes and granulocytes in low densities (19,34), in other cell types such as epithelial cells, CD95 expression is well controlled and can be upregulated upon activation, for example during inflammation (35). In a series of elegant studies on the impact of CD95/CD95L signaling in fetal and neonatal lung development, De Paepe et al. showed in a mouse model that CD95L expression was regulated in a highly specific pattern and that overexpression of CD95L, especially during the late antenatal period, was associated with massive respiratory epithelial cell apoptosis. They related this to perinatal inflammatory conditions (35).
Recent data on the developing brain show that elevated CD95L levels in the cerebrospinal fluid of preterm infants may be correlated with progressive cystic white matter damage, suggesting that apoptosis via the CD95/CD95L pathway is involved in the pathogenesis of this disease (32).
Taken together, induction of apoptosis in inflamed tissues to control immune effector cells has to be balanced against the suceptibility of parenchymal cells toward apoptotic stimuli. This balance might be altered in the developing neonatal system as compared with conditions seen in adults. Diminished CD95L secretion of neonatal immune cells, e.g., monocytes, during inflammatory reactions as shown in our in vitro model might avert direct parenchymal damage on the one hand but might lead to damage through sustained inflammation on the other hand. A profound knowledge of the regulation of apoptosis induction in neonates and an understanding of the phenomenon of sustained inflammation are essential to develop new anti-inflammatory strategies.
Methods
Patients
The study protocol was approved by the ethics committees of Tuebingen University Hospital and Aachen University Hospital. All the mothers gave written consent before they went into labor. Randomly selected, unrelated adults donated blood and served as controls. All term neonates were delivered spontaneously and did not exhibit signs of infection, as defined by clinical status, white blood cell count, and C-reactive protein. Mothers with amnion infection and prolonged labor were excluded. Umbilical cord blood was placed in heparin-coated tubes (4 IU/ml blood), immediately following cord ligation.
Reagents
mAbs to CD14 (MFP9) were from BD Biosciences (Heidelberg, Germany), anti-CD95L-PE (MHCD95L04) from Caltag/Invitrogen (Karlsruhe, Germany), Ig-matched controls (IgG1, IgG2b) and Fix-and-Perm-Solution from BD Biosciences. SuperFasLigand was from Alexis (Lörrach, Germany). Blocking anti-CD95 mAb (ZB4) was from Beckman Coulter (Krefeld, Germany). Propidium iodide, isopropyl-β-D-thiogalactopyranosid, and antibiotics were purchased from Sigma (Munich, Germany), CD95L-enzyme-linked immunosorbent assay was from Ebiosciences (Ebiosciences-Natutec, Frankfurt, Germany). αCD3 mAb (OKT3) was from Ortho Diagnostics (Raritan, NJ), and 5-carboxyfluorescein diacetate succinimidyl ester (CFSE) was from Molecular Probes (Eugene, OR).
Cell Cultures
PBMCs and CBMCs were isolated by density gradient centrifugation (Biochrom AG, Berlin, Germany) as described (18). Washed cells were resuspended in VLE RPMI-1640 (Biochrom) containing 10% heat-inactivated fetal calf serum (Biochrom) at 2 × 106 cells/ml in flat-bottom 24 well cell culture plates (Costar, Bodenheim, Germany).
Purification of Monocytes
Monocytes were separated by negative selection using magnetic cell sorting Monocyte Isolation Kit II (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. The purity of the resulting population was >92% CD14-positive cells as detected by flow cytometry.
Mice
B6.MRL-Faslpr/J or C57BL/6 (Jackson Mouse Database, Jackson Laboratories, Bar Harbor, ME) mice were killed by dissection of the abdominal artery. Whole blood (2 ml) was collected in heparinized tubes and mononuclear cells were prepared using density gradient centrifugation as described (18). Amounts of gradient solutions were downscaled to the smaller sample volume.
Bacterial Culture
E. coli DH5α, an encapsulated K12 laboratory strain, carrying the GFP-mut2 gene (36), was used for most infection assays (E. coli-GFP) except for infection of CFSE-labeled cells, in which the wild-type E. coli DH5α (E. coli) was used. Bacteria were freshly grown in Lennox-L-Broth-medium (Invitrogen) until early logarithmic growth, resuspended in phosphate-buffered saline, and used immediately.
Phagocytosis Assay
The phagocytosis assay was carried out as previously described (18). Incubation was performed for 60 min at a multiplicity of infection of 50:1. For analysis of postphagocytic reactions, washed cells were cultured in RPMI supplemented with 10% fetal calf serum and gentamycin (Sigma; 200 µg/ml). To collect conditioned media, washed cells were cultured for 1 and 3 h; then the supernatant was removed and centrifuged at 2,000g for 5 min at room temperature. The supernatant was filtered through a 0.2 µm filter (Millipore, Schwalbach, Germany) and either used immediately or stored at −70 °C until use. For incubation, 50 µl supernatant was added to 2 × 105 allogeneic PBMOs in 50 µl fresh RPMI supplemented with 10% fetal calf serum to a final volume of 100 µl.
DNA Fragmentation
DNA fragmentation was assessed according to Nicoletti (19,37). Washed cells were slowly resuspended in 2 ml of −20 °C ethanol 70% under continuous vortexing and stored for 4 h at −20 °C. Cells were washed twice, resuspended in 50 µl phosphate-buffered saline containing 13 kunitz units RNase (DNase free; Sigma) and incubated for 15 min at 37 °C. Thereafter, 180 µl of propidium iodide (70 µg/ml) was added; the mixture was incubated for 20 min and immediately analyzed.
Enzyme-Linked Immunosorbent Assay
The CD95L enzyme-linked immunosorbent assay was purchased from Ebiosciences and used according to the manufacturer’s recommendations, as described previously (38).
T-Cell Proliferation Assay
T-cell proliferation was assessed by Vybrant CFDA SE Cell Tracer Kit (Molecular Probes, Eugene, OR), as described before (20).
Statistical Analysis
The primary study variable was the percentage of hypodiploide nuclei, which showed a right-skewed distribution. Hence, descriptive statistics such as median, minimum, and maximum as well as box-and-whisker plots were used to summarize results and present distributions. For statistical testing, the variable was log-transformed to achieve a normally distributed test variable. Means of this test variable were compared across factors using fixed-effects two-way ANOVA. Post hoc pair-wise comparisons were performed using Scheffé’s or Dunnett’s method where appropriate. A P value of <0.05 was considered statistically significant. Analyses were done with statistical software (IBM SPSS Statistics 20, Ehingen, Germany).
Statement of Financial Support
This work was supported by the Research Fund of the Medical Faculty of Tuebingen University (grant F. 1275143.2).
References
Benjamin DK Jr, Stoll BJ . Infection in late preterm infants. Clin Perinatol 2006;33:871–82; abstract x.
Stoll BJ, Hansen NI, Bell EF, et al.; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 2010;126:443–56.
Thomas W, Speer CP . Chorioamnionitis: important risk factor or innocent bystander for neonatal outcome? Neonatology 2011;99:177–87.
Adams-Chapman I, Stoll BJ . Neonatal infection and long-term neurodevelopmental outcome in the preterm infant. Curr Opin Infect Dis 2006;19:290–7.
Bartels DB, Kreienbrock L, Dammann O, Wenzlaff P, Poets CF . Population based study on the outcome of small for gestational age newborns. Arch Dis Child Fetal Neonatal Ed 2005;90:F53–9.
Hentschel J, Berger TM, Tschopp A, Müller M, Adams M, Bucher HU; Swiss Neonatal Network. Population-based study of bronchopulmonary dysplasia in very low birth weight infants in Switzerland. Eur J Pediatr 2005;164:292–7.
Schlapbach LJ, Aebischer M, Adams M, et al.; Swiss Neonatal Network and Follow-Up Group. Impact of sepsis on neurodevelopmental outcome in a Swiss National Cohort of extremely premature infants. Pediatrics 2011;128:e348–57.
Leviton A, Fichorova R, Yamamoto Y, et al. Inflammation-related proteins in the blood of extremely low gestational age newborns. The contribution of inflammation to the appearance of developmental regulation. Cytokine 2011;53:66–73.
Leviton A, Hecht JL, Allred EN, Yamamoto H, Fichorova RN, Dammann O; ELGAN Study Investigators. Persistence after birth of systemic inflammation associated with umbilical cord inflammation. J Reprod Immunol 2011;90:235–43.
Medzhitov R, Janeway C Jr . Innate immunity. N Engl J Med 2000;343:338–44.
Feig C, Peter ME . How apoptosis got the immune system in shape. Eur J Immunol 2007;37:Suppl 1:S61–70.
DeLeo FR . Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 2004;9:399–413.
Hotchkiss RS, Nicholson DW . Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol 2006;6:813–22.
Giamarellos-Bourboulis EJ, Routsi C, Plachouras D, et al. Early apoptosis of blood monocytes in the septic host: is it a mechanism of protection in the event of septic shock? Crit Care 2006;10:R76.
Brown SB, Savill J . Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J Immunol 1999;162:480–5.
Kiener PA, Davis PM, Rankin BM, et al. Human monocytic cells contain high levels of intracellular Fas ligand: rapid release following cellular activation. J Immunol 1997;159:1594–8.
Gille Ch, Leiber A, Mundle I, et al. Phagocytosis and postphagocytic reaction of cord blood and adult blood monocyte after infection with green fluorescent protein-labeled Escherichia coli and group B Streptococci. Cytometry B Clin Cytom 2009;76:271–84.
Gille C, Spring B, Tewes L, Poets CF, Orlikowsky T . A new method to quantify phagocytosis and intracellular degradation using green fluorescent protein-labeled Escherichia coli: comparison of cord blood macrophages and peripheral blood macrophages of healthy adults. Cytometry A 2006;69:152–4.
Gille C, Leiber A, Spring B, et al. Diminished phagocytosis-induced cell death (PICD) in neonatal monocytes upon infection with Escherichia coli. Pediatr Res 2008;63:33–8.
Gille C, Spring B, Bernhard W, et al. Differential effect of surfactant and its saturated phosphatidylcholines on human blood macrophages. J Lipid Res 2007;48:307–17.
Orlikowsky T, Dannecker GE, Wang Z, Horowitz H, Niethammer D, Hoffmann MK . Activation or destruction of T cells via macrophages. Pathobiology 1999;67:298–301.
Orlikowsky TW, Spring B, Dannecker GE, Niethammer D, Poets CF, Hoffmann MK . Expression and regulation of B7 family molecules on macrophages (MPhi) in preterm and term neonatal cord blood and peripheral blood of adults. Cytometry B Clin Cytom 2003;53:40–7.
Jendro MC, Deutsch T, Körber B, et al. Infection of human monocyte-derived macrophages with Chlamydia trachomatis induces apoptosis of T cells: a potential mechanism for persistent infection. Infect Immun 2000;68:6704–11.
Dockrell DH, Lee M, Lynch DH, Read RC . Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J Infect Dis 2001;184:713–22.
Nwakoby IE, Reddy K, Patel P, et al. Fas-mediated apoptosis of neutrophils in sera of patients with infection. Infect Immun 2001;69:3343–9.
Turrel-Davin F, Guignant C, Lepape A, Mougin B, Monneret G, Venet F . Upregulation of the pro-apoptotic genes BID and FAS in septic shock patients. Crit Care 2010;14:R133.
Baran J, Weglarczyk K, Mysiak M, et al. Fas (CD95)-Fas ligand interactions are responsible for monocyte apoptosis occurring as a result of phagocytosis and killing of Staphylococcus aureus. Infect Immun 2001;69:1287–97.
Janssen WJ, Barthel L, Muldrow A, et al. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med 2011;184:547–60.
Kuntz TB, Christensen RD, Stegner J, Duff P, Koenig JM . Fas and Fas ligand expression in maternal blood and in umbilical cord blood in preeclampsia. Pediatr Res 2001;50:743–9.
Protonotariou E, Rizos D, Malamitsi-Puchner A, Sarandakou A, Botsis D . Tissue polypeptide specific antigen and soluble Fas during normal pregnancy and early life.In Vivo 2006;20(6B):901–5.
May M, Marx A, Seidenspinner S, Speer CP . Apoptosis and proliferation in lungs of human fetuses exposed to chorioamnionitis. Histopathology 2004;45:283–90.
Schmitz T, Felderhoff-Mueser U, Sifringer M, Groenendaal F, Kampmann S, Heep A . Expression of soluble Fas in the cerebrospinal fluid of preterm infants with posthemorrhagic hydrocephalus and cystic white matter damage. J Perinat Med 2011;39:83–8.
Nagata S, Golstein P . The Fas death factor. Science 1995;267:1449–56.
Iwai K, Miyawaki T, Takizawa T, et al. Differential expression of bcl-2 and susceptibility to anti-Fas-mediated cell death in peripheral blood lymphocytes, monocytes, and neutrophils. Blood 1994;84:1201–8.
de Paepe ME, Haley SA, Lacourse Z, Mao Q . Effects of Fas-ligand overexpression on alveolar type II cell growth kinetics in perinatal murine lungs. Pediatr Res 2010;68:57–62.
Dehio M, Knorre A, Lanz C, Dehio C . Construction of versatile high-level expression vectors for Bartonella henselae and the use of green fluorescent protein as a new expression marker. Gene 1998;215:223–9.
Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C . A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 1991;139:271–9.
Hentschel N, Krusch M, Kiener PA, Kolb HJ, Salih HR, Schmetzer HM . Serum levels of sCD137 (4-1BB) ligand are prognostic factors for progression in acute myeloid leukemia but not in non-Hodgkin’s lymphoma. Eur J Haematol 2006;77:91–101.
Acknowledgements
We thank Ann-Sophie Müller for laying out the diagrams. Thanks to Dr Anne Wesner for her help with the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gille, C., Dreschers, S., Leiber, A. et al. The CD95/CD95L pathway is involved in phagocytosis-induced cell death of monocytes and may account for sustained inflammation in neonates. Pediatr Res 73, 402–408 (2013). https://doi.org/10.1038/pr.2012.196
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2012.196
This article is cited by
-
Expression of immune checkpoint molecules on adult and neonatal T-cells
Immunologic Research (2023)
-
Group B streptococci infection model shows decreased regulatory capacity of cord blood cells
Pediatric Research (2022)
-
Antecedents of epilepsy and seizures among children born at extremely low gestational age
Journal of Perinatology (2019)
-
MSCS in Scenarios of Infection and Inflammation: Focus on Neonatal Diseases
Current Stem Cell Reports (2016)
-
Intermittent or sustained systemic inflammation and the preterm brain
Pediatric Research (2014)
