
Abstract
Although neurofibromatosis type 1 (NF1) is a neurocutaneous disorder, skeletal abnormalities such as long-bone dysplasia, scoliosis, sphenoid wing dysplasia, and osteopenia are observed. To investigate the role of bone resorption as a mechanism for the bony abnormalities, we selected urinary pyridinium crosslinks (collagen degradation products excreted in urine) as a measure of bone resorption in NF1. Bone resorption was evaluated by quantitative assessment of the urinary excretion of pyridinium crosslinks [pyridinoline (Pyd) and deoxypyridinoline (Dpd)]. Total (free plus peptide-bound) pyridinium crosslinks from the first morning urines from 59 NF1 children (ages 5–19) were extracted and analyzed (17 children with a localized skeletal dysplasia, and 42 without). The data were compared with a healthy reference population without NF1 (n = 99). Multivariate analyses, controlling for age showed statistically significant increases for Dpd (p < 0.001) and the Dpd/Pyd ratio (p < 0.001) in NF1 individuals with and without a skeletal dysplasia. NF1 children have an increase in the urinary excretion of pyridinium crosslinks, reflecting increased bone resorption. The effects of NF1 haploinsufficiency likely contribute to abnormal bone remodeling, either directly or indirectly by aberrant Ras signaling, potentially predisposing NF1 individuals to localized skeletal defects.
Similar content being viewed by others

Main
Neurofibromatosis type 1 (NF1), a common autosomal dominant disorder affecting ≈1/3500 individuals worldwide, has variable expressivity. Clinical manifestations include café-au-lait macules, intertriginous freckling, Lisch nodules, neurofibromas, optic pathway tumors, and distinctive osseous lesions (1–3). The prototypical skeletal manifestations of NF1 are proportionate short stature, macrocephaly, long-bone dysplasia, progressive scoliosis, and sphenoid wing dysplasia. Long-bone dysplasia most often affects the tibia and presents with anterolateral bowing often leading to fracture and nonunion or pseudarthrosis (4,5). The long-bone dysplasia in NF1 is very distinctive, and the presence of tibial pseudarthrosis alone should raise the potential diagnosis of NF1, as 50–80% of individuals with pseudarthrosis have NF1 (6–8). Scoliosis is the most common orthopedic manifestation in NF1 with reports documenting between 10 and 33% of NF1 individuals having scoliosis (9). Other osseous manifestations of NF1 include bone cysts, spinal canal widening, vertebral body narrowing, rib-penciling, vertebral scalloping with dural ectasias, and decreased bone mineral density. In isolation, each skeletal abnormality associated with NF1 is rare, but, as a whole, the osseous defects are relatively frequent.
The NF1 gene, located on the long arm of chromosome 17, encodes the protein neurofibromin, which is a Ras-GAP protein (10). The “tumor suppressor” properties of this protein do not easily explain the mesodermally derived osseous manifestations observed in NF1. The frequent association of osseous dysplasias seen in NF1 suggests, however, that the neurofibromin-ras signal transduction pathway plays a role in the cellular processes of bone in NF1.
Several studies have reported decreased bone mineral density in NF1 individuals (11–16), thus pointing to an underlying disorder of skeletal homeostasis and mineralization in humans with NF1. One of these studies reported that all postmenopausal NF1 women in the cohort had either osteoporosis or osteopenia (13). Peripheral quantitative computed tomography modalities have also demonstrated that NF1 individuals display a different muscle mass, which may impact bone architecture (17). These generalized findings suggest that haploinsufficiency of the NF1 gene contributes to abnormal regulation of bone remodeling.
Bone is a dynamic organ system with a delicate balance of turnover and regeneration. The strength of bone is due in part to the crosslinking of collagen molecules. Inferences to the dynamics of bone can be obtained through the analysis of markers of bone resorption and formation. Many markers are readily available and have shown good correlation between predicted and measured bone mass (18,19). Pyridinoline (Pyd) and deoxypyridinoline (Dpd), which are hydroxylysine-derived crosslinks of mature collagen, are excreted in urine, both in free and peptide-bound forms, as a result of collagen degradation (20). They have been shown to be sensitive markers of bone resorption and correlate well with other established markers of bone resorption (21–23). Large increases have been shown in patients with high bone turnover states including patients with osteoporosis and osteoporotic fractures, femur fractures, Paget disease, and hyperparathyroidism (20–23). A specific abnormal pattern of urinary excretion of pyridinium crosslinks has also been observed in Ehlers-Danlos syndrome type VI, an inherited disorder of collagen metabolism with kyphoscoliotic changes (24,25). Little is known about the pathogenesis of the osseous abnormalities in NF1; the constellation of decreased bone mineralization and poor fracture healing suggest a disruption in normal bone remodeling. Therefore, we selected urinary pyridinium crosslink excretion analysis to indirectly assess bone resorption in individuals with NF1.
MATERIALS AND METHODS
Subjects.
Physical examinations and medical histories were obtained on a cohort of 59 individuals (ages 5–19 y) who fulfilled the diagnostic criteria for NF1 (1,2). NF1 individuals were recruited from an NF1 clinic at the University of Utah. NF1 individuals with conditions and circumstances known to influence bone health (e.g., recent fracture, illnesses requiring systemic steroids, anorexia, pregnancy, lactation, oral contraception, or hormone replacement therapy) were not included. Pyridinium crosslink data from a local regional cohort of healthy children (n = 99) without NF1 or known orthopedic conditions were used for comparison (ages 1–17 y).
NF1 individuals were classified into two groups: NF1 individuals with and without a skeletal dysplasia. A skeletal dysplasia in this study population was defined as the presence of long-bone dysplasia, scoliosis, and/or sphenoid wing dysplasia based on physical examination and review of medical records. All NF1 individuals were examined at the University of Utah General Clinical Research Center. Long-bone dysplasia was defined as anterolateral bowing of the tibia, fibula, radius, and/or ulna.
Institutional Review Board approval at the University of Utah was obtained. Written informed consent was obtained from the parents or guardians of the children who served as subjects and, when appropriate, assent or consent from the participants themselves.
Measurements.
Urine from two first morning voids was obtained from all individuals for the extraction and analysis of the total free and the peptide-bound Pyd and Dpd by high performance liquid chromatography (HPLC). To release the peptide-bound crosslinks, the urine samples were hydrolyzed in 6 M HCl, under vacuum, for 16 h at 150°C. Pyridinium crosslinks were then extracted from the hydrolysate by fractionation through a cellulose column, according to established procedures (26). The eluates containing Pyd and Dpd were lyophilized, reconstituted in 1% heptafluorobutyric acid, and analyzed by reverse-phase HPLC, using a Waters 2695 Alliance HPLC System, equipped with a fluorescence detector (Waters 2475; emission = 297 nm, excitation = 395 nm) and controlled by a computerized unit (Empower Software). The reverse phase column used for the separation of pyridinium crosslinks was a Waters Nova-Pak C18 column (4 μm; 15 cm × 3.9 mm). Eluant A was 0.18% n-heptafluorobutyric acid in 12% acetonitrile. Eluant B was 0.18% heptafluorobutyric acid in 100% acetonitrile. The column was equilibrated in 95% A and 5% B for 3 h before injection of the samples. The samples were eluted for 15 min with the same isocratic solvent composition at a flow rate of 0.7 mL/min. Pyridinium crosslink concentration was calculated using a 4-levels calibration curve obtained with an external standard. Standards for Pyd and Dpd were purchased from Quidel Corporation, San Diego, CA. Urinary pyridinium crosslink concentration was normalized to urinary creatinine, measured by a Beckman Creatinine Analyzer 2 and calculated as umol/mol creatinine. Two first-morning urine samples were obtained to adjust for potential variations in a single sample.
Data analysis.
Data for Pyd, Dpd, and Dpd/Pyd ratios, and age from the NF1 individuals with and without a skeletal dysplasia were compared with each other and with the control group using the unpaired t test for data with normal distributions, and with Mann-Whitney's U for data with non-normal distributions. Multivariate linear regression modeling was then used to model the comparison of the two NF1 groups and the controls on the dependent variables Pyd, Dpd, and Dpd/Pyd with control for the confounding effect of age. The resulting multivariate linear regression models were then used to generate least square means for all three groups. Additionally, full regression models (three groups and age) were compared with reduced models (age) to generate estimates of variance accounted for by the three groups. Statistical analyses were conducted using Stata 9.2 (College Station, TX).
RESULTS
Average age of individuals with NF1 was 10.6 y (median, 10.5 y; range, 5–19 y). Seventeen of 59 individuals with NF1 had a skeletal dysplasia (1 with sphenoid wing dysplasia; 1 with long-bone dysplasia; 12 with scoliosis; and 3 with scoliosis and long-bone dysplasia). The remaining 42 NF1 individuals did not have a history of long-bone dysplasia, scoliosis, or sphenoid wing dysplasia. Average age of controls was 9.1 y (median, 9 y; range, 1–17 y).
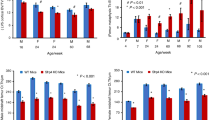
Multivariate linear regression analysis adjusting for age showed statistically significant differences (p < 0.001) in Dpd and the Dpd/Pyd ratio between groups (n = 17 for individuals with NF1 and a skeletal dysplasia; n = 42 for individuals with NF1 and no skeletal dysplasia; n = 99 for controls without NF1). Least square means, reported in Table 1, showed moderate increases of Pyd in NF1 individuals with and without a skeletal dysplasia compared with controls, but did not reach statistical significance when comparing all groups. Similar analysis of Dpd showed a more dramatic increase in NF1 individuals with and without a skeletal dysplasia compared with controls and an increase of Dpd in NF1 individuals with a skeletal dysplasia compared with NF1 individuals without a skeletal dysplasia (Table 1). The Dpd/Pyd ratio was increased by approximately 19% in individuals with NF1 without a skeletal dysplasia compared with the control group, and approximately 38% in individuals with NF1 with a skeletal dysplasia compared with the control group. These elevations in the Dpd/Pyd ratio were statistically significant (p < 0.001) and consistent with increased bone resorption in the NF1 patients (Table 1).
The comparisons of R2 values between the reduced and full multivariate model are shown in Table 2. The differences seen in the models for the dependent variables, Dpd and Dpd/Pyd ratio, show substantial increases in R2 values, indicating that the variable representing the grouping of NF1 individuals and the controls accounts for a significant portion of the variation present in the dependent variables.
DISCUSSION
Children and adolescents with NF1 had statistically significant increases in Dpd reflecting increased collagen degradation in children with NF1. In addition, the Dpd/Pyd ratio was elevated in the NF1 individuals consistent with increased bone turnover. Interestingly, individuals with Ehlers-Danlos syndrome type VI, who have normal amounts of pyridinium crosslinks but have a significantly increased Dpd/Pyd ratio (24), are in part differentiated from other types of Ehlers-Danlos syndrome based on the presence of skeletal abnormalities characterized by a progressive kyphoscoliosis usually present in the first year of life, osteoporosis, and clubfeet (27). Pyd is found in many tissues including both bone and cartilage, but Dpd is most abundant in bone and dentine and reflects more the status of bone (24,25,28,29). The observation that the Dpd/Pyd ratio is elevated in NF1 children and adolescents indicates a preferential increase in bone resorption rather than a generalized collagen breakdown.
Although decreased bone mineral density has been reported in individuals with NF1 (11–16), we are not aware that they display a substantially increased number of fractures compared with the general population, although long-bone fractures with poor bone healing is observed in patients with localized long-bone dysplasia. Potentially, decreased bone mineral density could predispose individuals with NF1 to clinically undetected microfractures. Microfractures are resorbed by osteoclasts and can alter trabecular architecture (30–32). The observed increase in the excretion of urinary pyridinium crosslinks in our cohort of children and adolescents with NF1 may be the result of microfractures leading to an abnormal microarchitecture.
There is evidence that somatic loss of the nonmutant NF1 allele in NF1 heterozygous individuals leads to localized skeletal abnormalities as double inactivation of NF1 was observed in pseudarthrosis tissue from patients with NF1 with tibial bowing and fracture with nonunion (33). In this cohort, 29% of individuals with NF1 had scoliosis, long-bone dysplasia, and/or sphenoid wing dysplasia. Although active fractures were not present, the contribution of scoliosis, long-bone dysplasia, and/or sphenoid wing dysplasia on the excretion of pyridinium crosslinks is unknown. Therefore, in this study NF1 individuals were divided into those with and without scoliosis, long-bone dysplasia, and/or sphenoid wing dysplasia. The NF1 individuals without a skeletal dysplasia still had increased Dpd and Dpd/Pyd ratios compared with controls, suggesting that NF1 haploinsufficiency contributes to increased bone resorption and represents a generalized abnormality of bone remodeling in the background of NF1. However, the precise role neurofibromin plays in the growth and development of bone in individuals with NF1 is not well understood.
Given the paucity of studies on bone remodeling in the NF1 human model, insights into the role of neurofibromin in osteoclast and osteoblast functioning must be taken from the murine model. It is known that neurofibromin directly impacts the Ras-signaling pathway, which interacts with multiple signaling pathways, several of which are important in bone. For example, transforming growth factor-beta increases neurofibromin mRNA (34), murine Nf1± mast cells secrete elevated concentrations of transforming growth factor-beta (35), fibroblast growth factors activate the Ras/mitogen-activated phosphorylation kinase pathway (36), and inactivation of the SHP2-Ras-mitogen-activated phosphorylation kinase pathway in mice results in enhanced bone formation after an increase in osteoclast activity suggesting a dissociation of the intercellular communications between osteoclasts and osteoblasts (37,38). Specific investigations using the Nf1 haploinsufficient transgenic mouse model has shown abnormalities in myeloid cells. Yang et al. (39) showed that monocytes from Nf1± mice have an increased potential to mature into multinucleated osteoclasts, and that the osteoclasts have increased adhesive and lytic properties, which was also observed in a small cohort of two individuals with NF1. This increase in osteoclast formation and lytic activity in the murine model and the two NF1 individuals (39) is in concert with our findings of increase bone resorption in children and adolescents with NF1.
Because of the complexities of bone remodeling, the observed increase in bone resorption in our cohort and the previously described abnormalities in the myeloid lineages do not fully explain the pathophysiology of the skeletal defects of NF1. Additional animal studies have shown that deficiency of neurofibromin also impacts differentiation of mesenchymal progenitor cells. Yu et al. (40) reported that Nf1± committed osteoprogenitors exhibited premature apoptosis and higher proliferation, and Wu et al. (41) showed impaired osteoblast differentiation in Nf1± mesenchymal stem/progenitor cells. The effects of neurofibromin deficiency on skeletal morphogenesis and remodeling likely vary depending on timing of expression, potentially impacted by early somatic inactivation of NF1 in the normal allele in a subset of mesenchymal cell lineages. This is evidenced by the discordant phenotypes observed in various Nf1 transgenic mouse models. For example, mice lacking neurofibromin in osteoblasts (Nf1ob−/−) show increased bone formation without long-bone bowing (42), whereas mice lacking neurofibromin in undifferentiated mesenchymal cells of the developing limb (Nf1Prx1) display tibial bowing with a high degree of porosity (36). The Nf1ob−/− mice also showed increased urinary excretion of pyridinium crosslinks (42), which is consistent with our results in children.
Investigation of the effects of downstream targets of the neurofibromin-Ras signal transduction pathway on osteoprogenitor cells will be important in selecting targeted therapies for the localized skeletal defects of NF1. With the advancement of transgenic mouse models that partially recapitulates the human NF1 skeletal phenotype such as the long-bone bowing in the Nf1Prx1 mouse model described by Kolanczyk et al. (36), human clinical trials with therapeutic agents identified through animal studies are likely to develop. Given that decreased bone mineral density has been reported previously in individuals with NF1, and the herein reported data show evidence of increased bone resoprtion, currently used therapeutic agents within the general population such as bisphosphonates are potential candidates. Schindeler et al. (43) used bone morphogenetic protein and bisphosphonate combination therapy in the mouse model showing an increase of net bone production in vivo in Nf1± mice. Therefore, single agent therapy may not be the most appropriate treatment strategy, and further studies will be necessary to characterize the human NF1 skeletal phenotype more accurately, understand the effects of increased Ras signaling on bone remodeling, and understand the safety efficacy of each candidate agent before proceeding with clinical trials.
Pyridinium crosslinks may prove to be a good surrogate marker for future clinical trials. In addition, the analysis of pyridinium crosslinks may prove useful to identify individuals with NF1 who are at risk for clinical osseous complications. However, prospective studies measuring urinary pyridinium crosslinks before the development of an osseous complication will be needed to assess their clinical utility in individuals with NF1.
Abbreviations
- Dpd:
-
deoxypyridinoline
- HPLC:
-
high performance liquid chromatography
- NF1:
-
neurofibromatosis type 1
- Pyd:
-
pyridinoline
References
1988 Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 45: 575–578.
Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, Rubenstein A, Viskochil D 1997 The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 278: 51–57
Crawford AH, Schorry EK 1999 Neurofibromatosis in children; the role of the orthopaedist. J Am Acad Orthop Surg 7: 217–230
Friedman JM, Birch PH 1997 Type 1 neurofibromatosis: A descriptive analysis of the disorder in 1728 patients. Am J Med Genet 70: 138–143
Stevenson DA, Birch PH, Friedman JM, Viskochil DH, Balestrazzi P, Buske A, Korf BR, Niimura M, Pivnick E, Schorry E, Short P, Tenconi R, Tonsgard J, Carey JC 1999 Descriptive analysis of tibial pseudarthrosis in patients with neurofibromatosis 1. Am J Med Genet 84: 413–419
Gilbert A, Brockman R 1995 Congenital pseudarthrosis of the tibia. Clin Orthop Relat Res 314: 37–44
Morrissy RT, Riseborough EJ, Hall JE 1981 Congenital pseudarthrosis of the tibia. J Bone Joint Surg Br 63: 367–375
Sofield HA 1971 Congenital pseudarthrosis of the tibia. Clin Orthop Relat Res 76: 33–42
Vitale MG, Guha A, Skaggs DL 2002 Orthopaedic manifestations of neurofibromatosis in children: an update. Clin Orthop Relat Res 401: 107–118
Viskochil D 2002 Genetics of neurofibromatosis 1 and the NF1 gene. J Child Neurol 17: 562–570
Illes T, Halmai V, de Jonge T, Dubousset J 2001 Decreased bone mineral density in neurofibromatosis-1 patients with spinal deformities. Osteoporos Int 12: 823–827
Yilmaz K, Ozmen M, Bora Goksan S, Eskiyurt N 2007 Bone mineral density in children with neurofibromatosis 1. Acta Paediatr 96: 1220–1222
Kuorilehto T, Pöyhönen M, Bloigu R, Heikkinen J, Väänänen K, Peltonen J 2005 Decreased bone mineral density and content in neurofibromatosis type 1: lowest local values are located in the load-carrying parts of the body. Osteoporos Int 16: 928–936
Lammert M, Kappler M, Mautner VF, Lammert K, Störkel S, Friedman JM, Atkins D 2005 Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int 16: 1161–1166
Stevenson DA, Moyer-Mileur LJ, Murray M, Slater H, Sheng X, Carey JC, Dube B, Viskochil DH 2007 Bone mineral density in children and adolescents with neurofibromatosis type 1. J Pediatr 150: 83–88
Dulai S, Briody J, Schindeler A, North KN, Cowell CT, Little DG 2007 Decreased bone mineral density in neurofibromatosis type 1: results from a pediatric cohort. J Pediatr Orthop 27: 472–475
Stevenson DA, Moyer-Mileur LJ, Carey JC, Maxwell S, Quick JL, Hoff CJ, Viskochil DH 2005 Case–control study of the muscular compartments and osseous strength in neurofibromatosis type 1 using peripheral quantitative computed tomography. J Musculoskelet Neuronal Interact 5: 145–149
Eastell R, Blumsohn A 1997 The value of biochemical markers of bone turnover in osteoporosis. J Rheumatol 24: 1215–1217
Cranney A, Tugwell P, Cummings S, Sambrook P, Adachi J, Silman AJ, Gillespie WJ, Felson DT, Shea B, Wells G 1997 Osteoporosis clinical trials endpoints: candidate variables and clinimetric properties. J Rheumatol 24: 1222–1229
McLaren AM, Hordon LD, Bird HA, Robins SP 1992 Urinary excretion of pyridinium crosslinks of collagen in patients with osteoporosis and the effects of bone fracture. Ann Rheum Dis 51: 648–651
Rosen HN, Dresner-Pollak R, Moses AC, Rosenblatt M, Zeind AJ, Clemens JD, Greenspan SL 1994 Specificity of urinary excretion of cross-linked N-telopeptides of type I collagen as a marker of bone turnover. Calcif Tissue Int 54: 26–29
Delmas PD, Schlemmer A, Gineyts E, Riis B, Christiansen C 1991 Urinary excretion of pyridinoline crosslinks correlates with bone turnover measured on iliac crest biopsy in patients with vertebral osteoporosis. J Bone Miner Res 6: 639–644
Robins SP, Black D, Paterson CR, Reid DM, Duncan A, Seibel MJ 1991 Evaluation of urinary hydroxypyridinium crosslink measurements as resorption markers in metabolic bone diseases. Eur J Clin Invest 21: 310–315
Pasquali M, Still MJ, Vales T, Rosen RI, Evinger JD, Dembure PP, Longo N, Elsas LJ 1997 Abnormal formation of collagen cross-links in skin fibroblasts cultured from patients with Ehlers-Danlos Syndrome Type VI. Proc Assoc Am Physicians 109: 33–41
Pasquali N, Still MJ, Dembure PP, Elsas LJ 1995 Pyridinium cross-links in heritable disorders of collagen. Am J Hum Genet 57: 1508–1510
Uebelhart D, Gineyts E, Chapuy MC, Delmas PD 1990 Urinary excretion of pyridinium crosslinks: a new marker of bone resporption in metabolic bone disease. Bone Miner 8: 87–96
Wenstrup RJ, Hoechstetter LB 2005 Ehlers-Danlos syndromes. In: Cassidy SB, Allanson JE (eds) Management of Genetic Syndromes. Wiley, New York, pp 211–223
Eyre DR, Koob TJ, Van Ness KP 1984 Quantification of hydroxypyridinium crosslinks in collagen by high-performance liquid chromatography. Anal Biochem 137: 380–388
Bettica P, Moro L, Robins SP, Taylor AK, Talbot J, Singer FR, Baylink DJ 1992 Bone-resorption markers galactosyl hydroxylysine, pyridinium crosslinks, and hydroxyproline compared. Clin Chem 38: 2313–2318
Hernandez CJ, Tang SY, Baumbach BM, Hwu PB, Sakkee AN, van der Ham F, DeGroot J, Bank RA, Keaveny TM 2005 Trabecular microfracture and the influence of pyridinium and non-enzymatic glycation-mediated collagen cross-links. Bone 37: 825–832
Parfitt AM 1984 The cellular basis of bone remodeling: the quantum concept reexamined in light of recent advances in the cell biology of bone. Calcif Tissue Int 36: S37–S45
Mosekilde L 1990 Consequences of the remodelling process for vertebral trabecular bone structure: a scanning electron microscopy study (uncoupling of unloaded structures). Bone Miner 10: 13–35
Stevenson DA, Zhou H, Ashrafi S, Messiaen LM, Carey JC, D'Astous JL, Santora SD, Viskochil DH 2006 Double inactivation of NF1 in tibial pseudarthrosis. Am J Hum Genet 79: 143–148
Yla-Outinen H, Aaltonen V, Bjorkstrand AS, Hirvonen O, Lakkokorpi J, Vaha-Kreula M, Laato M, Peltonen J 1998 Upregulation of tumor suppressor protein neurofibromin in normal human wound healing and in vitro evidence for platelet derived growth factor (PDGF) and transforming growth factor-beta1 (TGF-beta1) elicited increase in neurofibromin mRNA steady-state levels in dermal fibroblast. J Invest Dermatol 110: 232–237
Yang FC, Chen S, Clegg T, Li X, Morgan T, Estwick SA, Yuan J, Khalaf W, Burgin S, Travers J, Parada LF, Ingram DA, Clapp DW 2006 Nf1+/− mast cells induce neurofibroma like phenotypes through secreted TFG-beta signaling. Hum Mol Genet 15: 2421–2437
Kolanczyk M, Kossler N, Kühnisch J, Lavitas L, Stricker S, Wilkening U, Manjubala I, Fratzl P, Sporle R, Herrmann BG, Parada LF, Kornak U, Mundlos S 2007 Multiple roles for neurofibromin in skeletal development and growth. Hum Mol Genet 16: 874–886
Mundy GR, Elefterious F 2006 Boning up on ephrin signaling. Cell 126: 441–443
Martin TJ, Sims NA 2005 Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med 11: 76–81
Yang FC, Chen S, Robling AG, Yu Z, Nebesio TD, Yan J, Morgan T, Li X, Yuan J, Hock J, Ingram DA, Clapp DW 2006 Hyperactivation of p21ras and PI3K cooperate to alter murine and human neurofibromatosis type 1 – haploinsufficient osteoclast functions. J Clin Invest 116: 2880–2891
Yu X, Chen S, Potter OL, Murthy SM, Li J, Pulcini JM, Ohashi N, Winata T, Everett ET, Ingram D, Clapp WD, Hock JM 2005 Neurofibromin and its inactivation of Ras are prerequisites for osteoblast functioning. Bone 36: 793–802
Wu X, Estwick SA, Chen S, Yu M, Ming W, Nebesio TD, Li Y, Yuan J, Kapur R, Ingram D, Yoder MC, Yang FC 2006 Neurofibromin plays a critical role in modulating osteoblast differentiation of mesenchymal stem/progenitor cells. Hum Mol Genet 15: 2837–2845
Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, Parada LF, Karsenty G 2006 ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab 4: 441–451
Schindeler A, Ramachandran M, Godfrey C, Morse A, McDonald M, Mikulec K, Little DG 2008 Modeling bone morphogenetic protein and bisphosphonate combination therapy in wild-type and Nf1 haploinsufficient mice. J Orthop Res 26: 65–74
Acknowledgements
The authors thank the study participants for their help. They thank the research coordinators and laboratory assistants (Jeanne Siebert, Susan Geyer, Meredith Winn, Janice Davis, Diane Hartford, Bronte Clifford, Ann Rutherford, Heather Hanson, and Amy Watkins). They also thank Dr. Nicola Longo for his insights and guidance, and the International NF1 Bone Study Group (INBSG): (Judith Adams, Linlea Armstrong, Patricia Birch, John Carey, Alvin Crawford, Jacques D'Astous, Janice Davis, Judith Eelloo, Gareth Evans, Jan Friedman, Susan Geyer, Heather Hanson, Susan Huson, Harry Joe, Heidi Kalkwarf, Bernie LaSalle, Heather MacDonald, Heather McKay, Scott Miller, Laurie Moyer-Mileur, Zulf Mughal, Mary Murray, Kathleen Murray, Marzia Pasquali, Elizabeth Schorry, Jill Shea, Jeanne Siebert, Hillarie Slater, David Stevenson, David Viskochil, Martha Walker, Meredith Winn) for their discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by the Shriners Research Foundation; grant M01-RR00064 from the National Center for Research Resources; research grants K23 NS052500 and R01 NS050509-01A1 from the National Institute of Neurological Disorders and Stroke; the Children's Health Research Center, Clinical Genetics Research Program, and Center for Pediatric Nutrition Research at the University of Utah, and the Primary Children's Research Foundation.
Rights and permissions
About this article
Cite this article
Stevenson, D., Schwarz, E., Viskochil, D. et al. Evidence of Increased Bone Resorption in Neurofibromatosis Type 1 Using Urinary Pyridinium Crosslink Analysis. Pediatr Res 63, 697–701 (2008). https://doi.org/10.1203/PDR.0b013e31816fee45
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31816fee45
This article is cited by
-
Bone Status According to Neurofibromatosis Type 1 Phenotype: A Descriptive Study of 60 Women in France
Calcified Tissue International (2021)
-
Aberrant Myeloid Differentiation Contributes to the Development of Osteoporosis in Neurofibromatosis Type 1
Current Osteoporosis Reports (2016)
-
Spinal bone defects in neurofibromatosis type I with dural ectasia: stress fractures or dysplastic? A case series
European Radiology (2013)
-
High bone turnover and accumulation of osteoid in patients with neurofibromatosis 1
Osteoporosis International (2010)
