
Abstract
In this study, selective expression of therapeutic transgenes was evaluated in neuroblastoma cells. Promoter fragments of the genes for neuron-specific enolase (NSEp), tyrosine hydroxylase (THp), and dopamine-β-hydroxylase (DBHp) were studied in neuroblastoma and nonneuronal cell lines by transient transfection experiments using fluorescence-activated cell sorting (FACS) analysis of enhanced green fluorescent protein (egfp) and luciferase (luc +) assay. Both reporter gene assays revealed a neuroblastoma-selective expression mediated by NSEp and THp, whereas DBHp was active only in a murine neuroblastoma cell line. Reporter gene expression by NSEp in neuroblastoma cells was markedly higher than expression by THp, but NSEp also showed considerable background activity in nonneuronal cells. THp-driven expression of egfp was 35-fold higher in human neuroblastoma MHH-NB11 compared with nonneuronal HeLa cells. Thus, THp was chosen for a neuroblastoma-selective suicide gene therapy approach using the herpes simplex virus type 1 thymidine kinase (HSV-tk)/ganciclovir (GCV) system. A retrovirus vector that contained an expression cassette of a HSV-tk/egfp fusion gene and THp in antisense orientation was generated. Stably transduced human neuroblastoma cells and nonneuronal cell lines were generated, and HSV-tk/egfp expression was measured by FACS and GCV cytotoxicity assay. There was a 2.2-fold difference in green fluorescence and a 1.4-fold difference in cell killing between the human neuroblastoma MHH-NB11 and HeLa cells after HSV-tk/egfp gene transfer. The overall difference in THp-HSV-tk/egfp–mediated cell killing between neuroblastoma and nonneuronal tumor cell lines was statistically significant (P = 0.001). In conclusion, the present study demonstrated the feasibility of a neuroblastoma-selective gene therapy approach using the THp/ HSV-tk/egfp expression cassette.
Similar content being viewed by others


Main
Suicide gene therapy is a novel experimental paradigm for tumor treatment. It is based on enzyme gene transfer–mediated prodrug activation in target cells (1). A central aim of suicide gene therapy is to limit prodrug activation and, therefore, toxicity to a specific cell population.
For the most commonly used herpes simplex virus type 1 thymidine kinase (HSV-tk)/ganciclovir (GCV) suicide gene therapy approach, target cell selectivity has already been achieved in the treatment of brain tumors (2). Because brain tumor cells represent the predominant dividing cell population in the brain, any gene therapy approach that uses retroviral vectors targeted to dividing cells will be selective for tumor cells or neoplastic capillaries and will spare normal glia and neuronal cells (3). This restriction, however, does not necessarily apply to tumors located outside the CNS because these tumors may be surrounded by other proliferating normal cell types.
Here, a possible strategy to focus transgene expression to tumor cells may be the use of tissue- or cell-specific transcriptional elements, i.e. promoters or enhancers (4). Promising candidates for mediating tumor-selective transgene expression are promoters of tumor marker genes. Tumor marker genes are defined as being predominantly expressed by distinct tumor cell types. They may also be found—to a lower grade of activity—in normal cell types. Thus, to use a tumor marker promoter for tumor-selective expression of therapeutic genes, not only does promoter activity have to be present in tumor cells, but also regular background activity in normal cells has to be low.
In the present study, the feasibility of a tumor-selective gene therapy approach was studied in neuroblastoma cells. Neuroblastoma is a neoplastic disease of very young children that derives from the peripheral sympathetic nervous system and can occur as adrenal gland or sympathetic chain tumors with widespread metastases, always in close neighborhood to normal cells and tissues. Neuroblastomas are—in their most malignant form—very resistant to conventional treatment regimens and have a poor prognosis, with a <20–30% 5-year survival rate. Thus, there is certainly an urgent demand for new treatment strategies, and although it is usually a systemic disease, local treatment approaches to reduce tumor burden seem to be crucial for the overall therapeutic success.
In this respect, suicide gene therapy may have an impact on neuroblastoma therapy as a possible local treatment approach, especially when other local therapies, e.g. radiation therapy, have already failed. However, possible toxic side effects of suicide gene therapy on normal tissues should be minimized by targeting suicide gene expression to neuroblastoma cells.
Several tumor markers are known for neuroblastomas, and for some of them, associated transcriptional elements that may be used as promoter systems to restrict suicide gene expression to neuroblastoma cells are also identified. Among those candidate promoter systems are the promoter fragments of the genes for neuron-specific enolase (NSEp) (5,6) tyrosine hydroxylase (THp) (7), and dopamine-β-hydroxylase (DBHp) (8,9). The main aim of the present study was to identify the most suitable of these promoter systems for neuroblastoma-selective gene therapy. The most promising promoter system should display an acceptable low background activity in non-neuronal cells. However, low background activity in nonneuronal cells will not automatically exclude toxicity to normal neuronal cells, especially because all neuroblastoma marker genes are usually expressed in normal neuronal cells. Thus, our further approach in the present study was to integrate the most promising neuroblastoma-selective promoter system into a retroviral vector backbone to ensure an additional selectivity for proliferating cells, therefore sparing also normal neuronal cells from toxicity.
METHODS
The present study was performed according to the German Law for Genetic Engineering and has been approved by the biosafety review board of the local government.
Cell lines.
The mouse fibroblast cell line L929, the human cervix carcinoma cell line HeLa, the human osteosarcoma cell line MG-63, the neuroblastoma cell lines NEURO-2A (mouse) and SK-N-SH (human), and the retroviral packaging cell lines FNX-E and PG13 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, U.S.A.). The human fibrosarcoma cell line HT1080 and the human neuroblastoma cell line MHH-NB11 were supplied by the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). The MHH-NB11 and L929 cell lines were cultured in RPMI with 1 mM of sodium pyruvate, 2 mM of glutamine, and 10% FCS (Life Technologies, Karlsruhe, Germany). The cell lines HeLa, SK-N-SH, NEURO-2A, HT1080, and FNX-E were cultured in Dulbec-co's modified Eagle's medium with 4.5 mM of glucose, 1 mM of glutamine, and 10% FCS. The cell line PG13 was cultured in Iscove's modified Dulbecco's medium (Sigma Chemical Co., Deisenhofen, Germany) with 1 mM of sodium pyruvate, 2 mM of glutamine, and 10% FCS. All cell lines were grown at 37°C in an atmosphere of 95% air, 5% CO2.
Plasmid construction.
Plasmids pGL3-basic, pGL3-control, and pRL-SV40 were obtained from Promega (Mannheim, Germany). The various pGFP3 constructs were generated by replacing the luciferase (luc) coding sequence in pGL3-basic or pGL3control, respectively, through the cDNA for enhanced green fluorescent protein (egfp) from plasmid pEGFP-N1 (Clontech, Heidelberg, Germany). The 308 bp rat NSE promoter fragment (NSEp) spanning from −255 to +53 relative to the transcription start site was cloned by PCR amplification from plasmid pNSE-lacZ (10). The 522 bp TH promoter fragment (THp) spanning from −491 to 31 as well as the 498 bp DBH promoter fragment (DBHp) spanning from −474 to 24 were cloned by PCR amplification from human genomic DNA. The promoter fragments—NSEp, THp, DBHp—were cloned into the promoter- and enhancerless plasmids pGL3-basic and pGFP3-basic, respectively, to drive reporter gene expression without any interference by additional transcriptional elements within the construct.
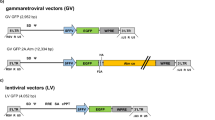
For generating retroviral vectors expressing a truncated version of the CD4 surface marker, the puromycin selection marker gene (puro) in the retroviral vector plasmid pBABE-puro (11) was replaced by the CD4 cDNA from plasmid pMACS4 (Miltenyi Biotech, Bergisch-Gladbach, Germany). The resulting retroviral vector plasmid was designated as pBABE-CD4. The retroviral vector pBABE-TKEG-CD4 was obtained by cloning an HSV-tk/egfp-fusion gene (12) into the multiple cloning site of pBABE-CD4. For generating the neuroblastoma-selective retroviral vector pBrev-TH-TKEG-CD4, the THp/HSV-tk/egfp expression cassette was inserted in anti-sense orientation to the retroviral LTR promoter into the multiple cloning site of pBABE-CD4. The final retroviral vector constructs pBABE-TKEG-CD4 and pBrev-TH-TKEG-CD4 are schematically represented in Fig. 1.

Schematic representation of the proviral structure of the retroviral vectors used for TH promoter–mediated (pBrev-TH-TKEG-CD4) or LTR promoter–mediated (pBABE-TKEG-CD4) expression of HSV-TK/EGFP fusion protein (TKEG). The untranslated leader region contains the packaging signal (Ψ). As selectable marker, both vectors contain a truncated cDNA of the CD4 marker gene under transcriptional control of the SV40 promoter.
Luciferase reporter gene assay.
Luciferase reporter gene expression was analyzed in neuroblastoma (MHH-NB11, NEURO-2A) and nonneuronal (HeLa, L929) cell lines using the Dual-Luciferase Reporter Assay System (Promega). Co-transfections of the various pGL3 constructs with the internal control pRL-SV40 were carried out with 0.5 μg of plasmid DNA each using FuGENE 6 transfection reagent (Roche, Mannheim, Germany). For ruling out possible promoter interferences within the dual system, the pGL3 constructs were also transfected alone without co-transfecting pRL-SV40 using 1 μg of plasmid DNA. Luciferase activities were determined 48 h after transfection. The activity of the firefly luciferase was adapted to the activity of the Renilla luciferase within each cell line to rule out possible bias as a result of different transfection efficiencies. The single pGL3 plasmid transfections were adapted according to the protein concentration of each sample, which was determined by the Bio-Rad DC Protein Assay (BioRad Laboratories, Munich, Germany).
Enhanced green fluorescent protein reporter gene assay.
Transfections of the pGFP3 constructs were performed in duplicate with 1 μg of plasmid DNA. EGFP expression/green fluorescence was flow-cytometrically measured 48 h after transfection using a FACScan (Becton Dickinson, Heidelberg, Germany). Data were analyzed on an Apple Power Macintosh G3 with CellQuest software (Becton Dickinson).
Generation of retrovirus stocks.
Ecotropic FNX-E cells were transfected with retroviral vector plasmids pBABE-TKEG-CD4 and pBrev-TH-TKEG-CD4 using FuGENE 6 transfection reagent. After overnight incubation, medium was replaced with 10 mL Iscove's modified Dulbecco's medium, and cells were incubated at 37°C for 24 h before supernatants were harvested, filtered using 0.45-μm sterile filters (Sartorius, Göttingen, Germany), immediately frozen, and finally stored at −80°C until further usage. Transduction of PG13 cells with FNX-E supernatants and subsequent sorting of CD4+ cells using magnetic activated cell sorting (MACS) technology (see below) resulted in homogeneous (>99%) CD4+ PG13 producer cell lines. PG13 supernatants were harvested as described above.
Retrovirus titer determination.
One day before infection, HT1080 cells were plated in 12-well plates at a density of 8 × 104 cells/well. Cells were infected in duplicate by a total volume of 0.5 mL of final solution each containing serial dilutions of supernatant stocks in the presence of 5 μg/mL of protamine (Sigma Chemical Co.). One day after infection, retroviral supernatants were replaced by fresh medium. After another 24 h, cells were harvested for FACS analysis of EGFP and CD4 expression using a PE-coupled mouse anti-CD4 antibody (Coulter Immunotech, Krefeld, Germany). Retroviral vector titers were determined by multiplying the number of HT1080 cells with the percentage of CD4+ cells. For all calculations, a correction factor neutralizing the bias by the expected increase of HT1080 cell numbers between plating and infection was taken into account. Resulting final retroviral titers were indicated as transducing units (TU) per milliliter.
Transduction of neuroblastoma and nonneuronal cell lines.
One day before infection, the various human cell lines (MHH-NB11, SK-N-SH, HeLa, and MG-63) were plated in six-well plates at a density of 2–5 × 105 cells/well. Cell lines SK-N-SH and MG-63 were added to these experiments as additional positive and negative controls because the murine cell lines NEURO-2A and L929 had been persistently resistant to an effective retroviral transduction. To increase transduction efficiency, viral supernatants were concentrated 1:10 by centrifugation for 30 min at 10,000 rpm and 4°C. Infections were performed as described above in a final volume of 1 mL/well. In parallel, transduced cells were analyzed for EGFP and CD4 expression and further expanded for magnetic sorting of CD4+ cells as described below.
MACS.
A total of 1 × 106–107 cells were used for MACS-mediated enrichment of CD4+ cells. After incubation with a PE-coupled mouse anti-CD4 antibody (Coulter Immunotech), cells were washed with PBS and incubated with a microbead-coupled anti-PE antibody (Miltenyi Biotech) according to the manufacturer's guidelines. CD4+ cells were then separated using a MiniMACS magnetic cell separator and MS+/RS+ columns (Miltenyi Biotech). After enrichment, the increase of CD4 (and also EGFP) expression within the selected population was determined by FACS analysis.
Cell viability assay.
To evaluate the cytotoxicity of ganciclovir (GCV) treatment, cells were plated in 96-well dishes in quintuplets at a density of 4 ×103 to 1 ×104 cells/well and incubated for 4 d in a total volume of 100 μL of medium without GCV or containing 1 μg/mL of GCV (Cymevene; Synthex, Aachen, Germany). Cell viability was determined using the CellTiter96 AQueous NonRadioactive Cell Proliferation Assay (Promega). The cytotoxic effect was indicated as percentage of surviving cells (ratio of surviving cells after treatment and without treatment).
For statistical evaluation, percentages of nonsurviving pBrev-TH-TKEG-CD4 cells were adapted to percentages of nonsurviving corresponding pBABE-TKEG-CD4 cells, which were defined for each individual experiment by a GCV cyto-toxicity factor of 1.0. This adaptation was done to exclude cell line–dependent variations in GCV-mediated cytotoxicity and retroviral transduction efficiencies. Resulting GCV cytotoxicity factors of the various pBrev-TH-TKEG-CD4 cell lines in four independent experiments were statistically evaluated (t test for independent samples; SPSS for Windows 10.0, SPSS GmbH Software, Munich, Germany) to prove significant differences in THp-mediated cell killing between neuroblastoma and nonneuronal cell lines. Statistical significance was defined by P < 0.05.
RESULTS
Neuroblastoma-selective promoter activity in transiently transfected cells measured by luciferase and egfp reporter gene assays.
To investigate the feasibility of various candidate promoter systems for neuroblastoma-selective gene expression, we generated constructs containing the reporter genes egfp and luc under control of the rat NSEp and the human THp or DBHp. We studied reporter gene expression by the various promoter fragments in transient transfection experiments using human and mouse neuroblastoma cell lines as well as fibro-blasts and nonneuronal tumor cell lines as negative controls. A construct that contained the constitutive SV40 promoter and a promoterless construct were used as additional controls. To investigate the feasibility of the reporter gene egfp for promoter studies, we compared promoter activities determined by flow-cytometric detection of EGFP expression with the results from the luciferase reporter gene assay, which is commonly used for promoter studies.
The luciferase reporter gene assays were performed both as single and as dual luciferase assay. In the dual assay, activity of the constitutively expressed Renilla luciferase was used as internal control to adapt the activity of the specifically expressed firefly luciferase to minimize the possible bias as a result of variations in transfection efficiencies. The dual lucif-erase assay results are summarized in Table 1. The indicated values of the promoterless and selective promoters that contained plasmids represent percentages relative to the values of the corresponding control plasmid with the nonselective SV40p (= 100%). The additionally performed single luciferase assays revealed similar results, thus indicating that the activities obtained with the dual assay were not altered by promoter interferences (data not shown). Qualitatively similar results were also obtained by the newly established egfp reporter gene assay (Table 1), supporting its feasibility for promoter studies. In our assay, determination of promoter-dependent EGFP expression was performed in single-transfection experiments followed by flow-cytometric quantification of mean EGFP fluorescence intensities after 48 h.
Both egfp and luciferase reporter gene assays revealed a neuroblastoma-selective expression by NSEp and THp, whereas DBHp was active only in the murine neuroblastoma cell line NEURO-2A. Reporter gene expression by NSEp in neuroblastoma cells was markedly higher than expression by THp, but NSEp also showed a considerably high background activity in nonneuronal cells, which was especially in the luciferase assay as strong as the activity of the nonselective viral promoter. In contrast, THp and DBHp mediated constantly low reporter gene expression levels in the nonneuronal cell lines. Actual differences in reporter gene expression between human and murine neuroblastomas and their nonneuronal counterparts are summarized for each promoter system in Table 2.
Cloning and production of retroviral vectors expressing the HSV-tk/egfp fusion gene and the truncated CD4 marker gene.
On the basis of the encouraging results of the transient transfection experiments, THp was chosen as the most promising candidate and was investigated further in the context of a neuroblastoma-selective suicide gene therapy approach using the HSV-tk/GCV system. The murine Moloney leukemia virus–based pBABE vector (11) was used as a backbone. The transduction capability of the pBABE vector is restricted exclusively to proliferating cells and provides an additional safety feature by reducing toxicity to normal nonproliferating neuronal cells. The newly generated retroviral vector contained an expression cassette of a HSV-tk/egfp fusion gene (12) and THp. For excluding possible promoter interference with retroviral promoter elements that might result in a reduced TH promoter specificity, the internal expression cassette was inserted in antisense orientation to the viral promoter elements within the 5′LTR. For monitoring transduction efficiency and stable cell selection, the retroviral vector also bore a truncated CD4-encoding cDNA (tCD4) regulated by an SV40 promoter in sense orientation (Fig. 1). The resulting retrovirus vector was designated as pBrev-TH-TKEG-CD4. The retroviral vector pBABE-TKEG-CD4 containing the HSV-tk/egfp fusion gene in sense orientation under transcriptional control of the viral 5′LTR promoter was also generated as a positive control for the retroviral vector studies (Fig. 1).
Infectious retroviral particles with ecotropic host range were obtained after transfection of retroviral plasmids pBABE-TKEG-CD4 and pBrev-TH-TKEG-CD4 into FNX-E packaging cells. For establishing stable producer cells for retrovirus vectors with amphotropic host range, retroviral vector packaging cells PG13 were transduced with the obtained FNX-E supernatants. The transduction efficiency as determined by FACS analysis of CD4+ cells was 96% for pBABE-TKEG-CD4 and 75.4% for pBrev-TH-TKEG-CD4. For producing highest infectious titers, transduced PG13 cells were enriched by magnetic sorting of CD4-expressing cells. The resulting stable pBABE-TKEG-CD4 producer cell line generated viral titers of 1.17 ± 0.2 × 106 TU/mL (determined by FACS analysis of CD4 expression), whereas the viral titer of the stable pBrev-TH-TKEG-CD4 producer cell line was significantly lower with 4.3 ± 1.6 105 TU/mL.
Generation of stably transduced human neuroblastoma and nonneuronal cell lines by retroviral transduction and magnetic cell sorting of infected cells.
To investigate the specificity of TH promoter-mediated suicide gene therapy, we established an in vitro model by stable retroviral transduction of human neuroblastoma and nonneuronal cell lines. The human neuroblastoma cell lines MHH-NB11 and SK-N-SH as well as the nonneuronal tumor cell lines HeLa and MG-63 were infected with supernatants of the pBABE-TKEG-CD4 and pBrev-TH-TKEG-CD4 PG13 producer cell clones. For obtaining stably and homogeneously transduced cell populations, the CD4 marker–expressing cell populations were enriched using MACS technology. After enrichment, CD4 and EGFP expression of the resulting cell populations was determined by FACS analysis. As shown in Fig. 2, the percentage of CD4-expressing cells in all cell populations was ≥92.9%. The EGFP expression within the enriched, pBABE-TKEG-CD4–transduced cell populations varied from 76.2 to 97.9%. Within the pBrev-TH-TKEG-CD4–transduced nonneuronal cell populations, the percentage of EGFP-expressing cells was 5.8% or 16.1%, respectively, whereas in the neuroblastoma cell lines, 30 to 35.6% of the cells were EGFP positive. The mean fluorescence intensity (= X-mean) of EGFP expression was significantly higher in the pBABE-TKEG-CD4–transduced MG-63 cells (947.4 relative units of fluorescence intensity) than in all other pBABE-TKEG-CD4–transduced cell populations (27.4 to 87 relative units of fluorescence intensity, respectively), thus indicating a strong LTR promoter activity in MG-63 cells.

EGFP and CD4 expression of MACS-purified pBABE-TKEG-CD4–(BABE) and pBrev-TH-TKEG-CD4-(Brev)–infected human cell lines: HeLa (cervix carcinoma; a and b), MG-63 (osteosarcoma; c and d), MHH-NB11 (neuroblastoma; e and f), and SK-N-SH (neuroblastoma; g and h). The figure shows a dot-plot representation of EGFP fluorescence versus phycoerythrin (PE) fluorescence of the conjugated CD4 antibody. The percentage of CD4+ cells indicates the purity of the transduced CD4-expressing populations after magnetic enrichment. The strength of EGFP expression mediated by either the LTR or THp is given by the percentage of EGFP+ cells within each population as well as the mean of EGFP fluorescence intensity (X-mean).
For comparing the levels of TH promoter-mediated trans-gene expression within the different cell lines, both the percentage and the mean fluorescence intensity (X-mean) of EGFP expression have to be considered as well as vector-related differences in transduction efficiencies. Differences in transduction efficiency between pBABE-TKEG-CD4 and pBrev-TH-TKEG-CD4 vectors are adequately described by the ratio of nonselective CD4 expression in pBABE- and pBrev-transduced cells. CD4-expression is—like EGFP expression—best characterized by the percentage and the mean fluorescence intensity (Y-mean) of positive cells. By taking all of these parameters into account, a selective EGFP expression factor was defined for each cell line as follows:
EQUATION

The difference in transduction efficiencies (Δ transduction efficiency pBrev: pBABE) seems to be inversely related to the ratio of selective to nonselective EGFP expression: a high selective EGFP expression by pBrev will even be more obvious when transduction efficiency of pBrev is low in comparison to pBABE.
Table 3 summarizes the various selective EGFP expression factors for each cell line investigated. These data demonstrated a 55- to 80- fold increase in EGFP expression between the non neuronal cell line MG-63 and the neuroblastoma cell lines. The selectivity of EGFP expression was not as strong when both neuroblastoma cell lines were compared with the non neuronal HeLa cell line. Here, only a 2-to 3-fold difference in selective EGFP expression was observed.
TH-Promoter mediates enhanced GCV sensitivity in neuroblastoma cells.
Finally, we investigated the THp-mediated cytotoxicity of GCV treatment in the various stably transduced neuroblastoma and nonneuronal cell lines. Survival of the enriched pBrev-TH-TKEG-CD4–and pBABE-TKEG-CD4–transduced cells was determined after incubation with the standard GCV dose of 10 μg/mL for 4 d.
Figure 3 shows that cell survival upon GCV treatment displayed a considerable variation within the different cell lines, even when the nonselective pBABE-TKEG-CD4 vector was used (mean cell survival after pBABE-TKEG-CD4 transduction: 8.8% SK-N-SH, 20.3% MG-63, 21.1% MHH-NB11, 29.7% HeLa). In both neuroblastoma cell lines, the TH promoter–dependent cytotoxicity was ∼5 to 10% lower than the 5′LTR promoter–dependent cytotoxicity (mean cell survival after pBrev-TH-TKEG-CD4 transduction: 12.7% SK-N-SH, 20.1% MHH-NB11). In nonneuronal cell lines, the cytotoxicity mediated by TH promoter–dependent HSV-tk expression was ∼25% lower than in pBABE-TKEG-CD4–transduced cells (mean cell survival after pBrev-TH-TKEG-CD4 transduction: 54.1% HeLa, 44.3% MG-63).

Survival of MACS-enriched pBABE-TKEG-CD4–(BABE) and pBrev-TH-TKEG-CD4–(Brev) infected cells after 4 d of incubation with 10 μg/mL of GCV. Indicated are mean values and SDs of four independently performed experiments. As expected, differences in survival between BABE- and Brev-mediated cell killing were statistically significant only for HeLa (*P < 0.05) and MG-63 (**P < 0.005) but not for the neuroblastoma cell lines. The overall difference in Brev-mediated cell killing between neuroblastoma (SK-N-SH, MHH-NB11) and nonneuronal tumor (HeLa, MG-63) cell lines was proved to be statistically significant (P = 0.001). By comparing each cell line with each other, the following P values were achieved:P = 0.035 for SK-N-SH vs HeLa (significant); P = 0.004 for SK-N-SH vs MG-63 (significant); P = 0.063 for MHH-NB11 vs HeLa (not significant); P = 0.010 for MHH-NB11 vs MG-63 (significant); P = 0.072 for SK-N-SH vs MHH-NB11 (not significant); and P = 0.639 for HeLa vs MG-63 (not significant). All statistical evaluations were performed using the t test.
GCV-induced killing of pBABE-TKEG-CD4 cells by GCV was defined as a cytotoxicity factor of 1.0 for each cell line to exclude cell line–dependent variations in drug sensitivity and transduction efficiency. The following GCV cytotoxicity factors of pBrev-TH-TKEG-CD4–transduced cells were found: 0.96 for SK-N-SH, 0.89 for MHH-NB11, 0.65 for HeLa, and 0.70 for MG-63. The overall difference in THp-mediated cell killing between neuroblastoma (SK-N-SH, MHH-NB11) and nonneuronal tumor (HeLa, MG-63) cell lines was proved to be statistically significant by performing t tests (P = 0.001). By comparing each cell line with each other, the following P values were achieved for differences in THp-mediated cell killing:P = 0.035 for SK-N-SH versus HeLa (significant); P = 0.004 for SK-N-SH versus MG-63 (significant); P = 0.063 for MHH-NB11 versus HeLa (not significant); P = 0.010 for MHH-NB11 versus MG-63 (significant); P = 0.072 for SK-N-SH versus MHH-NB11 (not significant); and P = 0.639 for HeLa versus MG-63 (not significant).
However, the overall selectivity of THp-mediated transgene expression was reduced from a 98- to 35-fold difference, respectively, between MHH-NB11 and HeLa cells in luciferase and egfp activity after transient transfection to a 2.2-fold difference in green fluorescence and a 1.4-fold difference in GCV-mediated cell killing after retroviral HSV-tk/egfp transfer. Similar results were found for retrovirus-mediated EGFP-expression and GCV cytotoxicity when MHH-NB11 and MG-63, SK-N-SH and MG-63, or SK-N-SH and HeLa cells were compared with each other. There was always a marked decrease between determination of pure reporter gene expression and actual cytotoxicity between neuroblastomas and nonneuronal cell lines. Table 4 summarizes all of these differences in neuroblastoma-selective transgene expression for the various assays performed in the present study.
DISCUSSION
The present study is the first one of its kind to systematically study and compare the feasibility of the promotor systems NSEp, THp, and DBHp for their potential use in cytotoxic gene therapy strategies to treat neuroblastomas. NSEp, THp, and DBHp all represent promoters of clinically relevant neuroblastoma-associated genes. They have been used before to achieve neuron-specific transgene expression in therapeutic models of Parkinson's disease and other neurodegenerative disorders, but only two studies have actually focused so far on their ability to direct transgene expression to neuroectodermal tumor cells: one report showed specific high-level reporter gene expression by THp in neuroblastoma cells (13), and the other demonstrated tumor cell–specific cytotoxicity of HSV-tk/GCV by NSEp in small lung carcinoma cells (14). No similar approaches have been reported to date for DBHp in tumor cells.
As a first step toward the development of a neuroblastoma-selective gene therapy approach, NSEp, THp, and DBHp were investigated and compared with each other for their potential to mediate neuroblastoma-selective transgene expression by two different assays to analyze promotor activities: besides the established luciferase assay, which was performed as a single and a dual luciferase assay (with Renilla luciferase as constitutively expressed control), the feasibility of FACS analyses of EGFP expression was studied. All assays revealed essentially similar results. Minor deviations, especially regarding THp activity in murine neuroblastoma cells, may have been due to variations based on cell numbers, transfection efficiencies, protein half-life, or simply that one assay was based on measuring an enzyme activity whereas the other one was detecting the protein itself. However, neuroblastoma-selective reporter gene expression was clearly shown for NSE and TH promoter fragments. Looking at absolute transgene activities, NSEp-mediated reporter gene expression in neuroblastoma cells was markedly higher than expression by THp, but nonselective reporter gene expression was also considerably high in the case of NSEp. Especially in the luciferase assays, NSEp activity in nonneuronal cell lines was as high as the activity of the nonselective viral promoter. In contrast, reporter gene expression by THp was low in all nonneuronal cells. DBHp activity was similarly reduced in nonneuronal cells but was also barely detectable in human MHH-NB11 neuroblastoma cells. Only in murine NEURO-2A neuroblastoma cells did DBHp mediate adequate reporter gene expression. These variations in DHBp activity certainly reflect that DBH is catalyzing only a minor pathway of catecholamine biosynthesis that is not found in all neuroblastomas (15). Its presence, therefore, cannot be compared with TH which — as a key enzyme of catecholamine biosynthesis — is expected to be active in the majority of neuroblastomas (15). Nevertheless, there are few neuroblastomas with no significantly increased catecholamine biosynthesis or TH activity, but neuroblastomas with potentially increased TH activity may be easily identified in a clinical setting, because investigation of increased catecholamine biosynthesis is essential during initial diagnosis of neuroblastoma (16).
Summarizing our reporter assay findings, the TH promoter seems to fulfill most essential features to direct transgene expression to neuroblastoma cells, i.e. high activity in these tumor cells and low activity in nonneuronal cells. However, THp activity may also be relatively high in normal (nonneoplastic) cells of neuroectodermal origin, especially in cells that belong to the sympathetic nervous system. Toxic side effects to these cells, therefore, cannot be ruled out by THp-mediated suicide gene therapy. Neuronal cells can be spared from toxicity when an additional mechanism is applied to confine the toxicity to proliferating cells, because normal neuronal cells have lost their mitotic capability. The HSV-tk/GCV system can represent such a proliferation-dependent safety mechanism as it is supposed to generate toxicity in proliferating cells only by interference with DNA synthesis (17).
In addition, conventional retroviral vectors serve as an additional safety mechanism because they direct their gene transfer exclusively to proliferating cells. Regarding this capability, retroviral vectors are unique among all vector systems currently used for gene therapy. Adenovirus, herpesvirus, lentivirus, and adeno-associated virus vectors all accomplish gene transfer independent of cell proliferation. Toxicity to neuronal cells cannot be excluded by application of these vector systems. However, retroviral vectors that bear the HSV-tk gene have been shown not to improve outcome in brain tumor patients after GCV treatment (18). This was probably due mainly to limited transduction efficiency of the vectors used. As potential alternatives, adenovirus and herpesvirus vectors provide markedly higher transduction efficiencies in experimental tumor models than retroviral vectors, but they, too, have not yet succeeded to prove their therapeutic significance in patients (19,20). Thus, despite lower transduction efficiencies, retroviral vectors still represented for us the most suitable choice for a neuroblastoma-selective suicide gene therapy approach.
We generated a retroviral vector (pBrev-TH-TKEG-CD4) with an HSV-tk/egfp fusion gene (12) under transcriptional control of the TH promoter. A nonselective (positive) control vector (pBABE-TKEG-CD4) with the HSV-tk/egfp fusion gene driven by the retroviral 5′LTR promoter was also constructed. For allowing selection of transduced cells, a second expression cassette that contained a truncated version of the CD4 gene was inserted into both vectors. Expression of the hematopoietic CD4 surface marker should allow magnetic enrichment of infected cells (21).
Titration of the supernatants from stable PG13 packaging cells revealed that the retrovirus vector with the neuroblastoma-selective THp-HSV-tk/egfp–expression cassette produced lower infectious titers than the retrovirus with LTR-mediated HSV-tk/egfp expression. Previous transgene expression studies based on retroviral vectors showed that any additional internal expression cassette often results in reduced transgene expression levels (22–26). Reduced transgene expression levels were due mostly to decreased overall transduction efficiencies (24).
To minimize the problem of a reduced retroviral transduction efficiency, we generated stably expressing cell pools by magnetic enrichment of transduced cells. For studying the actual neuroblastoma-selective potential of our retroviral suicide gene therapy approach, numbers of cells transduced by either the nonselective pBABE-TKEG-CD4 or the selective pBrev-TH-TKEG-CD4 vector had to be similar for a reliable comparison to take place. Similar transduction efficiencies of both vectors in neuroblastoma and nonneuronal cells after magnetic enrichment were subsequently confirmed by FACS analysis of CD4 expression, which was regulated in both vectors by the nonselective SV40 promoter. Thus, the CD4 gene was used not only to provide but also to monitor a sufficient transduction efficiency.
By securing an overall transduction efficiency ≥92.9% in infected cell lines, the selective THp-mediated and the nonselective 5′LTR–mediated EGFP expression was determined. FACS analysis revealed a 8.7- to 12.5-fold lower selective EGFP expression in the neuroblastoma cell lines. These results did not correlate with the EGFP expression levels found in the transient egfp reporter gene assays. Here, THp-mediated EGFP expression in the neuroblastoma cell line MHH-NB11 was only 2.7-fold lower than the nonselective SV40p-dependent expression. SV40p was not expected to be weaker than the retroviral 5′LTR promoter.
The reduced activity of THp in stably transduced cells might be due to interferences with retroviral promoter elements. In the past, various attempts have been made to overcome interferences of the 5′LTR with an additional internal promoter. Especially in the case of a selective transgene expression, overriding of the selective promoter by the nonselective LTR should be principally ruled out. One possibility to achieve this aim is the use of self-inactivating retroviral vectors whereby the LTR promoter is subsequently silenced by deletions after target cell transduction (27,28). The deletion of LTR elements usually resulted in markedly reduced retroviral titers (29). An interesting further development of this approach was to replace retroviral promoter and enhancer elements by tumor-selective promoter sequences (30,31). Infectious titers were also considerably reduced by this approach. Another possibility to exclude overriding by the LTR is to insert the selective expression cassette in antisense orientation (32–34). In a preliminary screening for the most suitable selective vector design (data not shown), we compared transduction efficiencies of self-inactivating and antisense vector constructs. Both vectors showed similar but markedly reduced transduction efficiencies. However, we chose the antisense principle for further investigations because its retroviral design seemed to be better and easier compared with to the pBABE control vector.
The development of stable transgene-expressing cell lines also allowed us to investigate the selectivity of THp-mediated cytotoxicity after HSV-tk/GCV suicide gene therapy. GCV treatment of pBABE-TKEG-CD4–transduced cells revealed that the HSV-tk/GCV gene therapy approach was similarly efficient in all cell lines when the viral LTR promoter was used for therapeutic transgene expression. Determination of survival within the pBrev-TH-TKEG-CD4–transduced cells showed a significant difference in GCV sensitivity between neuroblastoma and nonneuronal cells. However, THp-regulated HSV-tk expression still mediated a >45% killing rate in nonneuronal tumor cells. This considerable nonselective cytotoxicity was somewhat surprising because FACS analysis of THp-dependent EGFP expression had revealed relatively low expression levels in the nonneuronal cell lines. Especially remarkable was the 56% cell killing rate in MG-63 osteosarcoma, which had demonstrated only 6% HSV-TK/EGFP–expressing cells. These unexpected discrepancies between HSV-TK expression and GCV sensibility may have been due to the strong bystander effect in the investigated cell lines. Moolten and Wells (35) first observed the phenomenon that a low percentage of HSV-TK–expressing cells can expand GCV-mediated toxicity to a markedly increased cell population via cell-to-cell contacts with their nontransduced neighbors. As suggested by our findings, such bystander effects may counteract any tumor-selective suicide gene therapy approach using the HSV-tk/GCV system. Most of the known suicide gene therapy systems usually display a more or less effective bystander effect (36). However, the rabbit cytochrome P450 4B1/4-ipomeanol (37) and the human folylpolyglutamate synthetase/methotrexate systems (38) have been shown to lack substantial bystander effects. These systems may be good candidates to use in future tumor-selective suicide gene therapy approaches.
In conclusion, in reporter gene assays with transiently transfected cells, we have identified THp among three possible candidates as the most suitable neuroblastoma-selective promoter with a remarkable 35- to 98-fold difference in activity between human neuroblastoma and nonneuronal cells. By transferring a THp expression cassette into a retroviral vector, the selectivity of the reporter gene expression dropped to a 2.2-to 7.7-fold difference between neuroblastomas and nonneuronal cell lines. When HSV-tk/GCV suicide gene therapy was performed, there was only a 1.3- to 1.5-fold difference in THp-mediated cytotoxicity between neuroblastomas and non-neuronal neoplastic cell lines. Although this difference was still statistically significant, its marked reduction during the different stages of developing a neuroblastoma-selective suicide gene therapy demonstrates the obstacles of such an approach:1) there is still no fully convincing vector design available for an efficient neuroblastoma-selective gene therapy approach, and 2) any suicide gene therapy system with a strong bystander effect will reduce tumor selectivity if the chosen approach still shows a nonselective leakiness. However, in the present study, we have clearly demonstrated the feasibility of a neuroblastoma-selective suicide gene therapy approach using THp. Suitable THp expression cassettes can be transferred easily into any backbone of novel vector developments.
Abbreviations
- DBHp:
-
dopamine-β-hydroxylase gene promoter
- EGFP:
-
enhanced green fluorescent protein
- GCV:
-
ganciclovir
- HSV-tk:
-
Herpes simplex virus thymidine kinase
- luc:
-
luciferase
- NSEp:
-
neuron-specific enolase gene promoter
- pBABE:
-
murine Moloney leukemia virus–based vector
- pBREV:
-
pBABE vector with internal expression cassette inserted in antisense orientation
- THp:
-
tyrosine hydroxylase gene promoter
References
Kramm CM, Sena-Esteves M, Barnett FH, Rainov NG, Schuback DE, Yu JS, Pechan PA, Paulus W, Chiocca EA, Breakefield XO 1995 Gene therapy for brain tumors. Brain Pathol 5: 345–381.
Ram Z, Culver KW, Oshiro EM, Viola JJ, DeVroom HL, Otto E, Long Z, Chiang Y, McGarrity GJ, Muul LM, Katz D, Blaese RM, Oldfield EH 1997 Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med 3: 1354–1361.
Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM 1992 In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science 256: 1550–1552.
Brand K, Loser P, Arnold W, Bartels T, Strauss M 1998 Tumor cell-specific transgene expression prevents liver toxicity of the adeno-HSVtk/GCV approach. Gene Ther 5: 1363–1371.
Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG 1990 Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron 5: 187–197.
Twyman RM, Jones EA 1997 Sequences in the proximal 5′ flanking region of the rat neuron-specific enolase (NSE) gene are sufficient for cell type-specific reporter gene expression. J Mol Neurosci 8: 63–73.
Coker GT, Vinnedge L, O'Malley KL 1988 Characterization of rat and human tyrosine hydroxylase genes: functional expression of both promoters in neuronal and non-neuronal cell types. Biochem Biophys Res Commun 157: 1341–1347.
Kobayashi K, Kurosawa Y, Fujita K, Nagatsu T 1989 Human dopamine beta-hydroxylase gene: two mRNA types having different 3′-terminal regions are produced through alternative polyadenylation. Nucleic Acids Res 17: 1089–1102.
Shaskus J, Greco D, Asnani LP, Lewis EJ 1992 A bifunctional genetic regulatory element of the rat dopamine beta-hydroxylase gene influences cell type specificity and second messenger-mediated transcription. J Biol Chem 267: 18821–18830.
Andersen JK, Garber DA, Meaney CA, Breakefield XO 1992 Gene transfer into mammalian central nervous system using herpes virus vectors: extended expression of bacterial lacZ in neurons using the neuron-specific enolase promoter. Hum Gene Ther 3: 487–499.
Morgenstern JP, Land H 1990 Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18: 3587–3696.
Steffens S, Frank S, Fischer U, Heuser C, Meyer KL, Dobberstein KU, Rainov NG, Kramm CM 2000 Enhanced green fluorescent protein fusion proteins of herpes simplex virus type 1 thymidine kinase and cytochrome P450 4B1: applications for prodrug-activating gene therapy. Cancer Gene Ther 7: 806–812.
Gardaneh M, Gilbert J, Haber M, Norris MD, Cohn SL, Schmidt ML, Marshall GM 2000 Synergy between 5′ and 3′ flanking regions of the human tyrosine hydroxylase gene ensures specific, high-level expression in neuroblastoma cells. Neurosci Lett 292: 147–150.
Tanaka M, Inase N, Miyake S, Yoshizawa Y 2001 Neuron specific enolase promotor for suicide gene therapy in small cell lung carcinoma. Anticancer Res 21: 291–294.
LaBrosse EH, Comoy E, Bohuon C, Zucker JM, Schweisguth O 1976 Catecholamine metabolism in neuroblastoma. J Natl Cancer Inst 57: 633–638.
Chamberlain J 1994 Screening for neuroblastoma: a review of the evidence. J Med Screen 1: 169–175.
Elion GB 1983 The biochemistry and mechanism of action of acyclovir. J Antimicrob Chemother 12: suppl B): 9–17.
Rainov NG 2000 A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther 11: 2389–2401.
Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, Palmer CA, Feigenbaum F, Tornatore C, Tufaro F, Martuza RL 2000 Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther 7: 867–874.
Trask TW, Trask RP, Aguilar-Cordova E, Shine HD, Wyde PR, Goodman JC, Hamilton WJ, Rojas-Martinez A, Chen SH, Woo SL, Grossman RG 2000 Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol Ther 1: 195–203.
Miltenyi S, Muller W, Weichel W, Radbruch A 1990 High gradient magnetic cell separation with MACS. Cytometry 11: 231–238.
Apperley JF, Luskey BD, Williams DA 1991 Retroviral gene transfer of human adenosine deaminase in murine hematopoietic cells: effect of selectable marker sequences on long-term expression. Blood 78: 310–317.
Bowtell DD, Cory S, Johnson GR, Gonda TJ 1988 Comparison of expression in hemopoietic cells by retroviral vectors carrying two genes. J Virol 62: 2464–2473.
Correll PH, Colilla S, Karlsson S 1994 Retroviral vector design for long-term expression in murine hematopoietic cells in vivo. Blood 84: 1812–1822.
Emerman M, Temin HM 1984 Genes with promoters in retrovirus vectors can be independently suppressed by an epigenetic mechanism. Cell 39: 449–467.
Vile R, Miller N, Chernajovsky Y, Hart I 1994 A comparison of the properties of different retroviral vectors containing the murine tyrosinase promoter to achieve transcriptionally targeted expression of the HSVtk or IL-2 genes. Gene Ther 1: 307–316.
Yee JK, Moores JC, Jolly DJ, Wolff JA, Respess JG, Friedmann T 1987 Gene expression from transcriptionally disabled retroviral vectors. Proc Natl Acad Sci USA 84: 5197–5201.
Yu SF, von Ruden T, Kantoff PW, Garber C, Seiberg M, Ruther U, Anderson WF, Wagner EF, Gilboa E 1986 Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc Natl Acad Sci USA 83: 3194–3198.
Gilboa E, Eglitis MA, Kantoff PW, Anderson WF 1986 Transfer and expression of cloned genes using retroviral vectors. Biotechnology 4: 504–512.
Diaz RM, Eisen T, Hart IR, Vile RG 1998 Exchange of viral promoter/enhancer elements with heterologous regulatory sequences generates targeted hybrid long terminal repeat vectors for gene therapy of melanoma. J Virol 72: 789–795.
Mavria G, Jager U, Porter CD 2000 Generation of a high titre retroviral vector for endothelial cell-specific gene expression in vivo. Gene Ther 7: 368–376.
Belmont JW, MacGregor GR, Wager-Smith K, Fletcher FA, Moore KA, Hawkins D, Villalon D, Chang SM, Caskey CT 1988 Expression of human adenosine deaminase in murine hematopoietic cells. Mol Cell Biol 8: 5116–5125.
Chang BD, Roninson IB 1996 Inducible retroviral vectors regulated by lac repressor in mammalian cells. Gene 183: 137–142.
Ragheb JA, Couture L, Mullen C, Ridgway A, Morgan RA 1999 Inhibition of human immunodeficiency virus type 1 by Tat/Rev-regulated expression of cytosine deaminase, interferon alpha2, or diphtheria toxin compared with inhibition by transdominant Rev. Hum Gene Ther 10: 103–112.
Moolten FL, Wells JM 1990 Curability of tumors bearing herpes thymidine kinase genes transferred by retroviral vectors. J Natl Cancer Inst 82: 297–300.
Aghi M, Hochberg F, Breakefield XO 2000 Prodrug activation enzymes in cancer gene therapy. J Gene Med 2: 148–164.
Frank S, Steffens S, Fischer U, Tlolko A, Rainov NG, Kramm CM 2002 Differential cytotoxicity and bystander effect of the rabbit cytochrome P450 4B1 enzyme gene by two different prodrugs: implications for pharmacogene therapy. Cancer Gene Ther 9: 178–188.
Aghi M, Kramm CM, Breakefield XO 1999 Folylpolyglutamyl synthetase gene transfer and glioma antifolate sensitivity in culture and in vivo. J Natl Cancer Inst 91: 1233–1241.
Acknowledgements
We thank Prof. Dr. Ulrich Goebel for support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by grants from the German Federal Ministry of Research (BMBF BioRegio, FKZ 0311661/1402), Bonn, Germany, and the Elterninitiative Kinderklinik Duesseldorf e.V., Duesseldorf, Germany.
Rights and permissions
About this article
Cite this article
Steffens, S., Sandquist, A., Frank, S. et al. A Neuroblastoma-Selective Suicide Gene Therapy Approach Using the Tyrosine Hydroxylase Promoter. Pediatr Res 56, 268–277 (2004). https://doi.org/10.1203/01.PDR.0000132666.23103.EF
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000132666.23103.EF
This article is cited by
-
Expression mediated by three partial sequences of the human tyrosine hydroxylase promoter in vivo
Molecular Therapy - Methods & Clinical Development (2016)
-
Gene therapy as a potential tool for treating neuroblastoma—a focused review
Cancer Gene Therapy (2016)
-
Targeted expression of human folylpolyglutamate synthase for selective enhancement of methotrexate chemotherapy in osteosarcoma cells
Cancer Gene Therapy (2013)
