
Abstract
Schimke immuno-osseous dysplasia (SIOD) is an autosomal recessive disorder caused by loss-of-function mutations in SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a-like 1 (SMARCAL1), with clinical features of growth retardation, spondylo-epiphyseal dysplasia, nephrotic syndrome, and immunodeficiency. We report a patient with SIOD and SMARCAL1 splice mutation (IVS4-2 A>G) in a nonconsanguineous Ashkenazi family, who came to our attention at 1 mo of age due to renal malformation and only later developed signs compatible with Schimke. Interestingly, residual SMARCAL1 mRNA levels in the patient's peripheral blood were lower compared with those observed in both asymptomatic brothers' carrying the same bi-allelic mutation, whereas the latter had levels similar to those found in heterozygous carriers (parents and sister). Examination of the carrier frequency of the splice mutation in the Ashkenazi population demonstrated 1 carrier in 760 DNA samples. In situ localization of SMARCAL1 in human kidneys as well as analysis of its temporal expression during murine nephrogenesis and in the metanephric organ culture suggested a role in the early renal progenitor population and after renal maturation. Thus, disease severity within the same family might be modified by the splicing machinery. The renal expression pattern of SMARCAL1 explains a broader spectrum of renal disease in SIOD than previously described.
Similar content being viewed by others

Main
Schimke immuno-osseous dysplasia (SIOD) is characterized by autosomal recessive inheritance, spondyloepiphyseal dysplasia causing growth retardation, defective cellular immunity, progressive nephropathy leading to renal failure, hyperpigmented macules, and dysmorphic facial features (1–4). Half of SIOD patients also have hypothyroidism, half episodic cerebral ischemia, and a tenth bone marrow failure.
SIOD is caused by mutations in SWI/SNF2 related, matrix associated, actin dependent regulator of chromatin, subfamily a-like 1(SMARCAL1) (5). SNF2 related proteins participate in the DNA nucleosome restructuring which commonly occurs during gene regulation and DNA replication, recombination, methylation, and repair (6,7).
Recent analysis of detailed autopsies to correlate clinical and pathologic findings in SIOD identified T cell deficiency in peripheral lymphoid organs, defective chondrogenesis, focal segmental glomerulosclerosis (FSGS), cerebral ischemic lesions, and premature atherosclerosis (8).
Herein, we report on a male patient, 6 y old, who came to our attention after birth due to renal malformation and later on developed growth retardation, bone dysplasia, and steroid-resistant nephrotic syndrome and was shown to harbor a bi-allelic SMARCAL1 splice mutation. Investigation of his nonconsanguineous Ashkenazi family lead us to discover phenotypic diversity and variable expressivity of SMARCAL1 in the family along with low carrier frequency of the splice mutation in the Ashkenazi population. In addition, in situ localization of SMARCAL1 in the human kidney and developmental expression of the murine homologue during nephrogenesis and in the metanephric organ culture correlate with the broader renal phenotype were observed.
MATERIALS AND METHODS
Case report.
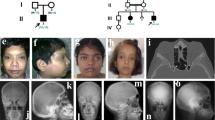
The index patient (III.1), the 6-y-old son of nonconsanguineous parents of Ashkenazi descent, was referred to our nephrology clinic at the age of 1 mo because of suspected renal agenesis. He was born at 36 wk of gestation in a cesarean section weighting 1730 g. At 31 wk of gestation, vaginal bleeding was observed and ultrasound revealed intrauterine growth retardation, single umbilical artery, relative oligohydramnios, and suspected single kidney. Prenatal course was normal. At 1 mo he had gained weight (2.6 kg) and physical exam including length was normal whereas renal ultrasound revealed the left kidney crossing the midline to the right underneath the right kidney; both renal parenchyma and collecting system were normal. On the second and third visits (3 and 8.5 mo), he was feeling well and gaining weight (5.5 kg and 8 kg, respectively). Dimercaptosuccinate acid scan showed no evidence of dysplasia with the left kidney attached to the right kidney leading to difficulty in discriminating and estimating relative function. A diagnosis of fused crossed ectopic kidney was made. He was later seen at the age of 3.5 y when growth failure was observed (height: 82.4 cm, −3.5 SD). On clinical examination, he had a disproportionately short stature with a short neck and trunk, thoracic kyphosis, lumbar lordosis, protruding abdomen, and minimal pretibial edema. Skin rash compatible with atopic dermatitis and several hyperpigmented spots were noted. Over the next months, he developed notable pretibial edema and laboratory results showed hypoalbuminemia (3.1→2.2 gr/dL), elevated cholesterol levels (223→384 mg/dL), nephrotic range proteinuria (105 mg/m2/h), and normal renal function. A 12-wk trial of corticosteroids (prednisone at a starting dose of 60 mg/m2/d) was given with no response. Renal biopsy performed at this point revealed changes consisting with FSGS (Fig. 1A). Endocrine work-up, including thyroid function tests, was normal. Skeletal radiographs revealed spondylo-epiphyseal dysplasia (Fig. 1A). There were low CD3 and CD4 cell counts (570/μL and 190/μL, respectively), reduced T cell proliferation and CD4/CD8 ratio was inverted (0.4). Both IgG levels and the specific immunoglobulins to Diphteria and Tetanus were low. The patient was diagnosed as having SIOD, and further immunosuppressive therapy was withheld. Treatment with the anticonverting enzyme-inhibitor enalapril plus the Angiotensin-II receptor antagonist losartan resulted in a minor reduction of proteinuria to 82 mg/m2/h. Recently he developed full blown nephrotic syndrome with massive proteinuria, hypoalbuminemia, dyslipidemia, and edema with no deterioration of renal function. He was treated with i.v. albumin and furoseamide with only partial improvement. He is currently treated with a combination of the anticonverting enzyme-inhibitors enalapril (0.6 mg/kg/d), aldospiron (1.5 mg/kg/d), and pravastatin with minor response. He still has nephrotic range proteinuria (protein to creatinine ratio 46), hypoalbuminemia (Albumin—1.9 gr/dL), dyslipidemia (triglycerides level 677 mg/dL, cholesterol level 248 mg/dL) with minimal pretibial edema and preserved renal function (Cr—0.15 mg/dL). The patient has not had any serious infections, migraines, transient ischemic attacks, or cerebrovascular complications. Physical examination and skeletal radiographs of parents (II.1 and II.2), two nonidentical younger twin brothers (III.2 and III.3) aged 5 y old and a younger sister aged 18 mo (III.4) were unremarkable. The twin brothers have been recently shown to have + 1 protein on albustix but no other signs of nephropathy. None of the other family members showed proteinuria or nephropathy.

Clinical characteristics of index patient. (A) Renal biopsy demonstrating focal segmental sclerosis. Bone abnormalities shown in (B) epiphyseal dysplasia of femur head and (C) skeletal kyphoscoliosis and abnormal thorax. Original magnification: (A) × 200, Scale bar = 5 μm.
Mutation analysis.
Informed consent and institutional approval (Schneider Medical Center) were obtained. Blood samples were collected and polymerase chain reaction (PCR) amplification of SMARCAL1 transcript was performed on patient's cDNA (RNA isolation and cDNA synthesis described below) using primers sets shown in Table 1. Amplicons typically overlapped by ∼100 bp. PCR amplicons were sequenced directly using Big Dye Termination chemistry (V1.1) on an ABI Prism 3100 sequencer (Applied Biosystems, Foster City, CA). Before the sequencing reaction was performed, PCR purification was done using Geneclean Spin Kit (Qbiogene) following the manufacture's instruction. PCR amplicon using primers corresponding cDNA nt 730–1262 (Table 1), turned-up to be shorter then expected, and sequencing analysis revealed that exon 5 was missing. Amplicons covering the rest of the SMARCAL1 coding sequence were also sequenced and no other change was found. Genomic DNA was extracted from the patient and his family peripheral blood using Puregene DNA Purification kit (Gentra). SMARCAL1 exon 5 was amplified using the following primers: F: 5′-CTAGTGCCTCTTGACCATGTC-3′ and R: 5′-GAAGAGGAGCAGAACAGGAAG-3′. PCR amplicons were purified and sequenced as described above. Sequencing both strands from the patient's amplicon revealed the presence of a splicing mutation at intron 4 acceptor site, IVS4-2 A>G. Segregation of this mutation in family members was investigated by sequencing the respective PCR product.
RNA isolation.
Total RNA from human and mouse samples was isolated from each sample using TRIZOL (Life Technologies, Invitrogen, Carlsbad, CA). Total RNA from rat metanephric mesenchymes (MM) was extracted using RNeasy mini kit (Qiagen, Valencia, CA) with on-column DNase digestion according to the instructions of the manufacturer. An Agilent Bioanalyzer was used to confirm RNA integrity.
Quantitative reverse transcription-PCR.
cDNA was synthesized using SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen) on total RNA. Real-time PCR of human and mouse samples was done using an ABI7900HT sequence detection system (Perkin-Elmer/Applied Biosystems, Foster City, CA) in the presence of SYBR green (SYBR green PCR kit; Qiagen, Hilden, Germany). This fluorochrome incorporates stoichiometrically into the amplification product, providing real-time quantification of double-stranded DNA PCR product. Primers were designed to amplify an 80–120 bp fragment with 50–65°C annealing temperature. Primers sequences are described in Table 2.
Analysis of SMARCAL1 IVS4-2 A>G in the Ashkenazi population.
Seven hundred sixty DNA samples from anonymous subjects of Ashkenazi descents were available for molecular diagnostic at the metabolic disease unit, Sheba Medical Center, Tel Hashomer. SMARCAL1 mutation was genotyped using a chip-based matrix-assisted laser desorption-time-of-flight (MALDI-TOF) mass spectrometer (Sequenom, San Diego, CA). PCR and extension reactions were designed using MassARRAY software (Sequenom). We designed the assay as Multiplex including screening for specific mutations in Schimke syndrome, Galactosemia, and Argininemia. PCR amplifications were carried out in standard 384-well plates, a 5 μL final volume containing 10 ng of template DNA, 0.15 U of Taq polymerase (HotStarTaq, Qiagen, Valencia, CA), 0.5 mM of each dNTP, 100 nmol of each primer, 1.625 mM MgCl2, and 1.25 × HotStar buffer. The following primers were used: Forward: 5′ ACGTTGGATGCCCCTGCTTTATCACTTCTG.
Reverse: 5′ ACGTTGGATGATTCCAGAGGCTGCAGGTTG.
The PCR thermal cycling was carried out in an ABI-9700 instrument for 15 min at 95°C, followed by 4 cycles of 20 s at 95°C, 30 s at 65°C, and 60 s at 72°C; 4 cycles of 20 s at 95°C, 30 s at 58°C, and 60 s at 72°C; and 38 cycles of 20 s at 95°C, 30 s at 53°C and 60 s at 72°C. This was followed by incubation with 0.3 U Shrimp alkaline phosphates in total volume of 7 μL for 20 min at 37°C and 5 min at 85°C. The MassEXTEND (Sequenom, San Diego, CA) assay was conducted in 9 μL final volume containing 1 μM extension primer (5′ CGATTCTTCTTCTTTGGC), 0.2 μL of termination mix (50 nM each of ddA, dG, ddT, and ddC), and 1.25 U ThermoSequenase (Sequenom, San Diego, CA) in 0.22 × PCR buffer. The cycling conditions were incubation for 2 min at 94°C followed by 99 cycles of 5 s at 94°C, 5 s at 52°C, and 5 s at 72°C. After this step, 3 μg MassEXTEND cleanup resin (Sequenom, San Diego, CA) and 25 μL DDW were added to remove extraneous salts.
Samsung nanodispenser was used to apply 15 nL of extension products from each well of the sample plate onto the SpectroChip. Mass spectra were recorded on a Bruker Biflex MALDI-TOF mass spectrometer operated in the linear mode, and were analyzed by MassARRAY Typer software (Sequenom, San Diego, CA).
The IVS4-2 A>G positive sample, as determined using the SEQUENOM MassArray, was confirmed as positive by direct sequencing.
Metanephric organ culture.
MM were dissected using minutien pins from E13.5 rats. Cross-contamination between ureteric buds and MM was ruled out by visual inspection and staining with dolichos bifloris lectin, which selectively labels the ureteric bud at this stage (10). For organ culture, rat MM were placed on filters (Corning Transwell, Corning, NY; collagen coated, 0.4-μm pore size) and grown in DMEM/F12 with insulin (5 μg/mL), transferrin (5 μg/mL), selenium (5 ng/mL), dexamethasone (5 μg/mL), prostaglandin (5 μg/mL), T3 (5 ng/mL; Sigma Chemical Co.), fibroblast growth factor-2 (3 nmol/L), transforming growth factor- α (3 nmol/L), and leukemia inhibitory factor (LIF) (50 ng/mL; all cytokines from R&D Systems, Minneapolis, MN).
Immunostaining for SMARCAL1.
Sections, 4-μm thick, were cut from whole blocks of fetal kidney, Wilms' tumor and adult kidney for immunohistochemistry and processed within 1 wk to avoid oxidation of antigens. Before immunostaining, sections were treated with 10 mM citrate buffer, pH 6.0 for 10 min at 97°C in a microwave oven for antigen retrieval, followed by treatment of 3% H2O2 for 10 min. The slides were subsequently stained by the labeled—(strept) avidin-biotin (LAB-SA) method using a histostain plus kit (Zymed, San Francisco, CA). Antihuman Smarcal1 polyclonal antibody (LifeSpan Biosciences, Inc. Seattle, WA), at a dilution of 1:50, was used. Controls were prepared by omitting the primary antibodies or by substituting the primary antibodies with goat IgG isotype. The immunoreaction was visualized by an HRP-based chromogen/substrate system, including DAB (brown) chromogen (liquid DAB substrate kit—Zymed).
RESULTS
SMARCAL1 IVS4-2 A>G splice mutation.
DNA analysis in our patient III.1 revealed a homozygous splicing mutation at intron 4 acceptor site (IVS4-2 A>G), causing an entire exon 5 skipping (Fig. 2A). Exon 5 skipping results in frameshift leading to truncated protein product in this patient. This mutation has been previously reported to cause severe disease (5). Segregation analysis was carried out by sequencing the respective PCR product from all family members and revealed the carrier status of both the parents (II.1, II.2) and sister (III.4) (Fig. 2B). Surprisingly the nonidentical twin brothers (III.2, III.3) were also found homozygous for the mutation without clinically apparent phenotype (Fig. 2B).

(A) The IVS 4-2 A>G mutation. This splice mutation results in exon 5 skipping as demonstrated in the electrophoresis gel of normal and mutated cDNA and in the upper diagram. SMARCAL1 sequence chromatograph of the intron 4/exon 5 junction of the index patient confirmed homozygocity for the splice mutation. (B) Family pedigree, both parents are of Ashkenazi origin. The index case III.1 is marked in black. Twin brothers found homozygous for the mutation with no clinical signs of the disease are marked in gray. Half full symbols indicate carrier status.
Reverse quantitative PCR of SMARCAL1 in peripheral blood.
Having identified that both diseased and asymptomatic brothers are homozygous for the splice mutation we wanted to determine mRNA levels of normal SMARCAL1. To measure SMARCAL1 mRNA levels in the peripheral blood of all family members we designed DNA primer sets on exon 5, which is deleted because of the splice mutation. In this case, only a full-length RNA transcript (functional) can give a detectable reverse quantitative (RQ)-PCR reaction. As shown in Figure 3, blood SMARCAL1 transcript levels were similar in heterozygous carriers. Levels in asymptomatic brothers were higher than that observed in the index patient and were very similar to the heterozygous state. Thus, in the presence of identical bi-allelic splice mutation transcript levels of functional SMARCAL1 were different.

Quantitative reverse transcription-PCR (qRT-PCR) analysis of SMARCAL1 mRNA in peripheral blood samples of index patient (III.1) and his family. Normalization was performed against control β-actin expression and RQ calculated relative to index patient (III.1). Normalization was performed against control β-actin expression and RQ calculated relative to adult mouse kidney.
Carrier frequency of IVS 4-2 A>G in the Ashkenazi population.
Taking into account both that the Ashkenazi family we analyzed was nonconsanguineous and the surprising finding of bi-allelic mutation in the unaffected siblings, we were interested in finding the carrier frequency of this specific mutation in the Ashkenazi population. For this purpose, we used the advanced sequenom method (see Materials and Methods section) and screened 760 DNA samples from anonymous subjects of Ashkenazi descents that were available for molecular diagnostic at the metabolic disease unit, Sheba Medical Center, Tel Hashomer. Only one carrier of the IVS4-2 A>G was detected indicating low carrier frequency in the Ashkenazi population and possible founder effect in this nonconsanguineous family.
SMARCAL1 gene expression in normal nephrogenesis.
Review of our previously published human kidney microarray database (9) for SMARCAL1 gene expression showed no significant difference in the hybridization signal between pooled human fetal and adult kidneys. Nevertheless, the microarray data does not include temporal expression. For this purpose, we initially analyzed developmental expression during mouse nephrogenesis, which continues 2 wk postnatal. mRNA levels of smarcal1 were determined by real-time PCR for the mouse fetal kidneys at embryonic days 13 (E13), 15 (E15), 17 (E17), 1 d, 2 wk, and in the adult kidney. Temporal expression pattern of smarcal1 showed peaks of expression in early nephrogenesis (E13–E15) as well as in neonatal (2 wk) and adult kidney, and down-regulation in between (E17—1 d) (Fig. 4). We next analyzed the expression of the smarcal1 in the organ culture model of epithelial differentiation of the isolated rat MM, which represents the early renal progenitor pool (10). For this purpose, freshly dissected whole rat MM from E13.5 embryos were grown in culture media supplemented with fibroblast growth factor-2, transforming growth factor-α, and LIF, which induce epithelial differentiation over a 7-day time course (11). As previously shown (9), Figures 5A–C illustrate this process by demonstrating differentiating mesenchymes after 2 d (A) and differentiated ones at 7 d (B). Epithelial differentiation at this time point is shown by tubular epithelia expressing E-cadherin whereas mesenchymal and proximal nephron cells are positive for Wilms' tumor transcription factor (WT1) (C). This process was followed by quantitative real-time PCR of smarcal1 (Fig. 5D). As shown, smarcal1 was expressed in the early progenitor compartment of the embryonic kidney (uninduced MM), initially down-regulated (LIF induction 24 h), only to return to baseline levels (LIF induction 48 h). Smarcal1 transcript levels were elevated after maturation and epithelization (LIF induction 7 d) (Fig. 5D). Thus, temporal analysis of smarcal1 transcript levels in both model systems showed similar results of early and late peaks of expression.

Quantitative reverse transcription-PCR (qRT-PCR) analysis of SMARCAL1 mRNA levels in the developing mouse kidney. Shown are representative examples of three independent experiments. Total RNA was isolated from pooled mouse embryonic (E14, E15, E17), newborn (1 d), neonatal (2 wk), and adult kidneys (n = 3–5 kidneys in each time point).

Regulation of SMARCAL1 during differentiation of metanephric mesenchyme. Freshly dissected rat metanephric mesenchymes were differentiated in culture medium supplemented with FGF-2, TGF-a, and LIF. Bright-field images from differentiating mesenchymes after 2 d (A) and 7 d (B) in culture. (D) At 7 d, immunofluorescence for E-cadherin (Cy2; green) reveals development of tubular epithelia, whereas mesenchymal and proximal nephron cells are positive for WT1 (Cy3; red) (9). (D), real-time reverse transcription-PCR (RT-PCR) for SMARCAL1 was performed on metanephric mesenchymes at baseline (0), and after 1, 2, and 7 d of culture. SMARCAL1 mRNA is initially down-regulated and then up-regulated with differentiation. Dunnett's multiple comparison test reveals significant differences (p < 0.05) of LIF24 and LIF7d compared with time point 0.
In situ localization of SMARCAL1.
Having established the temporal expression pattern for SMARCAL1 during nephrogenesis, we performed immunohistochemical analysis to determine what cell types in the kidney express SMARCAL1 (Fig. 6). Human fetal, adult kidneys, and Wilms tumor, the embryonic kidney malignancy originating from progenitor renal mesenchymal/blastemal cells that are arrested in differentiation (9), were all analyzed. We used human testis known to express the highest levels of SMARCAL1 as positive control (Figs. 6A and B).

Immunostaining of SMARCAL1 in human fetal kidney (FK), Wilms' tumor (WT), and adult kidney (AK). (A, B) Positive control staining in human testis, (C, D) human FK in low (C) and high (D) magnification shows expression in some uninduced mesenchymal cells of the nephrogenic cortex and in developing tubules toward the center; (E, F) WT demonstrates expression in malignant blastemal cells (E) and in tumorigenic rhabdomyoblasts (F); (G, H) human AK, SMARCAL1 is localized to distal tubules and collecting ducts (G) and glumerular podocytes (H). Original magnification: (A, C, E, F) × 10, Scale bar = 100 μm; (B, G) × 20, Scale bar = 50 μm; (D, H) × 40, Scale bar = 25 μm.
Immunostaining of mid-gestation human fetal kidney (19 wk) demonstrated expression of SMARCAL1 in metanephric mesenchymal cells localized in the outer part of the nephrogenic cortex and in early tubules observed toward the center (Figs. 6C and D). Fetal glomeruli were negative. In WT, we found expression in the malignant renal blastema (Fig. 6E) as well as in nonrenal elements such as early rhabdomyoblasts (Fig. 6F). Finally, in the adult kidney SMARCAL1 was localized to tubular cells, predominantly in distal tubular cells and collecting ducts (Fig. 6G), and glomerular cells including podocytes and parietal epithelia (Fig. 6H). Thus, expression shifts from one renal cell type to another with maturation.
DISCUSSION
Our study demonstrates phenotypic diversity in nonconsanguineous Ashkenazi family harboring SMARCAL1 IVS 4-2 A>G splice mutation. This mutation has been previously suggested to cause severe SIOD (5). However, until now, the index patient had no deterioration in renal function despite having overt nephrotic syndrome secondary to FSGS lasting almost 3 y already and no infectious or cerebrovascular symptoms. This, despite the lymphopenia, extremely reduced CD4 cells, inverted CD4/CD8 ratio, and low immunoglobulin production. Furthermore, his twin brothers' molecular diagnosis, rather than clinical findings, led the diagnosis of presumptive SIOD. The latter, which are now 5 y old, are minimally affected and do not conform to SIOD criteria. At this age, the index patient had already developed overt nephropathy. Taken together, these data suggest that the IVS 4-2 A>G splice mutation can cause a milder form of SIOD. In addition, it contradicts the previously reported constancy of presentation age and course of SIOD within a given pedigree (4). The carrier status of the specific mutation in the Ashkenazi population was estimated to be approximately 1:760. This finding correlates with an estimated prevalence of SIOD in North America, which is 1–3 × 106 live births.
How can one explain the intrafamilial phenotypic diversity? Boerkoel and coworkers have hypothesized that the variable expressivity among patients with biallelic SMARCAL1 mutations arises from environmental, genetic, or epigenetic modifiers (12). Splicing mutations can lead to a mixture of aberrantly and correctly spliced transcripts, by partial skipping of exons or inclusion of intronic sequences, or can change the ratio of programmed alternatively spliced isoforms (13). Disease severity correlates with the level of correctly spliced RNA transcribed from genes carrying splicing mutations with the ratio of alternatively spliced isoforms. Thus, in the presence of a splice mutation, splicing regulation has been suggested as a genetic modifier of disease severity (13). Accordingly, it has been recently shown in cells and animal models that over expression of various splicing factors modulates the splicing pattern of RNA that was transcribed from introduced minigenes carrying splicing mutations, affecting both the protein function and disease phenotype (13,14). We provide a possible example of such phenomenon; in the presence of a biallelic SMARCAL1 splice mutation, we observed intrafamilial variability in the functional SMARCAL1 transcript levels among carriers and homozygotes, correlating with disease severity. Functional SMARCAL1 transcript levels are likely to be determined by individual splicing factors. Thus, we hypothesize the splicing machinery as a possible modifier of the observed intrafamilial SIOD phenotypic diversity.
Although SMARCAL1 has been shown to be expressed in the fetal kidney, we provide in situ localization in human specimens as well as temporal expression during murine nephrogenesis and in the metanephric organ culture. Our findings suggest a role for SMARCAL1 in the early renal progenitor population and in the neonatal/adult kidney. Peak levels of SMARCAL1 in the neonatal/adult murine kidney and localization in glomerular podocytes can explain the renal phenotype in SIOD of proteinuria and progressive nephropathy that appears in childhood. Expression of SMARCAL1 in distal tubular cells and collecting ducts calls for investigation of tubular function in these patients. Although our finding of early expression peak of SMARCAL1, as well as localization in metanephric mesenchymal cells and in Wilms' tumor (early defect of nephrogenesis), correlates with the presence of renal malformation (crossed fused ectopic kidney) in the index patient, this phenotype has not been previously reported in SIOD and may represent an incidental finding. Indeed, the population incidence of renal ectopia, encompassing crossed fused ectopia, is approximately 1 in 10,000 patients, and in different autopsy series it is detected in 1 in 1,000 to 1 in 7,000, which is higher than SIOD (15). Nevertheless, we suggest this finding, which might have been overlooked in the past as part of the renal phenotype of SIOD. Others and we have shown by global gene profiling that the embryonic kidney is committed to cellular proliferation and growth early on (9,16) and only later on to terminal differentiation. Thus, the function of SMARCAL1 might be more redundant and dispensable in the early embryonic period and only later acquire a specific function, presumably in the podocyte. This would, in turn, influence the renal phenotype observed in SIOD.
In conclusion, we have identified a splice mutation in the SMARCAL1 gene associated with a diverse phenotype of SIOD that correlates with residual SMARCAL1 transcript levels. This may support the concept of the splicing machinery as a genetic modifier of disease severity.
It will be important to determine possible renal malformations and tubular function in SIOD to gain insight into the function of SMARCAL1 in early and late stages of kidney formation.
Abbreviations
- E:
-
embryonic days
- FSGS:
-
focal segmental glomerulosclerosis
- LIF:
-
leukemia inhibitory factor
- SIOD:
-
Schimke immuno-osseous dysplasia
- SMARCAL1:
-
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a-like 1
- WT:
-
Wilms' tumor
References
Boerkoel CF, O'Neill S, Andre JL, Benke PJ, Bogdanovic R, Bulla M, Burguet A, Cockfield S, Cordeiro I, Ehrich JH, Frund S, Geary DF, Ieshima A, Illies F, Joseph MW, Kaitila I, Lama G, Leheup B, Ludman MD, McLeod DR, Medeira A, Milford DV, Ormala T, Rener-Primec Z, Santava A, Santos HG, Schmidt B, Smith GC, Spranger J, Zupancic N, Weksberg R 2000 Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literature. Eur J Pediatr 159: 1–7
Spranger J, Hinkel GK, Stoss H, Thoenes W, Wargowski D, Zepp F 1991 Schimke immuno-osseous dysplasia: a newly recognized multisystem disease. J Pediatr 119: 64–72
Schimke RN, Horton WA, King CR 1971 Chondroitin-6-sulfaturia, defective cellular immunity, and nephrotic syndrome. Lancet 2: 1088–1089
Saraiva JM, Dinis A, Resende C, Faria E, Gomes C, Correia AJ, Gil J, da Fonseca N 1999 Schimke immuno-osseous dysplasia: case report and review of 25 patients. J Med Genet 36: 786–789
Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, Andre JL, Bogdanovic R, Burguet A, Cockfield S, Cordeiro I, Frund S, Illies F, Joseph M, Kaitila I, Lama G, Loirat C, McLeod DR, Milford DV, Petty EM, Rodrigo F, Saraiva JM, Schmidt B, Smith GC, Spranger J, Stein A, Thiele H, Tizard J, Weksberg R, Lupski JR, Stockton DW 2002 Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 30: 215–220
Havas K, Whitehouse I, Owen-Hughes T 2001 ATP-dependent chromatin remodeling activities. Cell Mol Life Sci 58: 673–682
Pazin MJ, Kadonaga JT 1997 SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions?. Cell 88: 737–740
Clewing JM, Antalfy BC, Lucke T, Najafian B, Marwedel KM, Hori A, Powel RM, Safo Do AF, Najera L, SantaCruz K, Hicks MJ, Armstrong DL, Boerkoel CF 2007 Schimke immuno-osseous dysplasia: a clinicopathological correlation. J Med Genet 44: 122–130
Dekel B, Metsuyanim S, Schmidt-Ott KM, Fridman E, Jacob-Hirsch J, Simon A, Pinthus J, Mor Y, Barasch J, Amariglio N, Reisner Y, Kaminski N, Rechavi G 2006 Multiple imprinted and stemness genes provide a link between normal and tumor progenitor cells of the developing human kidney. Cancer Res 66: 6040–6049
Schmidt-Ott KM, Yang J, Chen X, Wang H, Paragas N, Mori K, Li JY, Lu B, Costantini F, Schiffer M, Bottinger E, Barasch J 2005 Novel regulators of kidney development from the tips of the ureteric bud. J Am Soc Nephrol 16: 1993–2002
Barasch J, Yang J, Ware CB, Taga T, Yoshida K, Erdjument-Bromage H, Tempst P, Parravicini E, Malach S, Aranoff T, Oliver JA 1999 Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell 99: 377–386
Clewing JM, Fryssira H, Goodman D, Smithson SF, Sloan EA, Lou S, Huang Y, Choi K, Lucke T, Alpay H, Andre JL, Asakura Y, Biebuyck-Gouge N, Bogdanovic R, Bonneau D, Cancrini C, Cochat P, Cockfield S, Collard L, Cordeiro I, Cormier-Daire V, Cransberg K, Cutka K, Deschenes G, Ehrich JH, Frund S, Georgaki H, Guillen-Navarro E, Hinkelmann B, Kanariou M, Kasap B, Kilic SS, Lama G, Lamfers P, Loirat C, Majore S, Milford D, Morin D, Ozdemir N, Pontz BF, Proesmans W, Psoni S, Reichenbach H, Reif S, Rusu C, Saraiva JM, Sakallioglu O, Schmidt B, Shoemaker L, Sigaudy S, Smith G, Sotsiou F, Stajic N, Stein A, Stray-Pedersen A, Taha D, Taque S, Tizard J, Tsimaratos M, Wong NA, Boerkoel CF 2007 Schimke immunoosseous dysplasia: suggestions of genetic diversity. Hum Mutat 28: 273–283
Nissim-Rafinia M, Kerem B 2005 The splicing machinery is a genetic modifier of disease severity. Trends Genet 21: 480–483
Nissim-Rafinia M, Aviram M, Randell SH, Shushi L, Ozeri E, Chiba-Falek O, Eidelman O, Pollard HB, Yankaskas JR, Kerem B 2004 Restoration of the cystic fibrosis transmembrane conductance regulator function by splicing modulation. EMBO Rep 5: 1071–1077
Ignjatovic I, Stojkovic I, Dinic LJ, Jovanovic M, Ivanovic D 2007 Bilateral fused pelvic kidney ectopia with a single draining ureter solved by craniolateral displacement of the kidney and Boari-modified bladder-calyceal anastomosis with bladder augmentation. Int J Urol 14: 552–554
Stuart RO, Bush KT, Nigam SK 2001 Changes in global gene expression patterns during development and maturation of the rat kidney. Proc Natl Acad Sci USA 98: 5649–5654
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was partially supported by grants from the Israel Scientific Foundation Physician-Scientist Grant Award, Sheba Career Development Award and Moriss Kahn Career Development Award (BD).
Rights and permissions
About this article
Cite this article
Dekel, B., Metsuyanim, S., Goldstein, N. et al. Schimke Immuno-Osseous Dysplasia: Expression of SMARCAL1 in Blood and Kidney Provides Novel Insight Into Disease Phenotype. Pediatr Res 63, 398–403 (2008). https://doi.org/10.1203/PDR.0b013e31816721cc
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31816721cc
This article is cited by
-
Inborn errors of immunity with kidney and urinary tract disorders: a review
International Urology and Nephrology (2024)
-
Incomplete penetrance in primary immunodeficiency: a skeleton in the closet
Human Genetics (2020)
-
Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease
Human Genetics (2013)
