
Abstract
In the present study the relationship between exposure to the nitric oxide synthesis inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) and the induction of limb defects, with respect to stage specificity and dose dependency, was investigated in the mouse. ICR (CD-1) mice were dosed s.c with L-NAME at 50 or 90 mg/kg on gestation d 12, 13, 14, 15, or 16. A group of animals treated with vehicle on gestation d 14 served as control. Uterine contents were evaluated for teratogenesis on gestation d 18. A treatment-related disruption of limb development was noted. The effect was dose dependent and phase specific. L-NAME became teratogenically operational on gestation d 13 and elicited its maximum effect on gestation d 14, whereas no significant teratogenicity was observed when exposure occurred after gestation d 15. In utero exposure to L-NAME also reduced embryo viability relative to controls. When the higher dose was injected on gestation d 16, a significant number of dams delivered preterm. In a parallel study, the ability of hyperoxia to prevent limb teratogenesis was investigated. To this aim, a group of L-NAME-treated animals (90 mg/kg s.c. on gestation d 14) were exposed to 98 to 100% O2 for 12 h. L-NAME-treated mice breathing room air served as positive controls. In response to hyperoxia, a significant decrement of L-NAME-induced limb defects was found. This study characterizes for the first time the teratogenic capacity of L-NAME in the mouse. Results obtained with hyperoxia fit the hypothesis that hypoxic tissue damage may play a contributory role in L-NAME-induced limb defects.
Similar content being viewed by others

Main
NO is a colorless gaseous molecule involved in the regulation of diverse important physiologic processes, including vascular tone, platelet reactivity, smooth muscle contractility, neurotransmission, and the cytotoxic action of immune cells (1). NO is generated endogenously during the conversion of L-arginine to citrulline by a family of enzymes known as NOS. At least three isoforms of NOS have been isolated, including the constitutive eNOS and neuronal NOS isoforms, and the inducible isoform, iNOS (1). Pharmacologic manipulation of NOS activity can be achieved by several classes of NOS inhibitors including L-arginine analogs, which act by denying NOS its substrate in a competitive manner (2).
The L-arginine analog L-NAME, a nonselective inhibitor of NOS, has been shown to be teratogenic when administered to rats (3–8). Limb reduction defects with associated characteristic areas of macroscopic hemorrhage were the most common phenotypic alterations noted (3–8). L-NAME is not the only L-arginine analog known to cause dysmelia, as Nω-nitro-L-arginine also induced limb defects (9). Notably, disruptions similar to those induced by L-arginine analogs have been noted in knockout mice lacking eNOS (10, 11). The current state of knowledge on L-NAME teratogenicity also includes the following facts: 1) the period of fetal vulnerability has an onset relatively late in gestation (3–5); 2) both oral (3–6) and parenteral (5) treatments are effective; 3) limb dysmorphology is associated with reduced fetal viability (5); 4) both the teratogenic and fetus lethal effects are dose dependent (5); 5) defects are preceded by hemorrhages that appear in the limbs within hours of treatment (7, 8); 6) fetal growth restriction and decrement in placental weight have been observed by some authors (3, 4, 6), but not by others (5); 7) the compound is teratogenic when instilled in the amnion, suggesting that it exerts its activity within the vasculature of the limb or placenta (7); 8) limb defects were prevented by concurrent administration of L-arginine or the NO donors sodium nitroprusside and S-nitroso-acetyl-penicillamine (4), suggesting that L-NAME initiates teratology via inhibition of NO synthesis; and 9) limb defects were reduced by coadministration of α-phenyl-N-t-butylnitrone, a radical spin trap antioxidant, and allopurinol, a xanthine oxidase inhibitor (7, 8), suggesting a mechanistic role for radical oxygen species.
So far, teratogenic effects of L-arginine analogs have been investigated exclusively in the rat. Thus it is unknown whether or not the teratogenic response observed in this species exemplifies possible outcomes from the exposure of other animal species. In the present study the relationship between exposure to L-NAME and the induction of limb defects, with respect to stage specificity and dose dependency, was investigated in the mouse. In addition, because hypoxia and/or ischemia have been postulated by several authors as a possible mediator of L-NAME teratogenesis (3, 5, 7, 8), the ability of hyperoxia to prevent L-NAME-induced limb disruption was also investigated.
METHODS
Animals.
Pathogen-free ICR (CD-1) outbred mice were purchased from Harlan Italy (Udine). Animals were housed in suspended polycarbonate cages with stainless-steel tops containing heat-treated hardwood chips. Rodent laboratory food (Harlan Teklad) and tap water were provided ad libitum. The animal room was maintained at 22°C ± 1°C with a relative humidity of 55 ± 5% and a 12 h light/dark cycle, with the light on at 0800 h. Before the initiation of the study, mice were acclimatized to animal room conditions for a minimum of 7 d. Timed matings were obtained by overnight cohabitation of a single male with two to four nulliparous females during the dark cycle. The females were considered to be in gestation d 0 if a copulatory (vaginal) plug was found at the end of cohabitation (0800 h). Inseminated females were separated from the colony and housed singularly. All animal treatments and procedures were approved by the Institutional Review Board of the University “G. d'Annunzio” of Chieti.
Study 1 (L-NAME alone).
L-NAME (Research Biochemicals International, Natick, MA, U.S.A.) was dissolved in sterile saline solution and injected s.c. at 50 or 90 mg/kg to pregnant animals on gestation d 12, 13, 14, 15, or 16. Because of the elevated risk of accidental injection into the pregnant uterus (considering that the treatment was carried out during relatively advanced phases of gestation), the compound was injected s.c. instead of i.p. (the most commonly parenteral route of drug administration used in teratology studies). The volume of each injection was 10 mL/kg. The injection procedure was performed under light ethyl ether anesthesia. Doses and timing of administration were selected on the basis of preliminary experiments. A group of animals injected with vehicle on gestation d 14 under light ether anesthesia served as a control group. Pregnancies were terminated on gestation d 18 by an overdose of anesthesia. Maternal and pregnant uterine weights were recorded. The uterine contents were inspected, and the number of implants, resorptions, and viable and dead fetuses recorded. Live fetuses were evaluated for body and placental weights, sex, and external morphology. All the fetuses were then fixed in 95% alcohol and processed for double-staining skeletal examination using the methods of Inouye (12) and Kimmel and Trammel (13) as modified by Kuczuk and Scott (14). Standardized nomenclature for malformations was obtained from Wise et al. (15).
Study 2 (L-NAME plus hyperoxia).
Because study 1 showed that maximal teratogenic response occurs when pregnant animals are injected with the higher dose of L-NAME on gestation d 14, these experimental conditions were selected to investigate the effect of hyperoxia on L-NAME teratogenicity. Hyperoxic exposure was carried out as reported previously (16, 17). After L-NAME treatment, cages containing the pregnant animals (n = 21) were housed in a sealed Plexiglas chamber specifically designed for oxygen exposure, and 98 to 100% O2 (760 mm Hg; normobaric hyperoxia) was supplied to the chamber for 12 h. The chamber air volume was recirculated with a pump. CO2 was removed from the chamber air with Baralyme and continuously monitored by a capnograph. During the entire period of exposures, mice were allowed free access to food and water. Animals were periodically observed for signs of toxicity. L-NAME-treated mice (n = 14) breathing room air and handled comparably to the hyperoxic animals served as positive controls. Teratologic assessments were carried out as described in study 1.
Statistics.
Continuous data were compared by ANOVA and post hoc Student-Newman-Keuls test for multiple comparisons or by t test. Binomial data were compared using the Χ2 test with Yates's correction. Differences were considered significant when p < 0.05.
RESULTS
Study 1 (L-NAME alone).
There were no apparent signs of maternal toxicity associated with administration of L-NAME at 50 or 90 mg/kg, and no maternal deaths occurred during the study (not shown). Treated mothers did not weigh significantly less than controls (Table 1). Several L-NAME-treated animals delivered before the scheduled sacrifice day (Table 1). This effect was noted in one (12.5%) of the animals treated with 50 mg/kg on gestation d 16, and in one (7%) and six (60%) of the animals treated with 90 mg/kg on gestation d 15 and 16, respectively (Table 1). In all instances, preterm parturition occurred on gestation d 17 (not shown). In most instances, maternal cannibalism prevented the inspection of pretermdelivered fetuses.
There was a treatment-related increase of embryonic or fetal loss (Table 1). When L-NAME was administered at 50 mg/kg, a statistically significant reduction of embryonic or fetal viability, in comparison to the control group, was noted among litters exposed on gestation d 15 and 16 (Table 1). After treatment with L-NAME at 90 mg/kg, a more homogeneous response was seen, resulting this level of exposure in approximately a 7-fold increase of embryo or fetal lethality over the control frequency in all the experimental groups (Table 1). Statistical analysis did not reveal significant treatment-related effects on fetal and placental weights (Table 1).
As major phenotypical stigmata, L-NAME caused limb reduction defects. At external inspection, affected limbs most commonly exhibited digits of reduced size or digits with terminal segments missing (Fig. 1B). With increasing severity, the entire autopod and more proximal segments of limbs were missing. Subcutaneous hematomas were often seen in the immediate area of defective structures (Fig. 1C). In some instances, hemorrhagic areas were also observed in the genital tubercle and tail. There were no significant sex-related differences in the occurrence of limb anomalies (not shown). Left and right limbs were equally affected (not shown). No morphologic anomalies were noted in the control group. The skeletal preparations revealed limb defects that were undetected at external examination, and allowed an accurate characterization of limb dysmorphology. A detailed summary of type and frequency of the skeletal limb defects produced by LNAME exposure is provided in Table 2. Shortening of phalangeal bones and missing distal phalanges were the most common skeletal findings (Table 2 and Fig. 2C). Shortening of long bones (sometimes associated with abnormal curvature) was noted less frequently (Table 2 and Fig. 2D). In sporadic occasions, phalangeal or phalangeal-metacarpal fusion were also recorded (Table 2 and Fig. 2B). The appearance of autopod ossification centers was also delayed (Fig. 2B). L-NAME-induced limb teratogenesis was phase specific. A trend toward dose dependency was also noted. The impact of the level of exposure and time of administration on the prevalence of limb defects in ICR (CD-1) fetuses is shown in Figure 3. Although two of the fetuses (1.9%) exposed to 90 mg/kg on gestation d 12 had limb defects, a statistically significant level of dysmelia (5.4%) was first seen when the higher dose was administered on gestation d 13. Administration of L-NAME (90 mg/kg) on gestation d 14 yielded the maximum response, with 35.6% of fetuses affected. With advancing gestation, limb sensitivity decreased, and although a maximum of 10% of fetuses exposed on gestation d 15 had limb defects, there were no limb malformations after treatment on gestation d 16. Besides impacting on the frequency of limb defects, the timing of treatment also influenced defect location, with a trend toward a shift from forelimb to hindlimb becoming apparent with progression of gestation (Fig. 3, inset). Overall, the forelimb was more frequently affected than the hindlimb (Table 2).

Effect of L-NAME on the external limb morphology of ICR (CD-1) mouse fetuses on gestation d 18. A, normal right forelimb from a control fetus. B, treated right forelimb from a fetus dosed on gestation d 14 (90 mg/kg) with severe brachydactyly. C, treated right hindlimb from a fetus dosed on gestation d 15 (90 mg/kg) showing hemorrhagic lesions involving the second, third, and fourth digits.

Gestation d 18 limbs stained with alizarin red S (bone) and Alcian blue (cartilage) from fetuses exposed to L-NAME. A, normal left forelimb from a control fetus. B, treated left forelimb from a fetus dosed on gestation d 13 (50 mg/kg) showing absence of interphalangeal joints and complete lack of phalangeal ossification. C, treated right forelimbs from fetus dosed on gestation d 14 (90 mg/kg) with absence of distal phalanges. D, left, right forelimb from a control fetus; right, left forelimb from a fetus treated on gestation d 14 (90 mg/kg) showing severe shortening of the humerus, radius, and ulna.

Effects of the NOS inhibitor L-NAME on limb development in the ICR (CD-1) mouse. L-NAME (50 or 90 mg/kg) was administered s.c. on gestation d (GD) 12, 13, 14, 15, or 16. Control animals were injected s.c. with vehicle on gestation d 14. Teratologic assessments were carried out on gestation d 18. Numbers in parentheses refer to the number of affected fetuses the total number of fetuses examined. *p < 0.05 by Χ2 test vs control group. **p < 0.001 by Χ2 test vs other experimental groups. Inset, relative contribution of hind- and forelimb defects to the overall frequency of limb malformations.
Study 2 (L-NAME plus hyperoxia).
Mice subjected to the combined treatment L-NAME plus hyperoxia did not exhibit clinical signs of toxicity. However, one animal died 2 d after hyperoxic exposure. Hyperoxia provided protection from L-NAME-induced limb disruption by significantly (p < 0.001) reducing the frequency of fetuses with defective limbs caused by L-NAME treatment from 29% to 8% (Fig. 4). On the other hand, hyperoxia failed to prevent L-NAME-induced embryonic or fetal lethality, as inferred by the fact that comparable levels of postimplantation loss (27.94% versus 23.88% between control and hyperoxia-exposed fetuses, respectively) were noted in the two experimental groups (not shown).

Effects of hyperoxia on L-NAME-induced limb defects in the ICR (CD-1) mouse. On gestation d 14, animals (n = 21) were injected s.c. with L-NAME at 90 mg/kg and exposed to 98 to 100% O2 (760 mm Hg; normobaric hyperoxia) for 12 h. L-NAME-treated mice breathing room air (n = 14) served as positive controls. Teratologic assessments were carried out on gestation d 18. Numbers in parentheses refer to the number of affected fetuses the total number of fetuses examined. *p < 0.001 by Χ2 test vs control group.
DISCUSSION
The present study provides for the first time information regarding the teratogenic capacity of L-NAME in the mouse. Inhibitors of NO synthesis are currently under experimental evaluation for the treatment of several conditions associated with an overproduction of NO, including migraine, septic shock, inflammation, and neurodegenerative disorders (2). The need for an accurate characterization of the teratogenic potential of pharmacologic agents inhibiting NO synthesis is thus raised.
Studies with rats pointed out that the in vivo teratogenicity of L-NAME is confined largely to limbs (3–8). This was also our evidence in the mouse. In terms of severity, the rat appears to display a greater sensitivity to the teratogenic effects of LNAME than the mouse. Massive limb disruptions have been commonly observed in the rat (3–8), whereas defects have been in most occasions limited to digits in the present study. Another relevant interspecies difference relates to the topography of the defects, with forelimbs being the preferential site of disruption in the mouse and the opposite in the rat (3–6). However, it must be noted that, as shown by Fantel et al. (5), the experimental conditions used (parenteral versus oral administration) appear to play an important role in determining the site of limb disruption. Double-staining skeletal technique, a well-suited tool to investigate skeletal morphology, has been systematically applied in the present study (as this was not done in previous investigations) to fully characterize limb dysmorphology. Skeletal preparations revealed that, besides gross limb disruptions associated with hemorrhage, L-NAME can induce subtle dysmorphic features, including phalangeal-metacarpal hypoplasia, shortening of long bones of the limb, and fusion of skeletal elements. These findings expand the knowledge about the spectrum of limb anomalies induced by L-NAME.
Mouse limb organogenesis starts around gestation d 9 with the appearance of the forelimb bud, and is completed in most of its processes at about gestation d 15 (18). Only minor developmental events, including formation of nails (claws) and joint structures, occur during later stages, when phenomena like ossification of skeletal elements and growth of the differentiated limb structures predominate (18). As expected on the basis of the previous studies carried out in the rat (3–5), we found that the teratogenic sensitivity to L-NAME was restricted to the advanced stages of mouse limb development. Indeed, L-NAME became teratogenically operational on gestation d 13 and elicited its maximum effect (peaking strikingly) on gestation d 14, whereas no significant teratogenicity was observed when exposure occurred after gestation d 15. By using a single treatment day regimen, Fantel et al. (5) showed that rat limb sensitivity to L-NAME first appears on gestation d 16, a gestational time that roughly corresponds to the mouse gestation d 14.5 (18), and peaks on gestation d 17 and 18. Thus, it would appear that in the mouse the developmental phase of limb susceptibility has an earlier stage of onset than in the rat. After treatment on gestation d 16, a significant number of animals delivered before term. This outcome correlates well with our previous findings (19), further supporting the concept that NO may play a role in the control of myometrial contractility. Interestingly, L-NAME has never been reported to evoke preterm parturition in the rat (3–6, 20), possibly reflecting the existence of species-specific differences with respect to the role played by NO in myometrial regulation between the rat and the mouse.
The biologic determinant establishing the window of vulnerability to L-NAME is unknown. It has been postulated that the developmental timing of cationic amino acid transporters, which carry both arginine and L-NAME across the cell membrane, may play a role (7). We found it intriguing that the onset of limb vulnerability to L-NAME (gestation d 13) coincides in the mouse with the stage when developing forelimbs start being supplied with exclusively oxygenated blood (18). Before that time, when the left subclavian artery originates caudally to the site where the ductus arteriosus enters the descending aorta, and the right subclavian artery originates from the right dorsal aorta (a transient vascular structure), forelimbs receive a mixture of oxygenated and deoxygenated blood (18). Thus it would seem that when limb oxygen supply and demand increase, then the teratogenicity of the vasoactive agent L-NAME is favored.
There is mounting evidence that NO has important vasodilatory action in the fetal circulation as it does in the adult (21). Despite the apparent role of both eNOS and iNOS in regulating the fetal circulation (21), initiation of L-NAME-induced limb disruption seems to be primarily dependent on eNOS inhibition. This idea is supported by the lack of teratogenic effects of selective iNOS inhibitors (4, 6, 21), and by the evidence that limb defects similar to those induced by L-NAME have been observed in knockout mice lacking eNOS (10, 11).
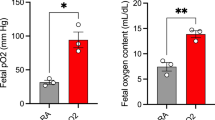
A remarkable aspect of our study relates to the fact that a 12-h exposure to hyperoxia (98 to 100% O2) significantly reduced the fetal frequencies of defective limbs caused by L-NAME. This observation, reported by us for the first time, appears to fit the postulation (3, 5, 7, 8) that tissue hypoxia plays a mechanistic role in L-NAME teratogenicity. It may be possible that L-NAME initiates limb defects by perturbing vascular homeostasis, thereby promoting reductions in vasculature perfusion at the utero-placental level or directly in the fetal vasculature, with resultant tissue hypoxia. The protection provided by hyperoxia may be interpreted as a compensatory effect. In keeping with this hypothesis, it is worth remembering that, as shown in studies carried out in sheep and humans, maternal hyperoxia can significantly increase fetal oxygen tension (22, 23). Coherently with a mechanistic involvement of hypoxia, hemorrhagic limb defects have been produced by subjecting fetal rats to a hypoxic environment (24) and, as pointed out in previous investigations (3–8), by exposing fetuses to vasoactive agents capable of uteroplacental perfusion reduction, including epinephrine (25, 26), cocaine (27), calcium-channel blockers (28, 29), phenytoin (30), and uterine vascular clamping (31, 32). Limb defects were also produced by multiple brief exposures of cultured rat embryos to hypoxia (33). On the other hand, maternal hyperoxia has been found to prevent hereditary congenital limb amputations and associated hemorrhages in the rabbit (34), and to reduce in the mouse the occurrence of craniofacial teratogenicity of phenytoin (35), an antiepileptic agent whose embryotoxicity has been causally linked to episodes of interrupted oxygen supply and possible generation of reactive oxygen species during reoxygenation (36).
Recent reports have shown that hyperoxic conditions can result in up-regulation of NOS (37–40). This notion raises the possibility that hyperoxia provided protection from L-NAME-induced teratogenesis by modulating NO synthesis, although evidence for oxygen-mediated NOS up-regulation in embryonic or fetal tissues is (to our knowledge) lacking. Interestingly, when the combined exposure L-NAME-hyperoxia took place beyond the prenatal period, deleterious effects, like increased mortality and pulmonary complications, have been observed (41). This suggests that hyperoxia may differently impact on the noxious effects of NOS inhibition during prenatal and postnatal life.
Unlike limb dysmorphology, hyperoxia did not impact on L-NAME-induced embryo or fetal lethality. This may suggest that reduced conceptal viability is not specific for hypoxic damage. In agreement with this idea, teratogenicity and embryo or fetal lethality showed significant differences with respect to the phase specificity, with in utero demise being only marginally modulated by the treatment timing.
The teratogenicity of L-NAME has been significantly reduced by coadministration of the radical spin trap antioxidant α-phenyl-N-t-butylnitrone, and the xanthine oxidase inhibitor allopurinol (7, 8). These notions, coupled with several other related findings (7, 8), recently led to the proposal that excessive generation of radical oxygen species represents the molecular mechanism whereby L-NAME teratogenicity is initiated. In the proposed chain of molecular events leading to free radical imbalance, hypoxia and possibly the subsequent reoxygenation would play a crucial role (8).
Abbreviations
- NO:
-
nitric oxide
- NOS:
-
nitric oxide synthase
- eNOS:
-
endothelial nitric oxide synthase
- iNOS:
-
inducible nitric oxide synthase
- L-NAME:
-
Nω-nitro-L-arginine methyl ester
REFERENCES
Murad F 1999 Cellular signaling with nitric oxide and cyclic GMP. Braz J Med Biol Res 32: 1317–1327
Hobbs AJ, Higgs A, Moncada S 1999 Inhibition of nitric oxide synthase as a potential therapeutic target. Annu Rev Pharmacol Toxicol 39: 191–220
Diket AL, Pierce MR, Munshi UK, Voelker CA, Eloby-Childress S, Greenberg SS, Zhang X-J, Clark DA, Miller MJS 1994 Nitric oxide inhibition causes intrauterine growth retardation and hind-limb disruption in rats. Am J Obstet Gynecol 171: 1243–1250
Pierce RL, Pierce MR, Liu H, Kadowitz PJ, Miller MJS 1995 Limb reduction defects after prenatal inhibition of nitric oxide synthase in rats. Pediatr Res 38: 905–911
Fantel AG, Nekahi N, Shepard TH, Cornel LM, Unis AS, Lemire RG 1997 The teratogenicity of NG-nitro-L-arginine methyl ester (L-NAME), a nitric oxide synthase inhibitor in rats. Reprod Toxicol 11: 709–717
Greenberg SS, Lancaster JR, Xie J, Sarphie TG, Zhao X, Hua L, Freeman T, Kapusta DR, Giles TD, Powers DR 1997 Effects of NO synthase inhibitors, arginine-deficient diet, and amiloride in pregnant rats. Am J Physiol 273:R1031–R1045
Fantel AG, Stamps LD, Tran TT, Mackler B, Person RE, Nekahi N 1999 Role of free radicals in the limb teratogenicity of L-NAME (NG-nitro-L-arginine methyl ester). Teratology 60: 151–160
Fantel AG, Person RE 2002 Further evidence for the role of free radicals in the limb teratogenicity of L-NAME. Teratology 66: 24–32
Salas SP, Altermatt F, Campos M, Giacaman A, Rosso P 1995 Effects of long-term nitric oxide synthesis inhibition in plasma volume expansion and fetal growth in the pregnant rat. Hypertension 26: 1019–1023
Gregg AR, Schauer A, Shi O, Liu Z, Lee CGL, O'Brien WE 1998 Limb reduction defects in endothelial nitric oxide synthase-deficient mice. Am J Physiol 275:H2319–H2324
Hefler LA, Reyes CA, O'Brien WE, Gregg AR 2001 Perinatal development of endothelial nitric oxide synthase-deficient mice. Biol Reprod 64: 666–673
Inouye M 1976 Differential staining of cartilage and bone in fetal mouse skeleton by Alcian blue and alizarin red S. Cong Anom 16: 171–173
Kimmel CA, Trammel C 1981 A rapid procedure for routine staining of cartilage and bone in fetal and adult animals. Stain Technol 56: 271–273
Kuczuk MJ, Scott WJ 1984 Potentiation of acetazolamide-induced ectrodactyly in SWV and C57/BL mice by cadmium sulphate. Teratology 29: 427–435
Wise LD, Beck SL, Beltrame D, Beyer BK, Chahoud I, Clark RL, Clark R, Druga AM, Feuston MH, Guittin P, Henwood SM, Kimmel CA, Lindstrom P, Palmer AK, Petrere JA, Solomon HM, Yasuda M, York RG 1997 Terminology of developmental abnormalities in common laboratory mammals (version 1). Teratology 55: 249–292
Amicarelli A, Di Ilio C, Masciocco L, Bonfigli A, Zarivi O, D'Andrea MR, Di Giulio C, Miranda M 1997 Aging and detoxifying enzymes responses to hypoxic or hyperoxic treatment. Mech Ageing Dev 97: 215–226
Spoto G, Di Giulio C, Contento A, Di Stilio M 1998 Hypoxic and hyperoxic effect on blood phosphodiesterase activity in young and old rats. Life Sci 63: 349–353
Kaufman MH, Bard JBL 1999 The Anatomical Basis of Mouse Development. Academic Press, San Diego, pp 93–108
Tiboni GM, Giampietro F 2000 Inhibition of nitric oxide synthesis causes preterm delivery in the mouse. Human Reprod 15: 1838–1842
Yalampalli C, Garfield RE 1993 Inhibition of nitric oxide synthesis in rats during pregnancy produced signs similar to those of preeclampsia. Am J Obstet Gynecol 169: 1316–1320
Bustamante SA, Pang Y, Romero S, Pierce MR, Voelker CA, Thompson JH, Sandoval M, Liu X, Miller MJS 1996 Inducible nitric oxide synthase and the regulation of central vessels caliber in the fetal rat. Circulation 94: 1948–1953
Rankin JHG, Meschia G, Makowski EL, Battaglia FC 1971 Relationship between uterine and umbilical PO2 in sheep. Am J Physiol 220: 1688–1692
Nicolaides KH, Campbell S, Bradley RJ, Bilardo CM, Soothill PW, Gibb D 1987 Maternal oxygen therapy for intrauterine growth retardation. Lancet 25: 942–945
Petter C, Bourbon J, Maltier JP, Jost A 1971 Production of hemorrhages of the extremities in the rat fetuses subjected to hypoxia in utero. C R Acad Sci Hebd Seances Acad Sci Ser D Sci Nat 272: 2488–2490
Jost A 1953 La dégénerescence des extémités du foetus de rat sous des actions hormonales (acroblapsie expérimentale) et la théorie des bulles myélencéphaliques de Bonnevie. Arch Franc Pediatr 10: 865–870
Jost A, Roffi J, Cowitat M 1969 Congenital amputations determined by the br gene and those induced by adrenaline injection in the rabbit fetus. In: Swinyard CA (eds) Limb Development and Deformity: Problems of Evaluation and Rehabilitation. Thomas, Springfield, pp 187–199
Webster WS, Brown-Woodman PDC, Lipson AH, Ritchie HE 1991 Fetal brain damage in the rat following prenatal exposure to cocaine. Neurotoxicol Teratol 13: 621–626
Danielsson BRG, Reiland S, Rundqvist E, Danielson M 1989 Digital defects induced by vasodilating agents: relationship to uteroplacental blood flow. Teratology 40: 351–358
Danielsson BRG, Danielson M, Reiland S, Rundqvist E, Dencker L, Regard CG 1990 Histological and in vitro studies supporting decreased uteroplacental blood flow as explanation for digital defects after administration of vasodilators. Teratology 41: 185–193
Danielsson BRG, Danielson M, Rundqvist E, Reiland S 1992 Identical phalangeal defects induced by phenytoin and nifedipine suggest fetal hypoxia and vascular disruption behind phenytoin teratogenicity. Teratology 45: 247–258
Brent RL, Franklin JB 1960 Uterine vascular clamping: new procedure for the study of congenital malformations. Science 132: 89–91
Leist KH, Grauwiler J 1974 Fetal pathology following uterine-vessel clamping on day 14 of gestation. Teratology 10: 55–68
Fantel AG, Barber CV, Carda MB, Tumbic RW, Mackler B 1992 Studies of the role of ischemia/reperfusion and superoxide anion radical production in the teratogenicity of cocaine. Teratology 46: 293–300
Petter C, Bourbon J, Maltier JP, Jost A 1971 Prevention of hereditary congenital amputations in rabbits with maternal hyperoxia. C R Acad Sci Hebd Seances Acad Sci Ser D Sci Nat 273: 2639–2642
Millicovsky G, Johnston MC 1981 Maternal hyperoxia greatly reduces the incidence of phenytoin-induced cleft lip and palate in A/J mice. Science 212: 671–672
Danielsson BR, Skold AC, Azarbayjani F 2001 Class III antiarrhythmics and phenytoin: teratogenicity due to embryonic cardiac dysrhythmia and reoxygenation damage, Curr Pharm Des 7: 787–802
Potter CF, Kuo NT, Farver CF, McMahon JT, Chang CH, Agani FH, Haxhiu MA, Martin RJ 1999 Effects of hyperoxia on nitric oxide synthase expression, nitric oxide activity, and lung injury in rat pups. Pediatr Res 45: 8–13
North AJ, Lau KS, Brannon TS, Wu LC, Wells LB, German Z, Shaul PW 1996 Oxygen upregulates nitric oxide synthase gene expression in ovine fetal pulmonary artery endothelial cells. Am J Physiol 270:L643–L649
Steudel W, Watanabe M, Dikranian K, Jacobson M, Jones RC 1999 Expression of nitric oxide synthase isoforms (NOS II and NOS III) in adult rat lung in hyperoxic pulmonary hypertension. Cell Tissue Res 295: 317–329
Chang L, Ma L, Zhang X, Chen Y 2001 The role of nitric oxide in hyperoxic lung injury in premature rats. J Tongji Med Univ 21: 78–81
Pierce MR, Voelker CA, Sosenko IRS, Bustamante SA, Olister SM, Zhang X-J, Clark DA, Miller MJS 1995 Nitric oxide synthase inhibition decreases tolerance to hyperoxia in newborn rats. Mediat Inflamm 4: 431–436
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tiboni, G., Giampietro, F. & di Giulio, C. The Nitric Oxide Synthesis Inhibitor Nω-Nitro-L-Arginine Methyl Ester (L-NAME) Causes Limb Defects in Mouse Fetuses: Protective Effect of Acute Hyperoxia. Pediatr Res 54, 69–76 (2003). https://doi.org/10.1203/01.PDR.0000069840.78984.76
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000069840.78984.76
This article is cited by
-
Prevention of neural tube defects by loss of function of inducible nitric oxide synthase in fetuses of a mouse model of streptozotocin-induced diabetes
Diabetologia (2009)
-
Impact of maternal endothelial nitric oxide synthase gene polymorphisms on blood pressure, protein excretion and fetal outcome in pregnancy
Journal of Human Hypertension (2008)
